The Role of Co-Stimulatory Molecules in Chagas Disease

Abstract
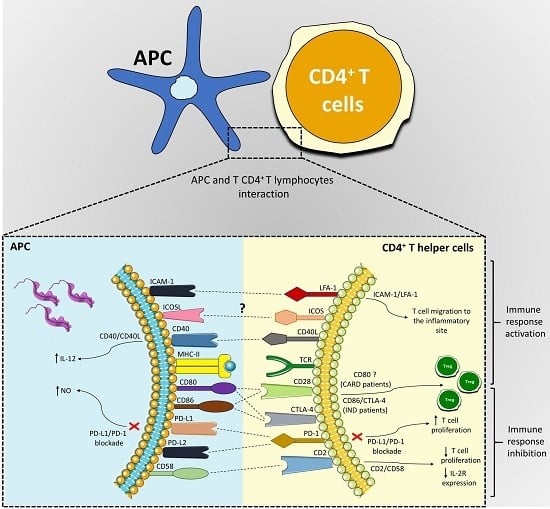
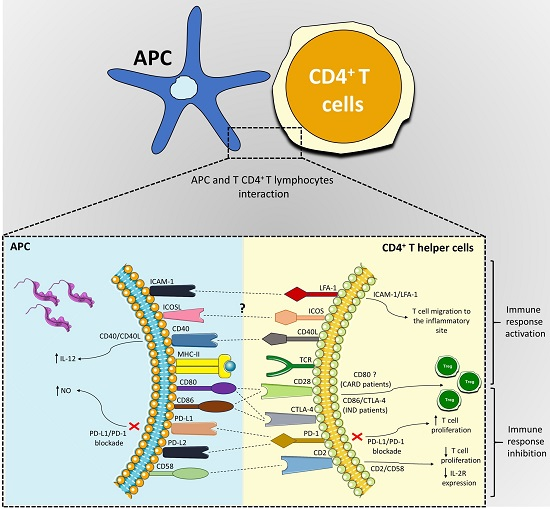
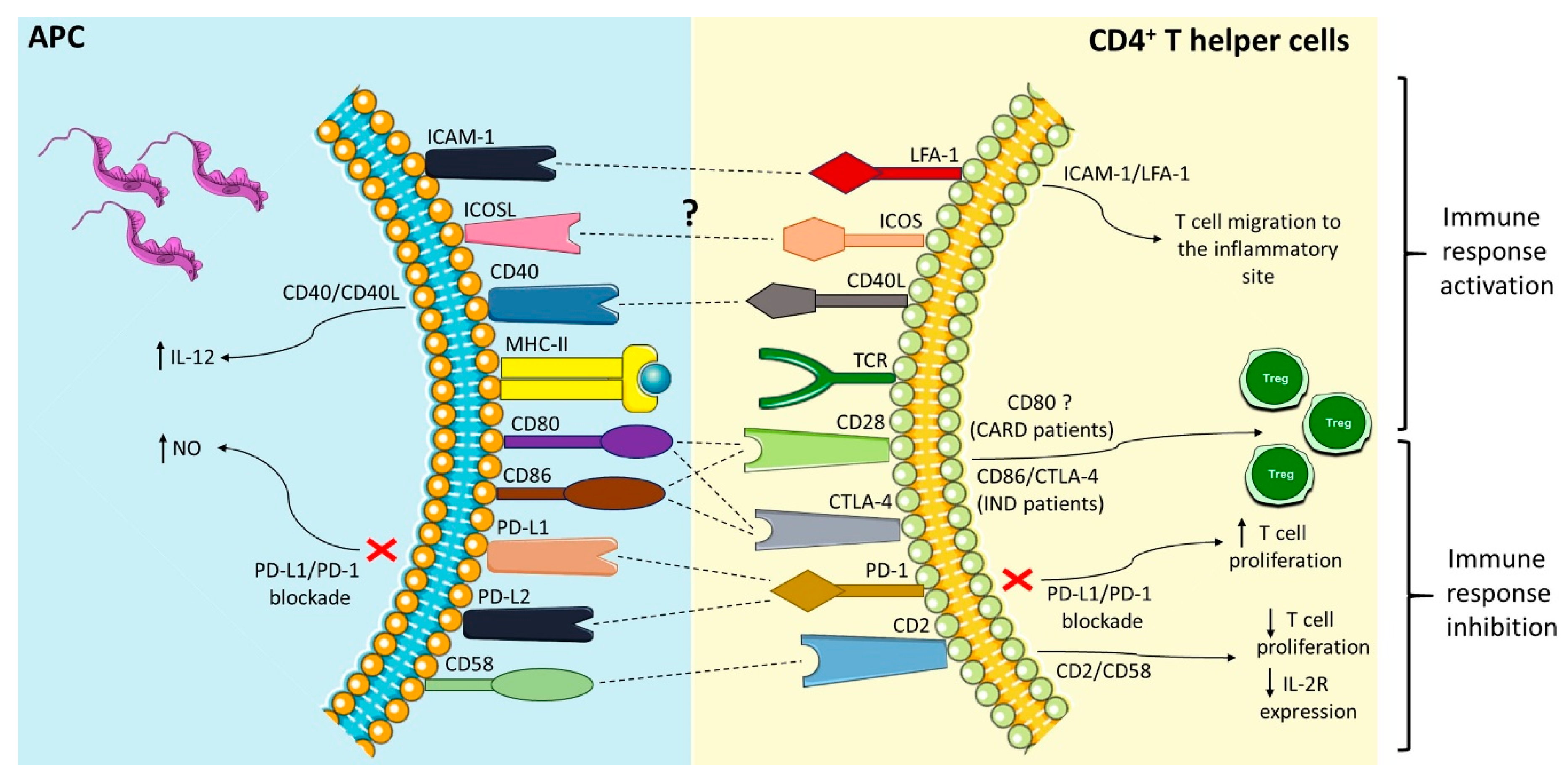
:
1. Introduction: A Brief Overview of Chagas Disease
2. Co-Stimulatory Molecules and Their Role in Chagas Disease
2.1. PD-L1, PD-L2, and PD-1 Receptors
2.2. ICOSL and ICOS Receptors
2.3. Tumor Necrosis Factor Family
2.4. CD2 and CD58 Receptors
2.5. LFA-1 and ICAM-1 Receptors
2.6. Role of CD28, CTLA-4, CD80, and CD86 Co-Stimulatory Molecules in the Pathogenesis of Chagas Disease
3. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Álvares, J.M.; Fonseca, R.; Silva, H.B.; Marinho, C.R.F.; Bortoluci, K.R.; Sardinha, L.R.; Epiphanio, S.; Lima, M.R.I. Chagas disease: Still many unsolved issues. Mediat. Inflamm. 2014, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Investing to Overcome the Global Impact of Neglected Tropical Diseases: Third WHO Report on Neglected Diseases 2015; World Health Organization: Geneva, Switzerland, 2015; pp. 1–211. [Google Scholar]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Seminar 2017. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Chagas Disease (American Trypanosomiasis). Available online: http://www.who.int/chagas/disease/en/ (accessed on 9 April 2018).
- Dias, J.C.P.; Ramos, A.N., Jr.; Gontijo, E.D.; Luquetti, A.; Shikanai-Yasusa, M.A.; Coura, J.R.; Torres, R.M.; Melo, J.R.C.; Alemida, E.A.; Oliveira, W., Jr.; et al. Consenso brasileiro em doença de Chagas. Epidemiol. Serv. Saúde 2016, 25, 7–86. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, Y.; Nakagawa, J. Control of Chagas disease. Adv. Parasitol. 2006, 61, 129–165. [Google Scholar] [CrossRef] [PubMed]
- Iniciativa de Salud del Cono Sur, Incosur. XIV Reunión de la Comisión Intergubernamental del Cono Sur para la Eliminación de Triatoma Infestans y la Interrupción de la Transmisión de la Tripanosomiasis Transfusional y Curso de Diagnóstico, Manejo y Tratamiento de Enfermedad de Chagas; OPS/MSF: Santa Cruz de la Sierra, Bolivia, 2005; ISBN 9974-7945-3-6. [Google Scholar]
- Schmunis, G.A.; Yadon, Z.E. Chagas disease: A latin american health problem becoming a world health problem. Acta Trop. 2010, 115, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Coura, J.R.; Viñas, P.A.; Junqueira, A.C.V. Ecoepidemiology, short history and control of Chagas disease in the endemic countries and the new challenge for non-endemic countries. Mem. Inst. Oswaldo Cruz 2014, 109, 856–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayo, C.M.; Dalalio, M.M.O.; Visentainer, J.E.L.; Reis, P.G.; Sippert, E.A.; Jarduli, L.R.; Alves, H.V.; Sell, A.M. Genetic Susceptibility to Chagas disease: An overview about the infection and about the association between disease and the immune response genes. Biomed. Res. Int. 2013, 2013, 284729. [Google Scholar] [CrossRef] [PubMed]
- Andrade, D.V.; Gollob, K.J.; Dutra, W.O. Acute Chagas disease: New global challenges for an old Neglected disease. PLoS Negl. Trop. Dis. 2014, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Prata, A. Classificação da infecção chagásica no homem. Rev. Soc. Bras. Med. Trop. 1990, 23, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Brener, Z.; Gazzinelli, R.T. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas’ disease. Int. Arch. Allergy Immunol. 1997, 114, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.R.L.; Hecht, M.M.; Guimaro, M.C.; Sousa, A.O.; Nitz, N. Pathogenesis of Chagas’ disease: Parasite persistence and autoimmunity. Clin. Microbiol. Rev. 2011, 24, 592–630. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.S.; Hardoim, D.J.; Souza, C.S.F.; Cardoso, F.O.; Mendes, V.G.; Previtalli-Silva, H.; Abreu-Silva, A.L.; Pelajo-Machado, M.; Costa, S.C.G.; Calabrese, K.S. Oral outbreak of Chagas disease in Santa Catarina, Brazil: Experimental evaluation of a patient’s strain. PLoS ONE 2015, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dario, M.A.; Rodrigues, M.S.; Barros, J.H.S.; Xavier, S.C.C.; D’Andrea, O.S.; Roque, A.L.R.; Jansen, A.M. Ecological scenario and Trypanosoma cruzi DTU characterization of a fatal acute Chagas disease case transmitted orally (Espírito Santo state, Brazil). Parasit. Vectors 2016, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ministério da Saúde. Doença de Chagas Aguda-Casos Confirmados Notificados no Sistema de Informação de Agravos de Notificação-Brasil. Available online: http://tabnet.datasus.gov.br/cgi/deftohtm.exe?sinannet/cnv/chagasbr.def (accessed on 27 March 2018).
- Ribeiro, A.L.P.; Nunes, M.P.; Teixeira, M.M.; Rocha, M.O. Diagnosis and management of Chagas disease and cardiomyopathy. Nat. Rev. Cardiol. 2012, 9, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Steverding, D. The history of Chagas disease. Parasit. Vectors 2014, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, A.L.P.; Rocha, M.O. Forma indeterminada da doença de Chagas: Considerações acerca do diagnóstico e do prognóstico. Rev. Soc. Bras. Med. Trop. 1998, 31, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Mathews, J.C. Valor de la Prueba de Esfuerzo Graduado (Ergometria) para Determinar la Capacidad Laboral del Cardiópata Chagásico Crônico. Ph.D. Thesis, Universidad Nacional de Córdoba, Córdoba, Argentina, 1973. [Google Scholar]
- Macêdo, V. Indeterminate form of Chagas disease. Mem. Inst. Oswaldo Cruz 1999, 94, 311–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, L.G.; Santos, R.H.B.; Fiorelli, A.I.; Mairena, E.C.; Benvenuti, L.A.; Bocchi, E.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E. Myocardial gene expression of T-bet, GATA-3, Ror-γt, FoxP3, and hallmark cytokines in chronic Chagas disease cardiomyopathy: An essentially unopposed TH1-Type Response. Mediat. Inflamm. 2014, 1–9. [Google Scholar] [CrossRef]
- Malik, L.H.; Singh, G.D.; Amsterdam, E.A. Chagas heart disease: An update. Am. J. Med. 2015, 128. [Google Scholar] [CrossRef] [PubMed]
- Andrade, Z.A. A Patologia da doença de Chagas no Homem. Ann. Soc. Belg. Med. Trop. 1985, 65, 15–30. [Google Scholar] [PubMed]
- Guedes, P.M.M.; Veloso, V.M.; Afonso, L.C.C.; Caliari, M.V.; Carneiro, C.M.; Diniz, L.F.; Marques-da-Silva, E.A.; Caldas, I.S.; Matta, M.A.V.; Souza, S.M.; et al. Development of chronic cardiomyopathy in canine Chagas disease correlates with high IFN-γ, TNF-α, and low IL-10 production during the acute infection phase. Vet. Immunol. Immunopathol. 2009, 130, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Frade, A.F.; Teixeira, P.C.; Ianni, B.M.; Pissetti, C.W.; Saba, B.; Wang, L.H.T.; Kuramoto, A.; Nogueira, L.G.; Buck, P.; Dias, F.; et al. Polymorphism in the alpha cardiac muscle actin 1 gene is associated to susceptibility to chronic inflammatory cardiomyopathy. PLoS ONE 2013, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Stanaway, J.D.; Roth, G. The burden of Chagas disease: Estimates and challenges. Glob. Heart 2015, 10, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Brener, Z.; Andrade, Z.A. Trypanosoma cruzi e doença de Chagas. In Epidemiologia; Koogan, G., Ed.; Livros de Texto: Rio de Janeiro, Brazil, 1979; pp. 89–151. [Google Scholar]
- Rezende, J.M.; Moreira, H. Forma digestiva da doença de Chagas. In Gastroenterologia; Castro, L.P., Coelho, L.G.V., Eds.; Medsi: Rio de Janeiro, Brazil, 2004; pp. 325–392. [Google Scholar]
- Malik, L.H.; Singh, G.D.; Amsterdam, E.A. The epidemiology, clinical manifestations, and management of chagas heart disease. Clin. Cardiol. 2015, 38, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.A.S.; Bahia-Oliveira, L.M.G.; Rocha, M.O.C.; Martins-Filho, O.A.; Gazzinelli, G.; Correa-Oliveira, R. Evidence that development of severe cardiomyopathy in human Chagas’ disease is due to a Th1-specific immune response. Infect. Immun. 2003, 71, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Savino, W.; Villa-Verdea, D.M.S.; Mendes-da-Cruza, D.A.; Silva-Monteiro, E.; Perez, A.R.; Aoki, M.P.; Bottasso, O.; Guiñazú, N.; Silva-Barbosa, S.D.; Gea, S. Cytokines and cell adhesion receptors in the regulation of immunity to Trypanosoma cruzi. Cytokine Growth Factor Rev. 2007, 18, 107–124. [Google Scholar] [CrossRef] [PubMed]
- Chaves, A.T.; Gomes, J.A.S.; Fiuza, J.A.; Carvalho, A.T.; Ferreira, K.S.; Fares, R.C.G.; Guimarães, P.H.G.; Fagundes, S.E.M.; Morato, M.J.; Fujiwara, R.T.; et al. Immunoregulatory mechanisms in Chagas disease: Modulation of apoptosis in T-cell mediated immune responses. BMC Infect. Dis. 2016, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.A.S.; Molica, A.M.; Keesen, T.S.L.; Morato, M.J.F.; De Araújo, F.F.; Fares, R.C.G.; Fiuza, J.Á.; Chaves, A.T.; Pinheiro, V.; Nunes, M.C.P.; et al. Inflammatory mediators from monocytes down-regulate cellular proliferation and enhance cytokines production in patients with Polar clinical forms of Chagas disease. Hum. Immunol. 2013, 75, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, C.; Caetano, B.; Bartholomeu, D.C.; Melo, M.B.; Ropert, C.; Rodrigues, M.M.; Gazzinelli, R.T. The endless race between Trypanosoma cruzi and host immunity: Lessons for and beyond Chagas disease. Expert Rev. Mol. Med. 2010, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Gravina, H.D.; Antonelli, L.; Gazzinelli, R.T.; Ropert, C. Differential use of TLR2 and TLR9 in the regulation of immune responses during the infection with Trypanosoma cruzi. PLoS ONE 2013, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bartholomeu, D.C.; Ropert, C.; Melo, M.B.; Parroche, P.; Junqueira, C.F.; Teixeira, S.M.; Sirois, C.; Kasperkovitz, P.; Knetter, C.F.; Lien, E.; et al. Recruitment and endo-lysosomal activation of TLR9 in dendritic cells infected with Trypanosoma cruzi. J. Immunol. 2008, 181, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Souza, P.E.A.; Rocha, M.O.C.; Menezes, C.A.S.; Coelho, J.S.; Chaves, A.C.L.; Gollob, K.J.; Dutra, W.O. Trypanosoma cruzi infection induces differential modulation of costimulatory molecules and cytokines by monocytes and T cells from patients with indeterminate and cardiac Chagas’ disease. Infect. Immun. 2007, 75, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Cunha-Neto, E.; Nogueira, L.G.; Teixeira, P.C.; Ramasawmy, R.; Drigo, S.A.; Goldberg, A.C.; Fonseca, S.G.; Bilate, A.M.; Kalil, J. Immunological and non-immunological effects of cytokines and chemokines in the pathogenesis of chronic Chagas disease cardiomyopathy. Mem. Inst. Oswaldo Cruz 2009, 104, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, D.B.R.; Reis, M.A.; Romano, A.; Pereira, S.A.L.; Teixeira, V.P.A.; Junior, S.T.; Rodrigues, V., Jr. In situ expression of regulatory cytokines by heart inflammatory cells in Chagas’ disease patients with heart failure. Clin. Dev. Immunol. 2012, 2012, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Basso, B. Modulation of immune response in experimental Chagas disease. World J. Exp. Med. 2013, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Araújo, F.F.; Corrêa-Oliveira, R.; Rocha, M.O.C.; Chaves, A.T.; Fiuza, J.A.; Fares, R.C.G.; Ferreira, K.S.; Nunes, M.C.P.; Keesen, T.S.; Damasio, M.P.S.; et al. Foxp3+CD25highCD4+ regulatory T cells from indeterminate patients with Chagas disease can suppress the effector cells and cytokines and reveal altered correlations with disease severity. Immunobiology 2012, 217, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.S.; Lorena, V.M.B.; Braz, S.C.M.; Docenab, C.; Gomes, Y.M. IL-10 and IFN-γ gene expression in chronic Chagas disease patients after in vitro stimulation with recombinant antigens of Trypanosoma cruzi. Cytokine 2012, 58, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, L.M.D.; Villani, F.N.A.; Nunes, M.C.P.; Gollob, K.J.; Rocha, M.O.C.; Dutra, W.O. High interleukin 17 expression is correlated with better cardiac function in human Chagas disease. J. Infect. Dis. 2013, 207, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Dutra, W.O.; Menezes, C.A.S.; Magalhães, L.M.D.; Gollob, K.J. Immunoregulatory networks in human Chagas disease. Parasite Immunol. 2014, 36, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.E.A.; Rocha, M.O.C.; Rocha-Vieira, E.; Menezes, C.A.S.; Chaves, A.C.L.; Gollob, K.J.; Dutra, W.O. Monocytes from patients with indeterminate and cardiac forms of Chagas’ disease display distinct phenotypic and functional characteristics associated with morbidity. Infect. Immun. 2004, 72, 5283–5291. [Google Scholar] [CrossRef] [PubMed]
- Dutra, W.O.; Menezes, C.A.; Villani, F.N.; da Costa, G.C.; da Silveira, A.B.; Reis, D.D.; Gollob, K.J. Cellular and genetic mechanisms involved in the generation of protective and pathogenic immune responses in human Chagas disease. Mem. Inst. Oswaldo Cruz 2009, 104, 208–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, B.F.; Medeiros, N.I.; Teixeira-Carvalho, A.; Eloi-Santos, S.M.; Fontes-Cal, T.C.M.; Rocha, D.A.; Dutra, W.O.; Correa-Oliveira, R.; Gomes, J.A.S. CD86 expression by monocytes influences an immunomodulatory profile in asymptomatic patients with chronic Chagas disease. Front. Immunol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.R.; Freeman, G.J. The B7–CD28 superfamily. Nat. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Salomon, B.; Bluestone, J.A. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 2001, 19, 225–252. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, A.R. Mechanisms of costimulation. Immunol. Rev. 2009, 229, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansom, D.M. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef]
- Nishimura, H.; Honjo, T. PD-1: An inhibitory immunoreceptor involved in peripheral tolerance. Trends Immunol. 2001, 22, 265–268. [Google Scholar] [CrossRef]
- Schachtele, S.J.; Hu, S.; Sheng, W.S.; Mutnal, M.B.; Lokensgard, J.R. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia 2014, 62, 1582–1594. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Walsh, A.M.; Canavan, M.; Wechalekar, M.D.; Cole, S.; Yin, X.; Scott, B.; Loza, M.; Orr, C.; McGarry, T.; et al. Immune checkpoint inhibitor PD-1 pathway is down-regulated in synovium at various stages of rheumatoid arthritis disease progression. PLoS ONE 2018, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Agata, Y.; Kawasaki, A.; Nishimura, H.; Ishida, Y.; Tsubata, T.; Yagita, H.; Honjo, T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int. Immunol. 1996, 8, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stempin, C.C.; Marquez, J.D.R.; Ana, Y.; Cerban, F.M. GRAIL and Otubain-1 are related to T cell hyporesponsiveness during Trypanosoma cruzi infection. PLoS Negl. Trop. Dis. 2017, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, T.A.; Silva, M.V.; Mendes, M.T.; Carvalho-Costa, T.M.; Batista, L.R.; Lages-Silva, E.; Rodrigues, V.; Oliveira, C.J.; Ramirez, L.E. Immunomodulation by Trypanosoma cruzi: Toward understanding the association of dendritic cells with infecting TcI and TcII populations. J. Immunol. Res. 2014, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dias, F.C.; Medina, T.S.; Mendes-Junior, C.T.; Dantas, R.O.; Pissetti, C.W.; Junior, V.R.; Dellalibera-Joviliano, R.; Marin-Neto, J.A.; Gutierrez, F.R.S.; Moreau, P.; et al. Polymorphic sites at the immunoregulatory CTLA-4 gene are associated with chronic Chagas disease and its clinical manifestations. PLoS ONE 2013, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lasso, P.; Mateus, J.; Pavía, P.; Rosas, F.; Roa, N.; Thomas, M.C.; López, M.C.; González, J.M.; Puerta, C.J.; Cuéllar, A. Inhibitory receptor expression on CD8+ T cells is linked to functional responses against Trypanosoma cruzi antigens in chronic chagasic patients. J. Immunol. 2015, 195, 3748–3758. [Google Scholar] [CrossRef] [PubMed]
- Borges, D.C.; Araújo, N.M.; Cardoso, C.R.; Chica, J.E.L. Different parasite inocula determine the modulation of the immune response and outcome of experimental Trypanosoma cruzi infection. Immunology 2012, 138, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, F.R.S.; Mariano, F.S.; Oliveira, C.J.F.; Pavanelli, W.R.; Guedes, P.M.M.; Silva, G.K.; Campanelli, A.P.; Milanezi, C.M.; Azuma, M.; Honjo, T.; et al. Regulation of Trypanosoma cruzi-induced myocarditis by programmed death cell receptor. Infect. Immun. 2011, 79, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Dulgerian, L.R.; Garrido, V.V.; Stempin, C.C.; Cerbán, F.M. Programmed death ligand 2 regulates arginase induction and modifies Trypanosoma cruzi survival in macrophages during murine experimental infection. Immunology 2011, 133, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Cassiano, G.C.; Santos, E.J.M.; Maia, M.H.T.; Furini, A.C.; Storti-Melo, L.M.; Tomaz, F.M.B.; Trindade, P.C.A.; Capobianco, M.P.; Amador, M.A.T.; Viana, G.M.R.; et al. Impact of population admixture on the distribution of immune response costimulatory genes polymorphisms in a Brazilian population. Hum. Immunol. 2015, 76, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Amatore, F.; Gorvel, L.; Olive, D. Inducible Co-Stimulator (ICOS) as a potential therapeutic target for anti-cancer therapy. Expert Opin. Ther. Target. 2018, 22, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Kovaleva, M.; Johnson, K.; Steven, J.; Barelle, C.J.; Porter, A. Therapeutic potential of shark Anti-ICOSL VNAR domains is exemplified in a murine model of autoimmune non-infectious uveitis. Front. Immunol. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brodie, D.; Collins, A.V.; Iaboni, A.; Fennelly, J.A.; Sparks, L.M.; Xu, X.-N.; van der Merwe, P.A.; Davis, S.J. LICOS, a primordial costimulatory ligand? Curr. Biol. 2000, 10, 333–336. [Google Scholar] [CrossRef]
- Yao, S.; Zhu, Y.; Chen, L. Advances in targeting cell surface signalling molecules for immune modulation. Nat. Rev. Drug Discov. 2013, 12, 130–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.; Juedes, A.E.; Temann, U.A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Villegas, E.N.; Elloso, M.M.; Reichmann, G.; Peach, R.; Hunter, C.A. Role of CD28 in the generation of effector and memory responses required for resistance to Toxoplasma gondii. J. Immunol. 1999, 163, 3344–3353. [Google Scholar] [PubMed]
- Villegas, E.N.; Lieberman, L.A.; Mason, N.; Blass, S.L.; Zediak, V.P.; Peach, R.; Horan, T.; Yoshinaga, S.; Hunter, C.A. A role for inducible costimulator protein in the CD28-independent mechanism of resistance to Toxoplasma gondii. J. Immunol. 2002, 169, 937–943. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, R.J.; McAdam, A.J.; Woude, D.V.; Satoskar, A.R.; Sharpe, A.H. Cutting edge: Inducible costimulator protein regulates both Th1 and Th2 responses to cutaneous leishmaniasis. J. Immunol. 2002, 168, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Dempsey, P.W.; Doyle, S.E.; He, J.Q.; Cheng, G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003, 14, 193–209. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Chung, J.Y.; Park, Y.C.; Ye, H.; Wu, H. All TRAFs are not created equal: Common and distinct molecular mechanisms of TRAF-mediated signal transduction. J. Cell Sci. 2002, 115, 679–688. [Google Scholar] [PubMed]
- Watts, T.H. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005, 23, 23–68. [Google Scholar] [CrossRef] [PubMed]
- Ward-Kavanagh, L.; Lin, W.W.; Šedý, J.S.; Ware, C.F. The TNF receptor superfamily in costimulating and coinhibitory responses. Immunity 2016, 44, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci. 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Vanarsdale, T.L.; Vanarsdale, S.L.; Force, W.R.; Walter, B.N.; Mosialos, G.; Kieff, E.; Reed, J.C.; Ware, C.F. Lymphotoxin-β receptor signaling complex: Role of tumor necrosis factor receptor-associated factor 3 recruitment in cell death and activation of nuclear factor kβ. Immunology 1997, 94, 2460–2465. [Google Scholar] [CrossRef]
- Kollias, G.; Kontoyiannis, D. Role of TNF/TNFR in autoimmunity: Specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev. 2002, 315–321. [Google Scholar] [CrossRef]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Shimozato, O.; Takeda, K.; Yagita, H.; Okumura, K. Expression of CD30 ligand (CD153) on murine activated T cells. Biochem. Biophys. Res. Commun. 1999, 256, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Gramaglia, I.; Cooper, D.; Miner, K.T.; Kwon, B.S.; Croft, M. Co-stimulation of antigen-specific CD4 T cells by 4-1BB ligand. Eur. J. Immunol. 2000, 30, 392–402. [Google Scholar] [CrossRef]
- Martínez Gómez, J.M.; Koh, V.H.; Yan, B.; Lin, W.; Ang, M.L.; Rahim, S.Z.; Pethe, K.; Schwarz, H.; Alonso, S. Role of the CD137 ligand (CD137L) signaling pathway during Mycobacterium tuberculosis infection. Immunobiology 2014, 219, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, K.; Borst, J. Targeting the T-cell co-stimulatory CD27/CD70 pathway in cancer immunotherapy: Rationale and potential. Immunotherapy 2015, 7, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Planelles, L.; Thomas, M.C.; Marañón, C.; Morell, M.; López, M.C. Differential CD86 and CD40 co-stimulatory molecules and cytokine expression pattern induced by Trypanosoma cruzi in APCs from resistant or susceptible mice. Clin. Exp. Immunol. 2003, 131, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ayala, M.A.; Casasco, A.; González, M.; Postan, M.; Corral, R.S.; Petray, P.B. Trypanosoma cruzi infection induces the expression of CD40 in murine cardiomyocytes favoring CD40 ligation-dependent production of cardiopathogenic IL-6. Parasitol. Res. 2016, 115, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Chaussabel, D.; Jacobs, F.; de Jonge, J.; de Veerman, M.; Carlier, Y.; Thielemans, K.; Goldman, M.; Vray, B. CD40 ligation prevents Trypanosoma cruzi infection through interleukin-12 upregulation. Infect. Immun. 1999, 67, 1929–1934. [Google Scholar] [PubMed]
- Abel, L.C.; Ferreira, L.R.; Cunha Navarro, I.; Baron, M.A.; Kalil, J.; Gazzinelli, R.T.; Rizzo, L.V.; Cunha-Neto, E. Induction of IL-12 production in human peripheral monocytes by Trypanosoma cruzi is mediated by glycosylphosphatidylinositol-anchored mucin-like glycoproteins and potentiated by IFN-γ and CD40-CD40L interactions. Mediat. Inflamm. 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Agarwal, R.; Murugaiyan, G.; Saha, B. CD40 expression levels modulate regulatory T cells in Leishmania donovani infection. J. Immunol. 2010, 185, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Kamanaka, M.; Yu, P.; Yasui, T.; Yoshida, K.; Kawabe, T.; Horii, T.; Kishimoto, T.; Kikutani, H. Protective role of CD40 in Leishmania major infection at two distinct phases of cell-mediated immunity. Immunity 1996, 4, 275–281. [Google Scholar] [CrossRef]
- Tuladhar, R.; Natarajan, G.; Satoskar, A.R. Role of co-stimulation in Leishmaniasis. Int. J. Biol. Sci. 2011, 7, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.A.; Ovendale, P.J.; Kennedy, M.K.; Fanslow, W.C.; Reed, S.G.; Maliszewski, C.R. CD40 ligand is required for protective cell-mediated immunity to Leishmania major. Immunity 1996, 4, 283–289. [Google Scholar] [CrossRef]
- Martins, G.A.; Vieira, L.Q.; Cunha, F.Q.; Silva, J.S. Gamma interferon modulates CD95 (Fas) and CD95 ligand (Fas-L) expression and nitric oxide-induced apoptosis during the acute phase of Trypanosoma cruzi infection: A possible role in immune response control. Infect. Immun. 1999, 67, 3864–3871. [Google Scholar] [PubMed]
- Gustafsson, A.B.; Gottlieb, R.A. Mechanisms of apoptosis in the heart. J. Clin. Immunol. 2003, 23, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Lula, J.F.; Rocha, M.O.; Nunes, M.C.P.; Ribeiro, A.L.; Teixeira, M.M.; Bahia, M.T.; Talvani, A. Plasma concentrations of tumour necrosis factor-alpha, tumour necrosis factor-related apoptosis-inducing ligand, and FasLigand/CD95L in patients with Chagas cardiomyopathy correlate with left ventricular dysfunction. Eur. J. Heart Fail. 2009, 11, 825–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tostes, S., Jr.; Rocha-Rodrigues, D.B.; de Araujo Pereira, G.; Rodrigues, V., Jr. Myocardiocyte apoptosis in heart failure in chronic Chagas’ disease. Int. J. Cardiol. 2005, 99, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Guillermo, L.V.; Silva, E.M.; Ribeiro-Gomes, F.L.; De Meis, J.; Pereira, W.F.; Yagita, H.; DosReis, G.A.; Lopes, M.F. The Fas death pathway controls coordinated expansions of type 1 CD8 and type 2 CD4 T cells in Trypanosoma cruzi infection. J. Leukocyte Biol. 2007, 81, 942–951. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.F.; Nunes, M.P.; Henriques-Pons, A.; Giese, N.; Morse, H.C., III; Davidson, W.F.; Araújo-Jorge, T.C.; Dos Reis, G.A. Increased susceptibility of Fas ligand-deficient gld mice to Trypanosoma cruzi infection due to a Th2-biased host immune response. Eur. J. Immunol. 1999, 29, 81–89. [Google Scholar] [CrossRef]
- Keesen, T.S.; Gomes, J.A.; Fares, R.C.; de Araújo, F.F.; Ferreira, K.S.; Chaves, A.T.; Rocha, M.O.; Correa-Oliveira, R. Characterization of CD4+ cytotoxic lymphocytes and apoptosis markers induced by Trypanossoma cruzi infection. Scand. J. Immunol. 2012, 76, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Killeen, N.; Stuart, S.G.; Littman, D.R. Development and function of T cells in mice with a disrupted CD2 gene. EMBO J. 1992, 11, 4329–4336. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; van der Merwe, P.A. The structure and ligand interactions of CD2: Implications for T-cell function. Immunol. Today 1996, 17, 177–187. [Google Scholar] [CrossRef]
- Hünig, T.; Tiefenthaler, G.; Meyer zum Büschenfelde, K.H.; Meuer, S.C. Alternative pathway activation of T cells by binding of CD2 to its cell-surface ligand. Nature 1987, 326, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Green, J.M.; Karpitskiy, V.; Kimzey, S.L.; Shaw, A.S. Coordinate regulation of T cell activation by CD2 and CD28. J. Immunol. 2000, 164, 3591–3595. [Google Scholar] [CrossRef] [PubMed]
- Skånland, S.S.; Moltu, K.; Berge, T.; Aandahl, E.M.; Taskén, K. T-cell co-stimulation through the CD2 and CD28 co-receptors induces distinct signalling responses. Biochem. J. 2014, 460, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Wingren, A.G.; Parra, E.; Varga, M.; Kalland, T.; Sjogren, H.O.; Hedlund, G.; Dohlsten, M. T Cell activation pathways: B7, LFA-3, and ICAM-1 shape unique T cell profiles. Crit. Rev. Immunol. 1995, 15, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; Dubey, C. Accessory molecule and costimulation requirements for CD4 T cell response. Crit. Rev. Immunol. 1997, 17, 89–118. [Google Scholar] [CrossRef] [PubMed]
- Shahinian, A.; Pfeffer, K.; Lee, K.P.; Kündig, T.M.; Kishihara, K.; Wakeham, A.; Kawai, K.; Ohashi, P.S.; Thompson, C.B.; Mak, T.W. Differential T cell costimulatory requirements in CD28-deficient mice. Science 1993, 261, 609–612. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Landskron, J.; Ask, E.H.; Enqvist, M.; Sohlberg, E.; Traherne, J.A.; Hammer, Q.; Goodridge, J.P.; Larsson, S.; Jayaraman, J.; et al. Critical role of CD2 co-stimulation in adaptive natural killer cell responses revealed in NKG2C-deficient humans. Cell Rep. 2016, 15, 1088–1099. [Google Scholar] [CrossRef] [PubMed]
- Murali, A.R.; Chandra, S.; Stewart, Z.; Blazar, B.R.; Farooq, U.; Ince, M.N.; Dunkelberg, J. Graft versus host disease after liver transplantation in adults: A case series, review of literature, and an approach to management. Transplantation 2016, 100, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Koumakis, E.; Bouaziz, M.; Dieudé, P.; Cauvet, A.; Ruiz, B.; Airò, P.; Cusi, D.; Matucci-Cerinic, M.; Salvi, E.; Cuomo, G.; et al. A candidate gene study identifies a haplotype of CD2 as novel susceptibility factor for systemic sclerosis. Clin. Exp. Rheumatol. 2016, 100, 43–48. [Google Scholar]
- Sharma, T.L.; Yeaney, G.A.; Soltanzadeh, P.; Li, Y.; Cotta, C.V. Intravascular T-cell lymphoma: A rare, poorly characterized entity with cytotoxic phenotype. Neuropathology 2017, 37, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Bolduan, S.; Koppensteiner, H.; Businger, R.; Rebensburg, S.; Kunze, C.; Brack-Werner, R.; Draenert, R.; Schindler, M. T cells with low CD2 levels express reduced restriction factors and are preferentially infected in therapy naïve chronic HIV-1 patients. J. Int. AIDS Soc. 2017, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tsitsikov, E.; Harris, M.H.; Silverman, L.B.; Sallan, S.E.; Weinberg, O.K. Role of CD81 and CD58 in minimal residual disease detection in pediatric B lymphoblastic leukemia. Int. J. Lab. Hematol. 2018, 40, 343–351. [Google Scholar] [CrossRef] [PubMed]
- El Menshawy, N.; Eissa, M.; Abdeen, H.M.; Elkhamisy, E.M.; Joseph, N. CD58; leucocyte function adhesion-3 (LFA-3) could be used as a differentiating marker between immune and non-immune thyroid disorders. Comp. Clin. Pathol. 2018, 27, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltz, L.A.; Kierszenbaum, F.; Sztein, M.B. Trypanosoma cruzi-induced suppression of human peripheral blood lymphocytes activated via the alternative (CD2) pathway. Infect. Immun. 1990, 58, 1114–1116. [Google Scholar] [PubMed]
- Reis, D.D.; Jones, E.M.; Tostes, S.; Lopes, E.R.; Chapadeiro, E.; Gazzinelli, G.; Colley, D.G.; McCurley, T.L. Expression of major histocompatibility complex antigens and adhesion molecules in hearts of patients with chronic Chagas’ disease. Am. J. Trop. Med. Hyg. 1993, 49, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Hogg, N.; Laschinger, M.; Giles, K.; McDowall, A. T-cell integrins: More than just sticking points. J. Cell. Sci. 2003, 116, 4695–4705. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, M.; Olotu, F.A.; Soliman, M.E. Allosteric inhibition abrogates dysregulated LFA-1 activation: Structural insight into mechanisms of diminished immunologic disease. Comput. Biol. Chem. 2018, 73, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Walling, B.L.; Kim, M. LFA-1 in T cell migration and differentiation. Front. Immunol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kai, H.; Chang, F.; Shibata, K.; Tahara-Hanaoka, S.; Honda, S.; Shibuya, A.; Shibuya, K. A critical role of LFA-1 in the development of Th17 cells and induction of experimental autoimmune encephalomyelytis. Biochem. Biophys. Res. Commun. 2007, 353, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Dempsey, E.; Long, A.; Davies, A.; Barry, S.P.; Fallon, P.G.; Volkov, Y.; Kelleher, D. Leukocyte function-associated antigen-1/intercellular adhesion molecule-1 interaction induces a novel genetic signature resulting in T-cells refractory to transforming growth factor-β signaling. J. Biol. Chem. 2012, 287, 27204–27216. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.K.; Fazil, M.H.U.T.; Ong, S.T.; Chalasani, M.L.S.; Low, J.H.; Kottaiswamy, A.; Praseetha, P.; Kizhakeyil, A.; Kumar, S.; Panda, A.K.; et al. LFA-1/ICAM-1 ligation in human T cells promotes Th1 polarization through a GSK3β signaling-dependent notch pathway. J. Immunol. 2016, 197, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tarleton, R.L. Persistent production of inflammatory and anti-inflammatory cytokines and associated MHC and adhesion molecule expression at the site of infection and disease in experimental Trypanosoma cruzi infections. Exp. Parasitol. 1996, 84, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, P.V.; Roffê, E.; Santiago, H.C.; Torres, R.A.; Marino, A.P.; Paiva, C.N.; Silva, A.A.; Gazzinelli, R.T.; Lannes-Vieira, J. Prevalence of CD8 (+) alpha beta T cells in Trypanosoma cruzi-elicited myocarditis is associated with acquisition of CD62L(Low)LFA-1(High)VLA-4(High) activation phenotype and expression of IFN-gamma-inducible adhesion and chemoattractant molecules. Microbes Infect. 2001, 3, 971–984. [Google Scholar] [CrossRef]
- Marino, A.P.; Azevedo, M.I.; Lannes-Vieira, J. Differential expression of adhesion molecules shaping the T-cell subset prevalence during the early phase of autoimmune and Trypanosoma cruzi-elicited myocarditis. Mem. Inst. Oswaldo Cruz 2003, 98, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.P.; Cariste, L.M.; Santos Virgílio, F.D.; Moraschi, B.F.; Monteiro, C.B.; Vieira Machado, A.M.; Gazzinelli, R.T.; Bruna-Romero, O.; Menin Ruiz, P.L.; Ribeiro, D.A.; et al. LFA-1 mediates cytotoxicity and tissue migration of specific CD8+ T cells after heterologous prime-boost vaccination against Trypanosoma cruzi infection. Front. Immunol. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Laucella, S.; Salcedo, R.; Castaños-Velez, E.; Riarte, A.; De Titto, E.H.; Patarroyo, M.; Orn, A.; Rottenberg, M.E. Increased expression and secretion of ICAM-1 during experimental infection with Trypanosoma cruzi. Parasite Immunol. 1996, 18, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T-cell responses to antigen. Annu. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.K.; Schwartz, R.H. Antigen presentation by chemically modified splenocytes induces antigen-specific T cell unresponsiveness in vitro and in vivo. J. Exp. Med. 1987, 165, 302–319. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, S.W.; Vandenberghe, P.; de Boer, M.; Ceuppens, J.L. CD80, CD86 and CD40 provide accessory signals in a multiple-step T-cell activation model. Immunol. Rev. 1996, 153, 47–83. [Google Scholar] [CrossRef] [PubMed]
- Ziller, C.; Stoeckel, F.; Boon, L.; Haegel-Kronenberger, H. Transient blocking of both B7.1 (CD80) and B7.2 (CD86) in addition to CD40–CD40L interaction fully abrogates the immune response following systemic injection of adenovirus vector. Gene Ther. 2002, 9, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsley, P.S.; Bradshaw, J.; Greene, J.; Peach, R.; Bennett, K.L.; Mittler, R.S. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 1996, 4, 535–543. [Google Scholar] [CrossRef]
- Linsley, P.S.; Greene, J.L.; Brady, W.; Bajorath, J.; Ledbetter, J.A.; Peach, R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1994, 1, 793–801. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Su, G.H.; Zuckerman, L.A.; Nabavi, N.; Jellis, C.L.; Gray, G.S.; Miller, J.; Bluestone, J.A. Expression and functional significance of an additional ligand for CTLA-4. Proc. Natl. Acad. Sci. USA 1993, 90, 11054–11058. [Google Scholar] [CrossRef] [PubMed]
- Hathcock, K.S.; Laszlo, G.; Pucillo, S.C.; Linsley, P.; Hodes, R.J. Comparative analysis of B7-1 and B7-2 costimulatory ligands: Expression and function. J. Exp. Med. 1994, 180, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Sigal, L.J.; Reiser, H.; Rock, K.L. The role of B7-1 and B7-2 costimulation for the generation of CTL responses in vivo. J. Immunol. 1998, 161, 2740–2745. [Google Scholar] [PubMed]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Green, J.M.; Noel, P.J.; Sperling, A.I.; Walunas, T.L.; Gray, G.S.; Bluestone, J.A.; Thompson, C.B. Absence of B7-dependent responses in CD-28 deficient mice. Immunity 1994, 1, 501–508. [Google Scholar] [CrossRef]
- Yu, X.; Fournier, S.; Allison, J.P.; Sharpe, A.H.; Hodes, R.J. The role of B7 costimulation in CD4/CD8 T cell homeostasis. J. Immunol. 2000, 164, 3543–3553. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Ledbetter, J.A.; Linsley, P.S.; Thompson, C.B. Role of the CD28 receptor in T-cell activation. Immunol. Today 1990, 11, 211–216. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Vermeiren, J.; Rafiq, K.; Lorr, K.; de Boer, M.; Ceuppens, J.L. Blocking CD40–CD154 and CD80/CD86–CD28 interactions during primary allogeneic stimulation results in T cell anergy and high IL-10 production. Eur. J. Immunol. 1999, 29, 2367–2375. [Google Scholar] [CrossRef]
- Zheng, Y.; Manzotti, C.N.; Liu, M.; Burke, F.; Mead, K.I.; Sansom, D.M. CD86 and CD80 differentially modulate the suppressive function of human regulatory T cells. J. Immunol. 2004, 172, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 29, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.; Jeffery, L.E.; Sansom, D.M. Understanding the CD28/CTLA-4 (CD152) pathway and its implications for costimulatory blockade. Am. J. Transplant. 2014, 14, 1985–1991. [Google Scholar] [CrossRef] [PubMed]
- Flörcken, A.; Johannsen, M.; Nguyen-Hoai, T.; Gerhardt, A.; Miller, K.; Dörken, B.; Pezzutto, A.; Westermann, J.; Jöhrens, K. Immunomodulatory molecules in renal cell cancer: CD80 and CD86 are expressed on tumor cells. Int. J. Clin. Exp. Pathol. 2017, 10, 1443–1454. [Google Scholar]
- Magalhães, L.M.D.; Viana, A.; Chiari, E.; Galvao, L.M.C.; Gollob, K.J.; Dutra, W.O. Differential activation of human monocytes and lymphocytes by distinct strains of Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2015, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Soares, A.K.D.; Neves, P.A.F.; Cavalcanti, M.; Marinho, S.M.; de Oliveira, W.; de Souza, J.R.; Barros de Lorena, V.M.; Gomes, Y.M. Expression of co-stimulatory molecules CD80 and CD86 is altered in CD14+HLA-DR+ monocytes from patients with Chagas disease following induction by Trypanosoma cruzi recombinant antigens. Rev. Soc. Bras. Med. Trop. 2016, 49, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Miyahira, Y.; Katae, M.; Kobayashi, S.; Takeuchi, T.; Fukuchi, Y.; Abe, R.; Okumura, K.; Yagita, H.; Aoki, T. Critical contribution of CD28–CD80/CD86 costimulatory pathway to protection from Trypanosoma cruzi infection. Infect. Immun. 2003, 71, 3131–3137. [Google Scholar] [CrossRef] [PubMed]
- Dutra, W.O.; Martins-Filho, O.A.; Cançado, J.R.; Pinto-Dias, J.C.; Brener, Z.; Gazzinelli, G.; Carvalho, J.F.; Colley, D.G. Chagasic patients lack CD28 expression on many of their circulating T lymphocytes. Scand. J. Immunol. 1996, 43, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Menezes, C.A.; Rocha, M.O.; Souza, P.E.; Chaves, A.C.; Gollob, K.J.; Dutra, W.O. Phenotypic and functional characteristics of CD28+ and CD28− cells from chagasic patients: Distinct repertoire and cytokine expression. Clin. Exp. Immunol. 2004, 137, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Martins, G.A.; Campanelli, A.P.; Silva, R.B.; Tadokoro, C.E.; Russo, M.; Cunha, F.Q.; Rizzo, L.V.; Silva, J.S. CD28 is required for T cell activation and IFN-gamma production by CD4+ and CD8+ T cells in response to Trypanosoma cruzi infection. Microbes Infect. 2004, 6, 1133–1144. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Co-Stimulatory Pathway | Cell Expression | Main Function | Main Findings in Chagas Disease | References |
|---|---|---|---|---|
| PD-1/PD-L1 or PD-L2 | PD-1 (T cells, B cells, and myeloid cells); PD-L1 (B cells, DCs, and monocytes) and PD-L2 (DCs) | Inhibition of the activation of T cells | Different strains were able to induce the expression of PD-L1 on DC; PD-1 is highly expressed in lymphocytes found in the hearts of mice infected with T. cruzi, and its blockade increased the proliferative response by T cells and mice mortality; macrophages of infected mice treated with anti-PD-1 or anti-PD-L increased NO production. | [50,56,58,59,61,65,66] |
| ICOSL/ICOS | ICOS (T cell) and ICOSL (B cells, macrophages, and DCs) | Activation and differentiation of T cells | To this date, no study has shown a role of the ICOS/ICOSL pathway in T. cruzi infection. | [52,67,68,69,72] |
| CD40/CD40L | CD40 (APCs) and CD40L (T cell) | Activation of APCs and provision of co-stimulatory signals to T cells | C57BL/6 mice infected with T. cruzi showed high levels of CD40 by APCs; CD40/CD40L interaction on lymphocytes in the heart can induce IL-6 production and contribute to tissue inflammation; blockage of CD40 on human monocytes infected with T. cruzi causes 50% of the inhibition of the IL-12 expression. | [52,83,84,89,90,92] |
| CD95/CD95L | Several cells and T and B lymphocytes | Induction of apoptosis | CD95/CD95L engagement promotes apoptosis of CD4+ T cells co-cultivated with macrophages infected with T. cruzi; the deficiency of this receptor in modified mice or its blockage can increase IL-10 and IL-4 production. | [97,99,101,102] |
| CD2/CD58 | CD2 (Red blood and T cells) and CD58 (APCs) | Activation of T cells | Co-cultures of human PBMCs infected with T. cruzi and stimulated with anti-CD2 antibodies showed a reduction in lymphocytes proliferative and IL-2R expression. | [106,107,108,119] |
| LFA-1/ICAM-1 | LFA-1 (T cells) and ICAM-1 (Endothelium and APCs) | Adhesion, migration of T cells and antigen presentation | Leukocytes of the inflammatory infiltrate, circulating mononuclear cells and cardiomyocytes increased ICAM-1 expression in the acute infection with T. cruzi. | [109,120,121,122,123,127,128,129,131] |
| CD80-CD86/CD28-CTLA-4 | CD80-CD86 (APCs) and CD28-CTLA-4 (CD4+ T cells) | CD80 or CD86 interaction with CD28 activates T cells, and with CTLA-4 inhibits the activation of lymphocytes | CD28-deficient mice and CD80/CD86 blockage increased susceptibility to T. cruzi infection; Chagas patients showed a higher frequency of CD80 monocytes; IND patients showed a high CD86 expression by monocytes and their subsets, CTLA-4 T CD4+ T lymphocytes frequency, and increased Treg proportion. | [39,49,135,137,138,139,151,152] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, B.F.; Medeiros, N.I.; Fontes-Cal, T.C.M.; Naziazeno, I.M.; Correa-Oliveira, R.; Dutra, W.O.; Gomes, J.A.S. The Role of Co-Stimulatory Molecules in Chagas Disease. Cells 2018, 7, 200. https://doi.org/10.3390/cells7110200
Pinto BF, Medeiros NI, Fontes-Cal TCM, Naziazeno IM, Correa-Oliveira R, Dutra WO, Gomes JAS. The Role of Co-Stimulatory Molecules in Chagas Disease. Cells. 2018; 7(11):200. https://doi.org/10.3390/cells7110200
Chicago/Turabian StylePinto, Bruna F., Nayara I. Medeiros, Tereza C. M. Fontes-Cal, Isabela M. Naziazeno, Rodrigo Correa-Oliveira, Walderez O. Dutra, and Juliana A. S. Gomes. 2018. "The Role of Co-Stimulatory Molecules in Chagas Disease" Cells 7, no. 11: 200. https://doi.org/10.3390/cells7110200
APA StylePinto, B. F., Medeiros, N. I., Fontes-Cal, T. C. M., Naziazeno, I. M., Correa-Oliveira, R., Dutra, W. O., & Gomes, J. A. S. (2018). The Role of Co-Stimulatory Molecules in Chagas Disease. Cells, 7(11), 200. https://doi.org/10.3390/cells7110200