Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens

{kind=link}
{kind=link}
Abstract
1. Oncolytic Virotherapy
Oncolytic Adenoviruses
2. Strategies to Increase the Efficacy of Oncolytic Virus Therapy with CRAds
2.1. Achieving More Effective Delivery of Oncolytic Adenovirus to Tumors
2.2. Improving Oncolytic Adenovirus Specificity by Employing microRNA-Dependent Replication
2.3. Strategies for Improving the Potency of Oncolytic Adenoviruses
3. Gene Suppression to Make Oncolytic Viruses More Effective
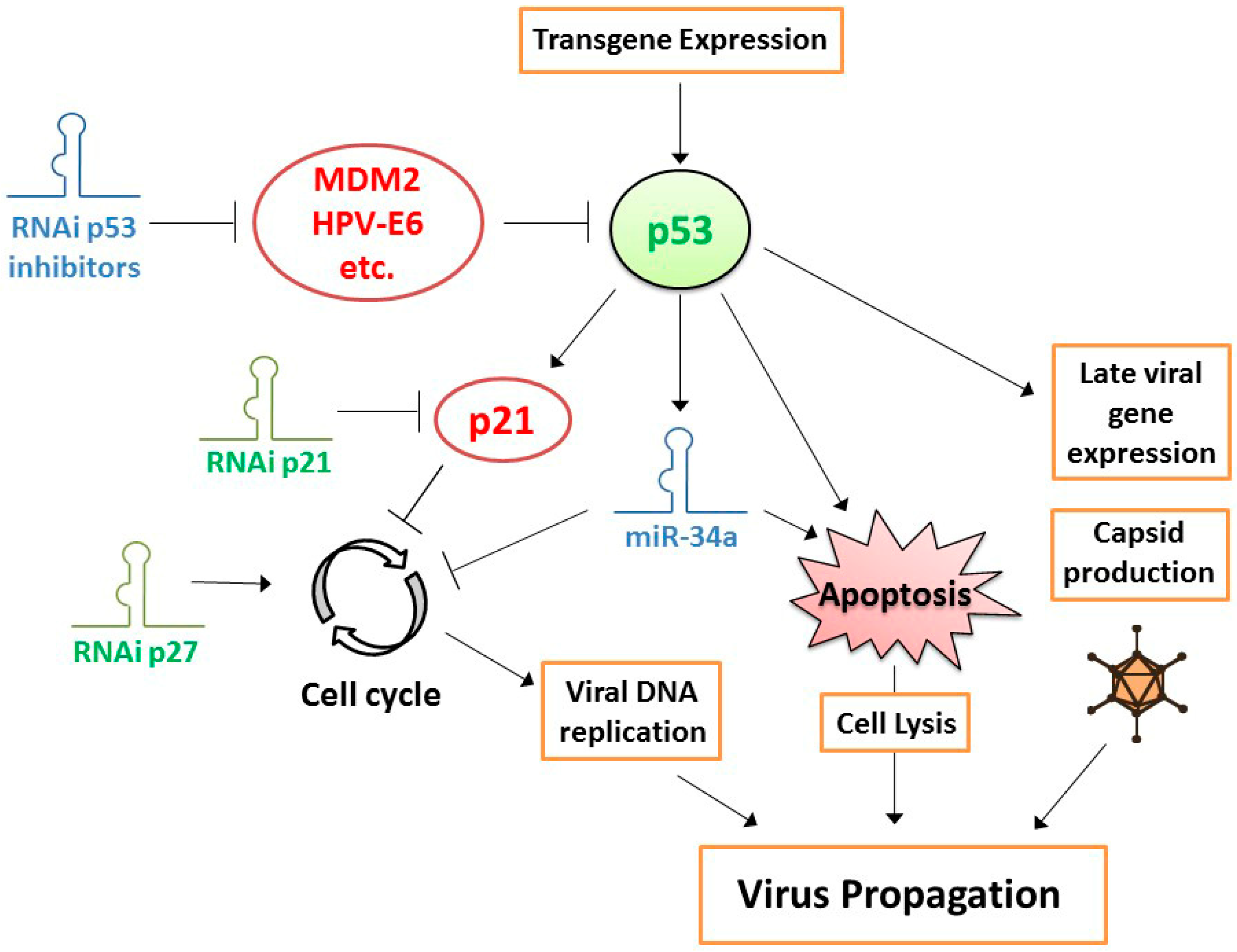
3.1. Combining OVT with Suppression of CRAd-Inhibitory Target Genes in Cancer Cells
3.2. Combining OVT with Targeting Immune Suppression
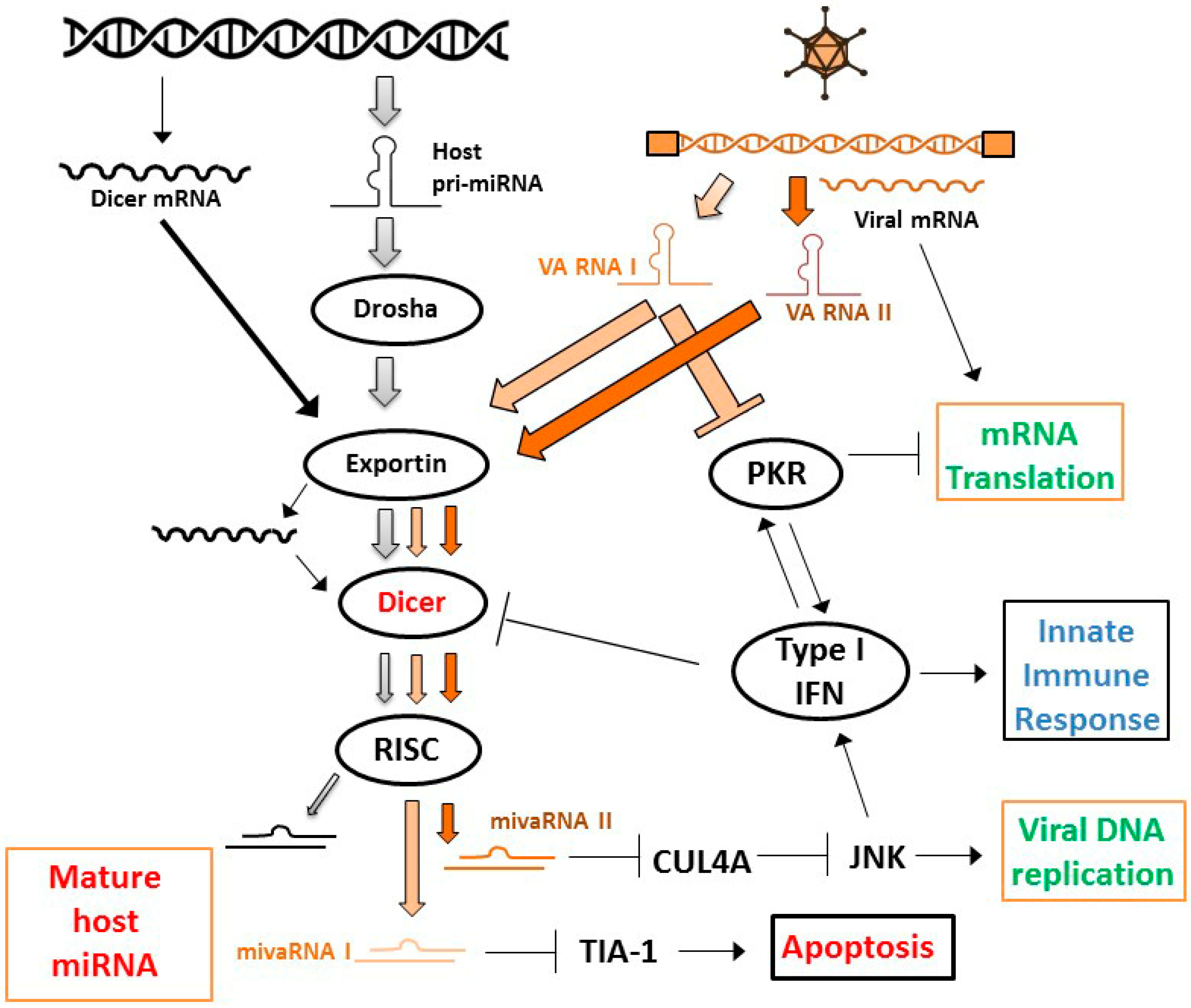
3.3. Exploiting Virus–Host Interactions via MicroRNAs
3.4. RNAi Screening for Inhibitors of Oncolytic Virus Efficacy in Cancer Cells
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Parato, K.A.; Senger, D.; Forsyth, P.A.J.; Bell, J.C. Recent progress in the battle between oncolytic viruses and tumours. Nat. Rev. Cancer 2005, 5, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Vähä-Koskela, M.J.V.; Heikkilä, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef] [PubMed]
- Sinkovics, J.G.; Horvath, J.C. Natural and genetically engineered viral agents for oncolysis and gene therapy of human cancers. Arch. Immunol. Ther. Exp. (Warsz.) 2008, 56, 1–59. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.L.A.; Tumilasci, V.F.; Singhroy, D.; Arguello, M.; Hiscott, J. The emergence of combinatorial strategies in the development of RNA oncolytic virus therapies. Cell. Microbiol. 2009, 11, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Diaconu, I.; Cerullo, V.; Hirvinen, M.L.M.; Escutenaire, S.; Ugolini, M.; Pesonen, S.K.; Bramante, S.; Parviainen, S.; Kanerva, A.; Loskog, A.S.I.; et al. Immune response is an important aspect of the antitumor effect produced by a CD40L-encoding oncolytic adenovirus. Cancer Res. 2012, 72, 2327–2338. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, T.D.; Janssen, A.B.; van Beusechem, V.W. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin. Biol. Ther. 2015, 15, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Buqué, A.; Aranda, F.; Bloy, N.; Cremer, I.; Eggermont, A.; Erbs, P.; Fucikova, J.; Galon, J.; Limacher, J.-M.; et al. Trial Watch—Oncolytic viruses and cancer therapy. Oncoimmunology 2016, 5, e1117740. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Fang, H. Clinical Trials with Oncolytic Adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.-W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, R.; Miest, T.; Shashkova, E.V.; Barry, M.A. Reprogrammed viruses as cancer therapeutics: Targeted, armed and shielded. Nat. Rev. Microbiol. 2008, 6, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Adenovirus Infections in Immunocompetent and Immunocompromised Patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed]
- Ben-Israel, H.; Kleinberger, T. Adenovirus and cell cycle control. Front. Biosci. 2002, 7, 1369–1395. [Google Scholar] [CrossRef]
- Robert-Guroff, M. Replicating and non-replicating viral vectors for vaccine development. Curr. Opin. Biotechnol. 2007, 18, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Bauerschmitz, G.J.; Barker, S.D.; Hemminki, A. Adenoviral gene therapy for cancer: From vectors to targeted and replication competent agents (review). Int. J. Oncol. 2002, 21, 1161–1174. [Google Scholar] [CrossRef] [PubMed]
- Doloff, J.C.; Waxman, D.J. Dual E1A oncolytic adenovirus: Targeting tumor heterogeneity with two independent cancer-specific promoter elements, DF3/MUC1 and hTERT. Cancer Gene Ther. 2011, 18, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Sugio, K.; Sakurai, F.; Katayama, K.; Tashiro, K.; Matsui, H.; Kawabata, K.; Kawase, A.; Iwaki, M.; Hayakawa, T.; Fujiwara, T.; et al. Enhanced Safety Profiles of the Telomerase-Specific Replication-Competent Adenovirus by Incorporation of Normal Cell-Specific microRNA-Targeted Sequences. Clin. Cancer Res. 2011, 17, 2807–2818. [Google Scholar] [CrossRef] [PubMed]
- Lou, W.; Chen, Q.; Ma, L.; Liu, J.; Yang, Z.; Shen, J.; Cui, Y.; Bian, X.; Qian, C. Oncolytic adenovirus co-expressing miRNA-34a and IL-24 induces superior antitumor activity in experimental tumor model. J. Mol. Med. 2013, 91, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M.; Ng, L.; Nye, J.A.; Sampson-Johannes, A.; Fattaey, A.; et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Khuri, F.; Ganly, I.; Arseneau, J.; Posner, M.; Vokes, E.; Kuhn, J.; McCarty, T.; Landers, S.; Blackburn, A.; et al. Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer. J. Clin. Oncol. 2001, 19, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Cascallo, M.; Alonso, M.M.; Rojas, J.J.; Perez-Gimenez, A.; Fueyo, J.; Alemany, R. Systemic Toxicity–Efficacy Profile of ICOVIR-5, a Potent and Selective Oncolytic Adenovirus Based on the pRB Pathway. Mol. Ther. 2007, 15, 1607–1615. [Google Scholar] [CrossRef] [PubMed]
- Fueyo, J.; Gomez-Manzano, C.; Alemany, R.; Lee, P.S.Y.; McDonnell, T.J.; Mitlianga, P.; Shi, Y.-X.; Levin, V.A.; Yung, W.K.A.; Kyritsis, A.P. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene 2000, 19, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Eager, R.M.; Nemunaitis, J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 2011, 18, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pong, R.C.; Bergelson, J.M.; Hall, M.C.; Sagalowsky, A.I.; Tseng, C.P.; Wang, Z.; Hsieh, J.T. Loss of adenoviral receptor expression in human bladder cancer cells: A potential impact on the efficacy of gene therapy. Cancer Res. 1999, 59, 325–330. [Google Scholar] [PubMed]
- Reeh, M.; Bockhorn, M.; Görgens, D.; Vieth, M.; Hoffmann, T.; Simon, R.; Izbicki, J.R.; Sauter, G.; Schumacher, U.; Anders, M. Presence of the Coxsackievirus and Adenovirus Receptor (CAR) in human neoplasms: A multitumour array analysis. Br. J. Cancer 2013, 109, 1848–1858. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Fueyo, J.; Krasnykh, V.; Reynolds, P.N.; Curiel, D.T.; Alemany, R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin. Cancer Res. 2001, 7, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Guse, K.; Ranki, T.; Ala-Opas, M.; Bono, P.; Särkioja, M.; Rajecki, M.; Kanerva, A.; Hakkarainen, T.; Hemminki, A. Treatment of metastatic renal cancer with capsid-modified oncolytic adenoviruses. Mol. Cancer Ther. 2007, 6, 2728–2736. [Google Scholar] [CrossRef] [PubMed]
- Kanerva, A.; Mikheeva, G.V.; Krasnykh, V.; Coolidge, C.J.; Lam, J.T.; Mahasreshti, P.J.; Barker, S.D.; Straughn, M.; Barnes, M.N.; Alvarez, R.D.; et al. Targeting adenovirus to the serotype 3 receptor increases gene transfer efficiency to ovarian cancer cells. Clin. Cancer Res. 2002, 8, 275–280. [Google Scholar] [PubMed]
- van Beusechem, V.W.; Mastenbroek, D.C.J.; van den Doel, P.B.; Lamfers, M.L.M.; Grill, J.; Würdinger, T.; Haisma, H.J.; Pinedo, H.M.; Gerritsen, W.R. Conditionally replicative adenovirus expressing a targeting adapter molecule exhibits enhanced oncolytic potency on CAR-deficient tumors. Gene Ther. 2003, 10, 1982–1991. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-W.; Lee, Y.S.; Yun, C.-O.; Kim, S.W. Polymeric oncolytic adenovirus for cancer gene therapy. J. Control. Release 2015, 219, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Power, A.T.; Bell, J.C. Taming the Trojan horse: Optimizing dynamic carrier cell/oncolytic virus systems for cancer biotherapy. Gene Ther. 2008, 15, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Yong, R.L.; Shinojima, N.; Fueyo, J.; Gumin, J.; Vecil, G.G.; Marini, F.C.; Bogler, O.; Andreeff, M.; Lang, F.F. Human Bone Marrow-Derived Mesenchymal Stem Cells for Intravascular Delivery of Oncolytic Adenovirus 24-RGD to Human Gliomas. Cancer Res. 2009, 69, 8932–8940. [Google Scholar] [CrossRef] [PubMed]
- Ong, H.T.; Hasegawa, K.; Dietz, A.B.; Russell, S.J.; Peng, K.-W. Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther. 2007, 14, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Power, A.T.; Wang, J.; Falls, T.J.; Paterson, J.M.; Parato, K.A.; Lichty, B.D.; Stojdl, D.F.; Forsyth, P.A.J.; Atkins, H.; Bell, J.C. Carrier cell-based delivery of an oncolytic virus circumvents antiviral immunity. Mol. Ther. 2007, 15, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ylosmaki, E.; Hakkarainen, T.; Hemminki, A.; Visakorpi, T.; Andino, R.; Saksela, K. Generation of a Conditionally Replicating Adenovirus Based on Targeted Destruction of E1A mRNA by a Cell Type-Specific MicroRNA. J. Virol. 2008, 82, 11009–11015. [Google Scholar] [CrossRef] [PubMed]
- Cawood, R.; Chen, H.H.; Carroll, F.; Bazan-Peregrino, M.; van Rooijen, N.; Seymour, L.W. Use of Tissue-Specific MicroRNA to Control Pathology of Wild-Type Adenovirus without Attenuation of Its Ability to Kill Cancer Cells. PLoS Pathog. 2009, 5, e1000440. [Google Scholar] [CrossRef] [PubMed]
- Leja, J.; Nilsson, B.; Yu, D.; Gustafson, E.; Åkerström, G.; Öberg, K.; Giandomenico, V.; Essand, M. Double-Detargeted Oncolytic Adenovirus Shows Replication Arrest in Liver Cells and Retains Neuroendocrine Cell Killing Ability. PLoS ONE 2010, 5, e8916. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Elamin, B.K.; D’Abundo, L.; Falzoni, S.; Donvito, G.; Moshiri, F.; Milazzo, M.; Altavilla, G.; Giacomelli, L.; Fornari, F.; et al. Anti-Tumor Activity of a miR-199-dependent Oncolytic Adenovirus. PLoS ONE 2013, 8, e73964. [Google Scholar] [CrossRef] [PubMed]
- Bofill-De Ros, X.; Gironella, M.; Fillat, C. MiR-148a- and miR-216a-regulated Oncolytic Adenoviruses Targeting Pancreatic Tumors Attenuate Tissue Damage Without Perturbation of miRNA Activity. Mol. Ther. 2014, 22, 1665–1677. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Guo, G.; Zhang, Q.; Fan, L.; Wu, N.; Bo, Y. The application of multiple miRNA response elements enables oncolytic adenoviruses to possess specificity to glioma cells. Virology 2014, 458, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Gürlevik, E.; Woller, N.; Schache, P.; Malek, N.P.; Wirth, T.C.; Zender, L.; Manns, M.P.; Kubicka, S.; Kühnel, F. p53-dependent antiviral RNA-interference facilitates tumor-selective viral replication. Nucleic. Acids Res. 2009, 37, e84. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Martínez-Quintanilla, J.; Puig, C.; Guedan, S.; Molleví, D.G.; Alemany, R.; Cascallo, M. Bioselection of a gain of function mutation that enhances adenovirus 5 release and improves its antitumoral potency. Cancer Res. 2008, 68, 8928–8937. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; van Ginkel, J.-W.H.; Au, K.Y.; Alemany, R.; Meulenberg, J.J.M.; van Beusechem, V.W. ORCA-010, a Novel Potency-Enhanced Oncolytic Adenovirus, Exerts Strong Antitumor Activity in Preclinical Models. Hum. Gene Ther. 2014, 25, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, I.; Harden, P.; Bauzon, M.; Chartier, C.; Nye, J.; Thorne, S.; Reid, T.; Ni, S.; Lieber, A.; Fisher, K.; et al. Directed Evolution Generates a Novel Oncolytic Virus for the Treatment of Colon Cancer. PLoS ONE 2008, 3, e2409. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Grases, D.; Rojas, J.J.; Gros, A.; Vilardell, F.; Vile, R.; Mercade, E.; Cascallo, M.; Alemany, R. GALV expression enhances the therapeutic efficacy of an oncolytic adenovirus by inducing cell fusion and enhancing virus distribution. Gene Ther. 2012, 19, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Rojas, J.J.; Gros, A.; Mercade, E.; Cascallo, M.; Alemany, R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol. Ther. 2010, 18, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, Y.S.; Kim, H.; Huang, J.H.; Yoon, A.R.; Yun, C.O. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J. Natl. Cancer Inst. 2006, 98, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.-K.; Lee, Y.-S.; Yoo, J.Y.; Yoon, A.-R.; Kim, H.; Kim, D.-S.; Seidler, D.G.; Kim, J.-H.; Yun, C.-O. Effect of decorin on overcoming the extracellular matrix barrier for oncolytic virotherapy. Gene Ther. 2010, 17, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Wildner, O. Therapy of Head and Neck Squamous Cell Carcinoma with an Oncolytic Adenovirus Expressing HSV-tk. Mol. Ther. 2000, 1, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Oosterhoff, D.; Pinedo, H.M.; Witlox, M.A.; Carette, J.E.; Gerritsen, W.R.; van Beusechem, V.W. Gene-directed enzyme prodrug therapy with carboxylesterase enhances the anticancer efficacy of the conditionally replicating adenovirus AdΔ24. Gene Ther. 2005, 12, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Li, Z.-Y.; Ni, S.; Steinwaerder, D.; Lieber, A. Induced Apoptosis Supports Spread of Adenovirus Vectors in Tumors. Hum. Gene Ther. 2001, 12, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Van Beusechem, V.W.; Van den Doel, P.B.; Grill, J.; Pinedo, H.M.; Gerritsen, W.R. Conditionally replicative adenovirus expressing p53 exhibits enhanced oncolytic potency. Cancer Res. 2002, 62, 6165–6171. [Google Scholar] [PubMed]
- Sauthoff, H.; Pipiya, T.; Heitner, S.; Chen, S.; Norman, R.G.; Rom, W.N.; Hay, J.G. Late Expression of p53 from a Replicating Adenovirus Improves Tumor Cell Killing and Is More Tumor Cell Specific than Expression of the Adenoviral Death Protein. Hum. Gene Ther. 2002, 13, 1859–1871. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Dix, B.R.; O’Carroll, S.J.; Braithwaite, A.W. p53-dependent cell death/apoptosis is required for a productive adenovirus infection. Nat. Med. 1998, 4, 1068–1072. [Google Scholar] [CrossRef] [PubMed]
- Royds, J.A.; Hibma, M.; Dix, B.R.; Hananeia, L.; Russell, I.A.; Wiles, A.; Wynford-Thomas, D.; Braithwaite, A.W. p53 promotes adenoviral replication and increases late viral gene expression. Oncogene 2006, 25, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, C.; Cao, H.; Li, K.; Chen, J.; Jiang, L.; Zhang, Q.; Wu, X.; Jia, X.; Liu, Y.; et al. A novel triple-regulated oncolytic adenovirus carrying p53 gene exerts potent antitumor efficacy on common human solid cancers. Mol. Cancer Ther. 2008, 7, 1598–1603. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Vassal, G.; Opolon, P.; Dirven, C.M.F.; Morizet, J.; Laudani, L.; Grill, J.; Giaccone, G.; Vandertop, W.P.; Gerritsen, W.R.; et al. Oncolytic Activity of p53-Expressing Conditionally Replicative Adenovirus AdΔ24-p53 against Human Malignant Glioma. Cancer Res. 2004, 64, 5753–5759. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; van Beusechem, V.W.; Opolon, P.; Morizet, J.; Laudani, L.; Lecluse, Y.; Barrois, M.; Idema, S.; Grill, J.; Gerritsen, W.R.; et al. Expression of p53, or targeting towards EGFR, enhances the oncolytic potency of conditionally replicative adenovirus against neuroblastoma. J. Gene Med. 2005, 7, 584–594. [Google Scholar] [CrossRef] [PubMed]
- van Beusechem, V.W.; van den Doel, P.B.; Gerritsen, W.R. Conditionally replicative adenovirus expressing degradation-resistant p53 for enhanced oncolysis of human cancer cells overexpressing murine double minute 2. Mol. Cancer Ther. 2005, 4, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Heideman, D.A.M.; Steenbergen, R.D.M.; van der Torre, J.; Scheffner, M.; Alemany, R.; Gerritsen, W.R.; Meijer, C.J.L.M.; Snijders, P.J.F.; van Beusechem, V.W. Oncolytic Adenovirus Expressing a p53 Variant Resistant to Degradation by HPV E6 Protein Exhibits Potent and Selective Replication in Cervical Cancer. Mol. Ther. 2005, 12, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Graat, H.C.A.; Carette, J.E.; Schagen, F.H.E.; Vassilev, L.T.; Gerritsen, W.R.; Kaspers, G.J.L.; Wuisman, P.I.J.M.; van Beusechem, V.W. Enhanced tumor cell kill by combined treatment with a small-molecule antagonist of mouse double minute 2 and adenoviruses encoding p53. Mol. Cancer Ther. 2007, 6, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Graat, H.C.A.; Schagen, F.H.E.; Abou El Hassan, M.A.I.; Gerritsen, W.R.; van Beusechem, V.W. Replication-dependent transgene expression from a conditionally replicating adenovirus via alternative splicing to a heterologous splice-acceptor site. J. Gene Med. 2005, 7, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Hannon, G.J. RNA interference. Nature 2002, 418, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Carmell, M.A.; Hannon, G.J. RNase III enzymes and the initiation of gene silencing. Nat. Struct. Mol. Biol. 2004, 11, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Huntzinger, E.; Izaurralde, E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat. Rev. Genet. 2011, 12, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Sharp, P.A. Specificity of microRNA target selection in translational repression. Genes Dev. 2004, 18, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Bushati, N.; Cohen, S.M. microRNA Functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Mao, Q.; Paulson, H.L.; Davidson, B.L. siRNA-mediated gene silencing in vitro and in vivo. Nat. Biotechnol. 2002, 20, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. RNA interference: Antiviral defense and genetic tool. Nat. Immunol. 2002, 3, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.M.; Pruss, G.J.; Vance, V.B. Plant viral suppressors of RNA silencing. Virus Res. 2004, 102, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Backes, S.; Langlois, R.A.; Schmid, S.; Varble, A.; Shim, J.V.; Sachs, D.; TenOever, B.R. The Mammalian response to virus infection is independent of small RNA silencing. Cell Rep. 2014, 8, 114–125. [Google Scholar] [CrossRef] [PubMed]
- tenOever, B.R. Questioning antiviral RNAi in mammals. Nat. Microbiol. 2017, 2, 17052. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Cullen, B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004, 78, 12868–12876. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, O.; Razquin, N.; Zaratiegui, M.; Narvaiza, I.; Fortes, P. Adenovirus Virus-Associated RNA Is Processed to Functional Interfering RNAs Involved in Virus Production. J. Virol. 2006, 80, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Bennasser, Y.; Chable-Bessia, C.; Triboulet, R.; Gibbings, D.; Gwizdek, C.; Dargemont, C.; Kremer, E.J.; Voinnet, O.; Benkirane, M. Competition for XPO5 binding between Dicer mRNA, pre-miRNA and viral RNA regulates human Dicer levels. Nat. Struct. Mol. Biol. 2011, 18, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Machitani, M.; Sakurai, F.; Wakabayashi, K.; Takayama, K.; Tachibana, M.; Mizuguchi, H. Type I Interferons Impede Short Hairpin RNA-Mediated RNAi via Inhibition of Dicer-Mediated Processing to Small Interfering RNA. Mol. Ther.-Nucleic Acids 2017, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Overmeer, R.M.; Schagen, F.H.E.; Alemany, R.; Barski, O.A.; Gerritsen, W.R.; Van Beusechem, V.W. Conditionally Replicating Adenoviruses Expressing Short Hairpin RNAs Silence the Expression of a Target Gene in Cancer Cells. Cancer Res. 2004, 64, 2663–2667. [Google Scholar] [CrossRef] [PubMed]
- Machitani, M.; Sakurai, F.; Katayama, K.; Tachibana, M.; Suzuki, T.; Matsui, H.; Yamaguchi, T.; Mizuguchi, H. Improving adenovirus vector-mediated RNAi efficiency by lacking the expression of virus-associated RNAs. Virus Res. 2013, 178, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Thimmappaya, B.; Weinberger, C.; Schneider, R.J.; Shenk, T. Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell 1982, 31, 543–551. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Machitani, M.; Tachibana, M.; Sakurai, F.; Mizuguchi, H. A microRNA derived from adenovirus virus-associated RNAII promotes virus infection via post-transcriptional gene silencing. J. Virol. 2018. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kamel, W.; Segerman, B.; Öberg, D.; Punga, T.; Akusjärvi, G. The adenovirus VA RNA-derived miRNAs are not essential for lytic virus growth in tissue culture cells. Nucleic Acids Res. 2013, 41, 4802–4812. [Google Scholar] [CrossRef] [PubMed]
- Machitani, M.; Sakurai, F.; Wakabayashi, K.; Tomita, K.; Tachibana, M.; Mizuguchi, H. Dicer functions as an antiviral system against human adenoviruses via cleavage of adenovirus-encoded noncoding RNA. Sci. Rep. 2016, 6, 27598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-A.; Nemunaitis, J.; Samuel, S.K.; Chen, P.; Shen, Y.; Tong, A.W. Antitumor Activity of an Oncolytic Adenovirus-Delivered Oncogene Small Interfering RNA. Cancer Res. 2006, 66, 9736–9743. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Gu, J.; Sun, L.; Qian, Q.; Qian, C.; Liu, X. Oncolytic adenovirus-mediated shRNA against Apollon inhibits tumor cell growth and enhances antitumor effect of 5-fluorouracil. Gene Ther. 2008, 15, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Kim, J.-H.; Kwon, Y.-G.; Kim, E.-C.; Kim, N.K.; Choi, H.J.; Yun, C.-O. VEGF-specific short hairpin RNA-expressing oncolytic adenovirus elicits potent inhibition of angiogenesis and tumor growth. Mol. Ther. 2007, 15, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Machitani, M.; Sakurai, F.; Wakabayashi, K.; Tachibana, M.; Fujiwara, T.; Mizuguchi, H. Enhanced Oncolytic Activities of the Telomerase-Specific Replication-Competent Adenovirus Expressing Short-Hairpin RNA against Dicer. Mol. Cancer Ther. 2017, 16, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Höti, N.; Chowdhury, W.; Hsieh, J.-T.; Sachs, M.D.; Lupold, S.E.; Rodriguez, R. Valproic Acid, a Histone Deacetylase Inhibitor, Is an Antagonist for Oncolytic Adenoviral Gene Therapy. Mol. Ther. 2006, 14, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Shiina, M.; Lacher, M.D.; Christian, C.; Korn, W.M. RNA interference-mediated knockdown of p21WAF1 enhances anti-tumor cell activity of oncolytic adenoviruses. Cancer Gene Ther. 2009, 16, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Höti, N.; Chowdhury, W.H.; Mustafa, S.; Ribas, J.; Castanares, M.; Johnson, T.; Liu, M.; Lupold, S.E.; Rodriguez, R. Armoring CRAds with p21/Waf-1 shRNAs: The next generation of oncolytic adenoviruses. Cancer Gene Ther. 2010, 17, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Idogawa, M.; Sasaki, Y.; Suzuki, H.; Mita, H.; Imai, K.; Shinomura, Y.; Tokino, T. A Single Recombinant Adenovirus Expressing p53 and p21-targeting Artificial microRNAs Efficiently Induces Apoptosis in Human Cancer Cells. Clin. Cancer Res. 2009, 15, 3725–3732. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Raya, A.; DeJesus, P.; Chao, S.-H.; Quon, K.C.; Caldwell, J.S.; Chanda, S.K.; Izpisua-Belmonte, J.C.; Schultz, P.G. Identification of p53 regulators by genome-wide functional analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 3456–3461. [Google Scholar] [CrossRef] [PubMed]
- Llanos, S.; Efeyan, A.; Monsech, J.; Dominguez, O.; Serrano, M. A High-Throughput Loss-of-Function Screening Identifies Novel p53 Regulators. Cell Cycle 2006, 5, 1880–1885. [Google Scholar] [CrossRef] [PubMed]
- Siebring-van Olst, E.; Blijlevens, M.; de Menezes, R.X.; van der Meulen-Muileman, I.H.; Smit, E.F.; van Beusechem, V.W. A genome-wide siRNA screen for regulators of tumor suppressor p53 activity in human non-small cell lung cancer cells identifies components of the RNA splicing machinery as targets for anticancer treatment. Mol. Oncol. 2017, 11, 534–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kang, D.; Choi, H.J.; Joo, Y.; Kim, J.-H.; Song, J.J. Prime-boost immunization by both DNA vaccine and oncolytic adenovirus expressing GM-CSF and shRNA of TGF-β2 induces anti-tumor immune activation. Oncotarget 2017, 8, 15858–15877. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Hong, Z.; Chen, C.; Gao, Q.; Luo, D.; Fang, Y.; Cao, Y.; Zhu, T.; Jiang, X.; Ma, Q.; et al. A novel oncolytic adenovirus selectively silences the expression of tumor-associated STAT3 and exhibits potent antitumoral activity. Carcinogenesis 2009, 30, 2014–2022. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.A.; Spencer, J.F.; La Regina, M.C.; Dhar, D.; Tollefson, A.E.; Toth, K.; Wold, W.S.M. Syrian Hamster as a Permissive Immunocompetent Animal Model for the Study of Oncolytic Adenovirus Vectors. Cancer Res. 2006, 66, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Särkioja, M.; et al. Oncolytic Adenovirus Coding for Granulocyte Macrophage Colony-Stimulating Factor Induces Antitumoral Immunity in Cancer Patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, X.; Wang, J.; Gao, D.; Li, Y.; Li, H.; Chu, Y.; Zhang, Z.; Liu, H.; Jiang, G.; et al. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat. Commun. 2017, 8, 1395. [Google Scholar] [CrossRef] [PubMed]
- Schütz, S.; Sarnow, P. Interaction of viruses with the mammalian RNA interference pathway. Virology 2006, 344, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and Host Interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S. Virus-encoded microRNAs. Virology 2011, 411, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Chiang, K.; Liu, H.; Rice, A.P. miR-132 enhances HIV-1 replication. Virology 2013, 438, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Potenza, N. Antiviral effects of human microRNAs and conservation of their target sites. FEBS Lett. 2011, 585, 2551–2555. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.G.; Haasnoot, P.C.J.; Xu, N.; Berenjian, S.; Berkhout, B.; Akusjarvi, G. Suppression of RNA Interference by Adenovirus Virus-Associated RNA. J. Virol. 2005, 79, 9556–9565. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.; Kato, Y.; Taira, K. Sequence-specific interference by small RNAs derived from adenovirus VAI RNA. FEBS Lett. 2006, 580, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, O.; Carnero, E.; Abad, X.; Razquin, N.; Guruceaga, E.; Segura, V.; Fortes, P. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res. 2010, 38, 750–763. [Google Scholar] [CrossRef] [PubMed]
- Hodzic, J.; Sie, D.; Vermeulen, A.; van Beusechem, V.W. Functional screening identifies human miRNAs that modulate adenovirus propagation in prostate cancer cells. Hum. Gene Ther. 2017, 2016, 143. [Google Scholar] [CrossRef] [PubMed]
- Lagos, D.; Pollara, G.; Henderson, S.; Gratrix, F.; Fabani, M.; Milne, R.S.B.; Gotch, F.; Boshoff, C. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 2010, 12, 513–519. [Google Scholar] [CrossRef] [PubMed]
- He, L.; He, X.; Lowe, S.W.; Hannon, G.J. microRNAs join the p53 network—Another piece in the tumour-suppression puzzle. Nat. Rev. Cancer 2007, 7, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.-D.; Mao, Z.; Liu, C.; Nie, Y.-S.; Sun, B.; Guo, M.; Su, C. Simultaneous overexpression of miR-126 and miR-34a induces a superior antitumor efficacy in pancreatic adenocarcinoma. OncoTargets Ther. 2017, 10, 5591–5604. [Google Scholar] [CrossRef] [PubMed]
- Santhakumar, D.; Forster, T.; Laqtom, N.N.; Fragkoudis, R.; Dickinson, P.; Abreu-Goodger, C.; Manakov, S.A.; Choudhury, N.R.; Griffiths, S.J.; Vermeulen, A.; et al. Combined agonist-antagonist genome-wide functional screening identifies broadly active antiviral microRNAs. Proc. Natl. Acad. Sci. USA 2010, 107, 13830–13835. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-B.; Bantounas, I.; Lee, D.-Y.; Phylactou, L.; Caldwell, M.A.; Uney, J.B. Twist-1 regulates the miR-199a/214 cluster during development. Nucleic Acids Res. 2009, 37, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Thornburg, N.J.; Hayward, S.L.; Crowe, J.E. Respiratory syncytial virus regulates human microRNAs by using mechanisms involving beta interferon and NF-κB. mBio 2012, 3, e00220-12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Huang, C.; Yang, Q.; Gao, L.; Liu, H.-C.; Tang, J.; Feng, W. MicroRNA-30c Modulates Type I IFN Responses To Facilitate Porcine Reproductive and Respiratory Syndrome Virus Infection by Targeting JAK1. J. Immunol. 2016, 196, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- Panda, D.; Cherry, S. Cell-based genomic screening: Elucidating virus–host interactions. Curr. Opin. Virol. 2012, 2, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, D.J.; Lefebvre, C.; Allan, K.; Brun, J.; Sanaei, C.A.; Baird, S.; Pearce, N.; Grönberg, S.; Wilson, B.; Prakesh, M.; et al. Virus-Tumor Interactome Screen Reveals ER Stress Response Can Reprogram Resistant Cancers for Oncolytic Virus-Triggered Caspase-2 Cell Death. Cancer Cell 2011, 20, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Ketela, T.; Moffat, J.; Cuddington, B.P.; Mossman, K.L. Genome-wide lentiviral shRNA screen identifies serine/arginine-rich splicing factor 2 as a determinant of oncolytic virus activity in breast cancer cells. Oncogene 2016, 35, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Teferi, W.M.; Dodd, K.; Maranchuk, R.; Favis, N.; Evans, D.H. A Whole-Genome RNA Interference Screen for Human Cell Factors Affecting Myxoma Virus Replication. J. Virol. 2013, 87, 4623–4641. [Google Scholar] [CrossRef] [PubMed]
- Homicsko, K.; Lukashev, A.; Iggo, R.D. RAD001 (Everolimus) Improves the Efficacy of Replicating Adenoviruses that Target Colon Cancer. Cancer Res. 2005, 65, 6882–6890. [Google Scholar] [CrossRef] [PubMed]
- Alonso, M.M.; Jiang, H.; Yokoyama, T.; Xu, J.; Bekele, N.B.; Lang, F.F.; Kondo, S.; Gomez-Manzano, C.; Fueyo, J. Delta-24-RGD in Combination With RAD001 Induces Enhanced Anti-glioma Effect via Autophagic Cell Death. Mol. Ther. 2008, 16, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Bagdassarian, E.; Ali, M.A.M.; McFadden, G. Identification of host DEAD-box RNA helicases that regulate cellular tropism of oncolytic Myxoma virus in human cancer cells. Sci. Rep. 2017, 7, 15710. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brachtlova, T.; Van Beusechem, V.W. Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens. Cells 2018, 7, 228. https://doi.org/10.3390/cells7120228
Brachtlova T, Van Beusechem VW. Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens. Cells. 2018; 7(12):228. https://doi.org/10.3390/cells7120228
Chicago/Turabian StyleBrachtlova, Tereza, and Victor W. Van Beusechem. 2018. "Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens" Cells 7, no. 12: 228. https://doi.org/10.3390/cells7120228
APA StyleBrachtlova, T., & Van Beusechem, V. W. (2018). Unleashing the Full Potential of Oncolytic Adenoviruses against Cancer by Applying RNA Interference: The Force Awakens. Cells, 7(12), 228. https://doi.org/10.3390/cells7120228