Carvacrol Attenuates Hippocampal Neuronal Death after Global Cerebral Ischemia via Inhibition of Transient Receptor Potential Melastatin 7


 and
and 
Abstract
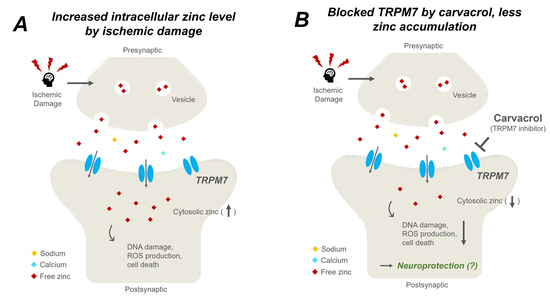
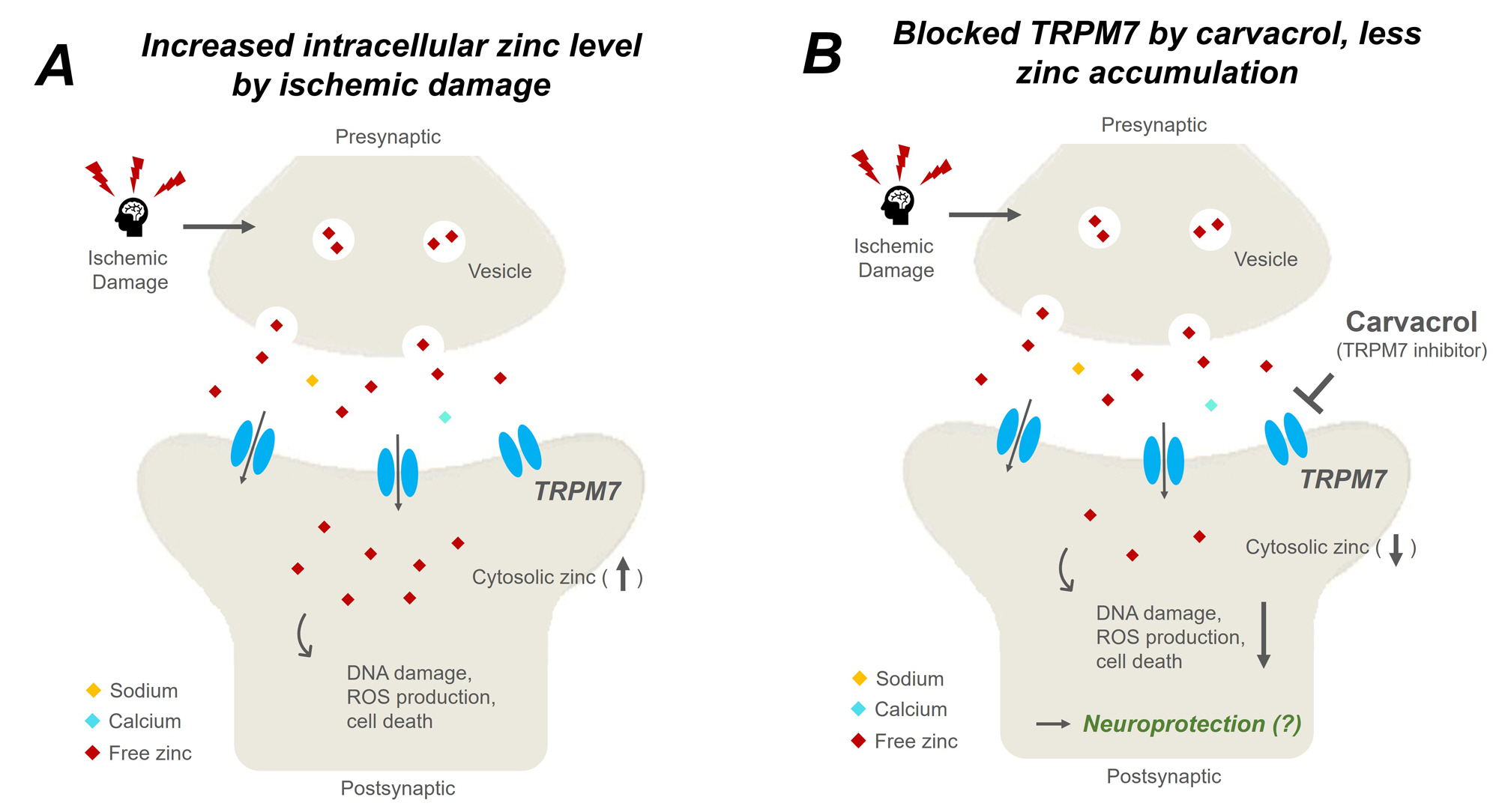
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
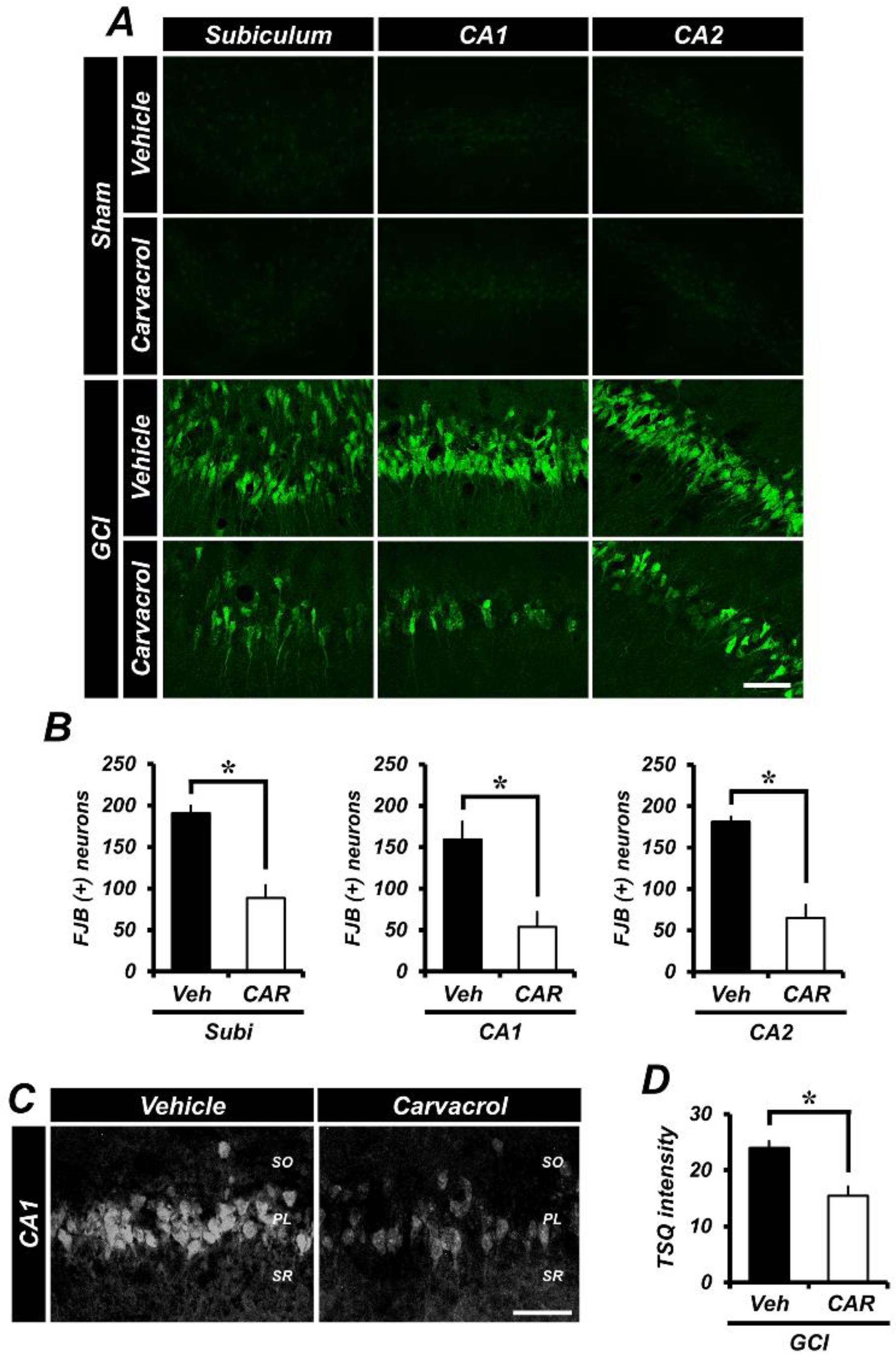
2.1. Carvacrol Attenuates Global Cerebral Ischemia-Induced Hippocampal Neuron Death
2.2. Carvacrol Administration Decreases Zinc Translocation to the Hippocampal Pyramidal Layer after Global Cerebral Ischemia
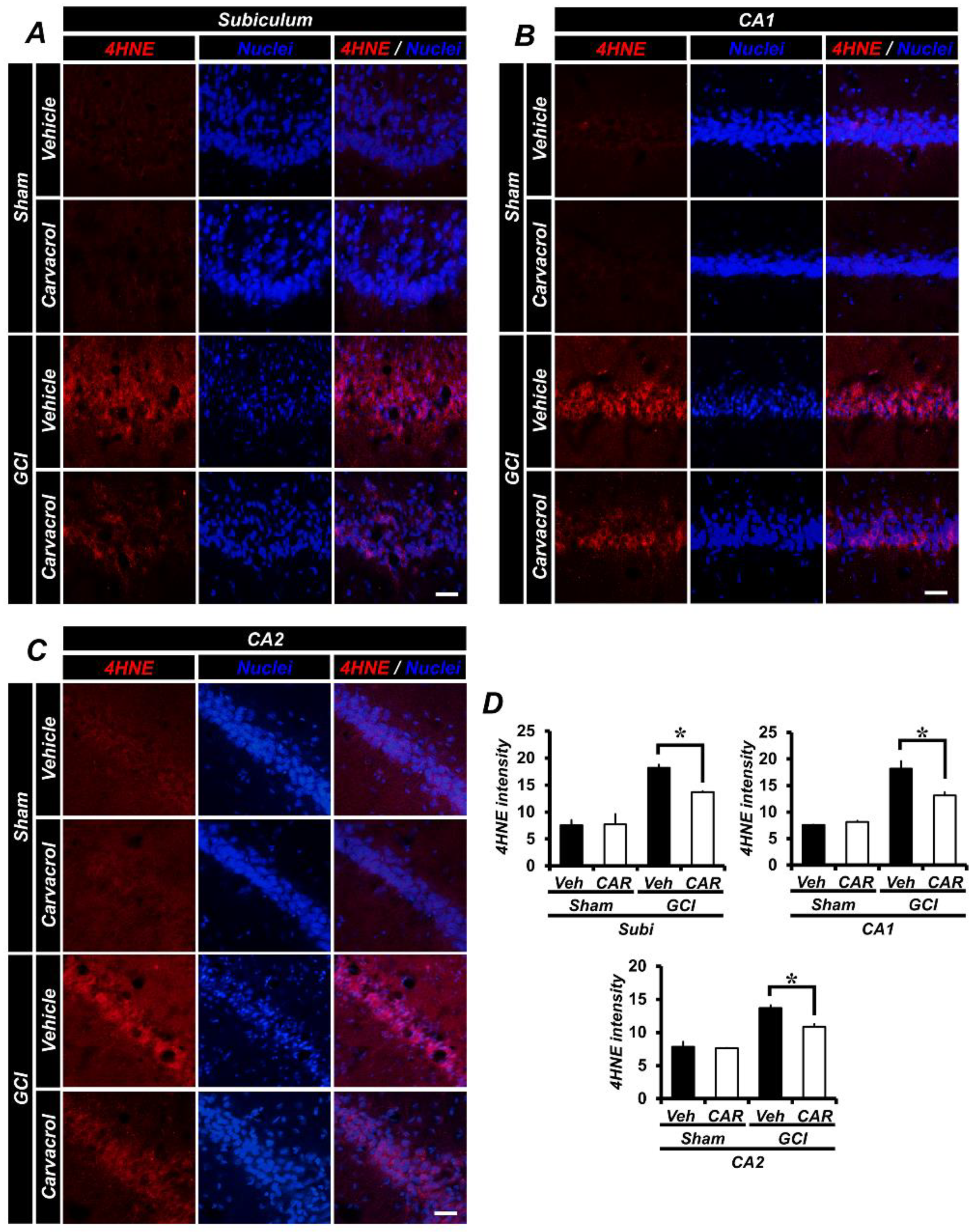
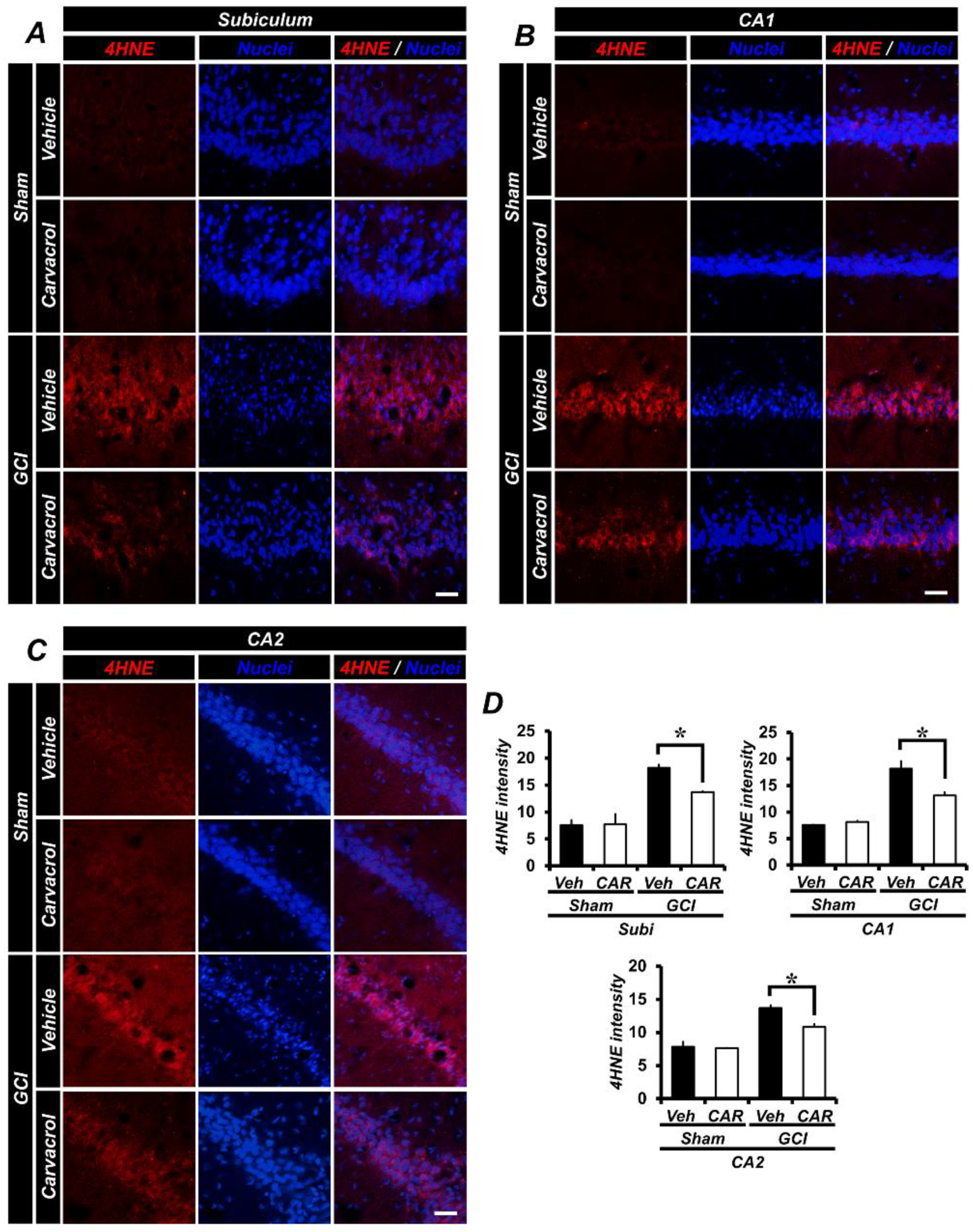
2.3. Carvacrol Administration Reduced Global Cerebral Ischemia-Induced Hippocampal Oxidative Damage
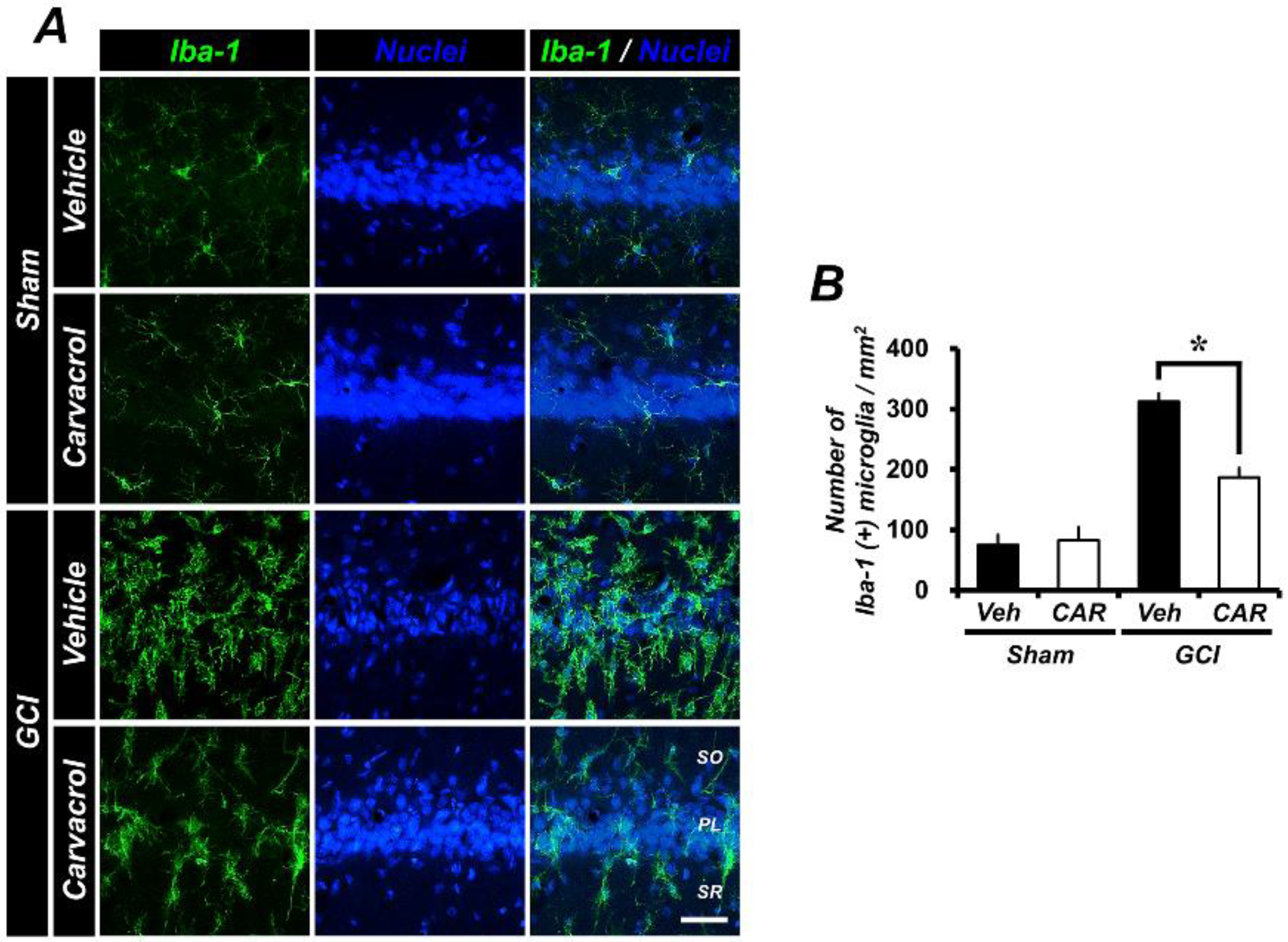
2.4. Carvacrol Administration Decreases Global Cerebral Ischemia-Induced Microglial Activation
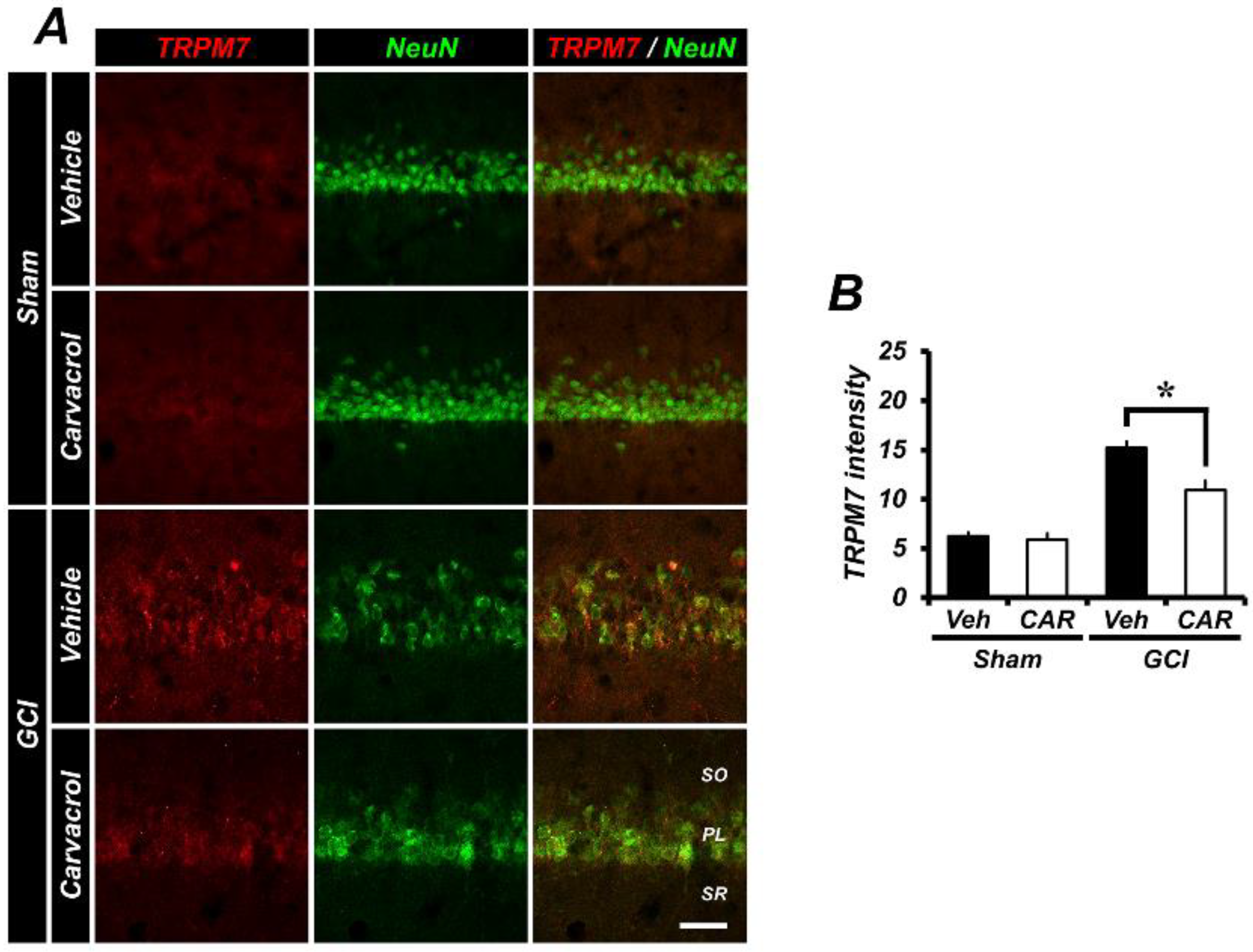
2.5. Carvacrol Administration Inhibits Expression of Transient Receptor Potential Melastatin 7 Channels after Global Cerebral Ischemia
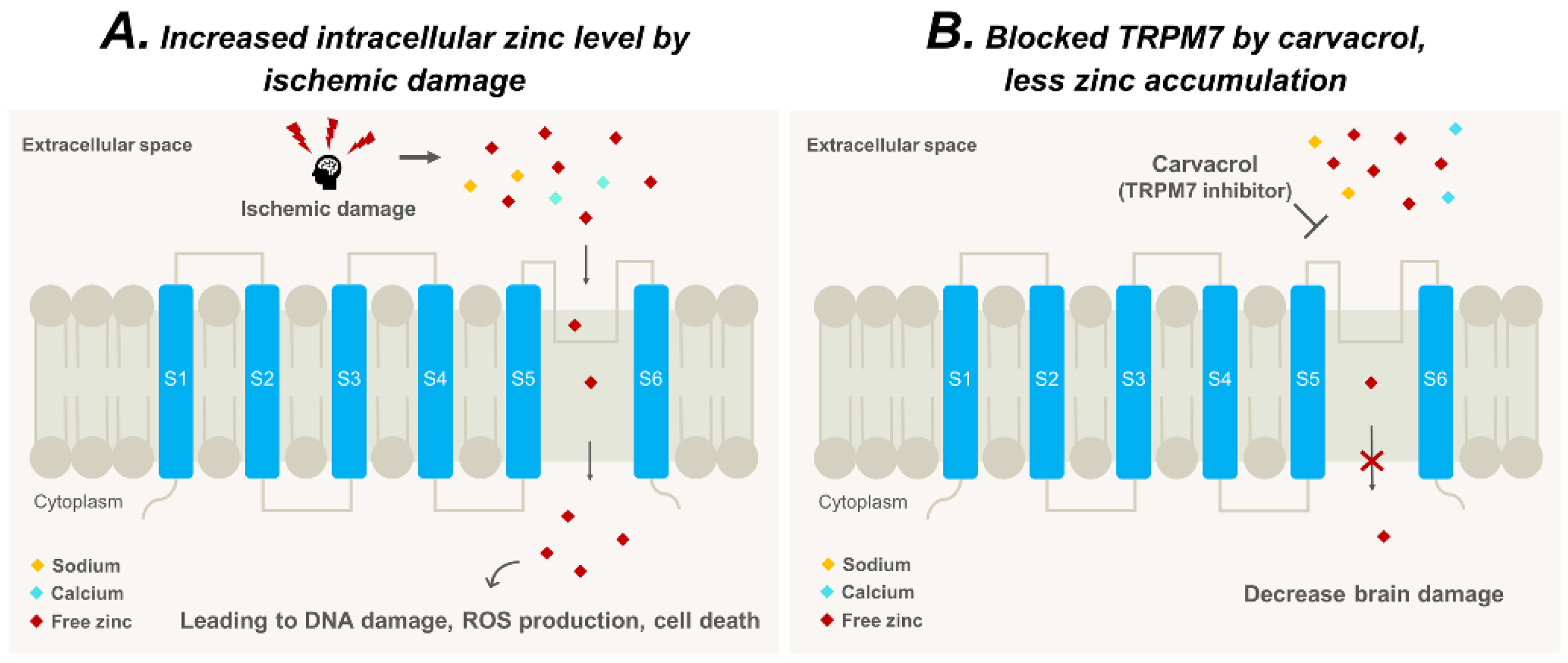
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Experimental Animals
4.3. Global Cerebral Ischemia Surgery
4.4. Carvacrol Administration
4.5. Brain Sample Preparation
4.6. Evaluation of Hippocampal Degenerating Neurons
4.7. Evaluation of Hippocampal Zinc Translocation
4.8. Evaluation of Hippocampal Lipid Peroxidation
4.9. Evaluation of Hippocampal Microglial Activation
4.10. Evaluation of Hippocampal Transient Receptor Potential Melastatin 7 Channel Regulation
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Leng, T.; Shi, Y.; Xiong, Z.G.; Sun, D. Proton-sensitive cation channels and ion exchangers in ischemic brain injury: New therapeutic targets for stroke? Prog. Neurobiol. 2014, 115, 189–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, R.H.C.; Lee, M.H.H.; Wu, C.Y.C.; Couto, E.S.A.; Possoit, H.E.; Hsieh, T.H.; Minagar, A.; Lin, H.W. Cerebral ischemia and neuroregeneration. Neural. Regener. Res 2018, 13, 373–385. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Alkayed, N.J.; Kirsch, J.R.; Hurn, P.D. Mechanisms of ischemic brain damage. Curr. Cardiol. Rep. 2003, 5, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Harukuni, I.; Bhardwaj, A. Mechanisms of brain injury after global cerebral ischemia. Neurol. Clin. 2006, 24, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bernard, S.A.; Gray, T.W.; Buist, M.D.; Jones, B.M.; Silvester, W.; Gutteridge, G.; Smith, K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. Eng. J. Med. 2002, 346, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.D.; Wityk, R.J.; Grega, M.A.; Borowicz, L.M.; Doty, J.R.; Petrofski, J.A.; Baumgartner, W.A. Stroke after cardiac surgery: short- and long-term outcomes. Ann. Thorac. Surg. 2001, 72, 1195–1201. [Google Scholar] [CrossRef]
- Siesjo, B.K. Pathophysiology and treatment of focal cerebral ischemia. Part I: Pathophysiology. J. Neurosurg. 1992, 77, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Traystman, R.J. Animal models of focal and global cerebral ischemia. ILAR J. 2003, 44, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Cheng, C.Y.; Tsai, T.H.; Lin, I.H.; Liu, C.H.; Chiang, S.Y.; Lin, J.G.; Lao, C.J.; Tang, N.Y. Paeonol reduced cerebral infarction involving the superoxide anion and microglia activation in ischemia-reperfusion injured rats. J. Ethnopharmacol. 2006, 106, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Maier, C.M.; Chan, P.H. Role of superoxide dismutases in oxidative damage and neurodegenerative disorders. Neuroscientist 2002, 8, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.K.; Kho, A.R.; Choi, B.Y.; Lee, S.H.; Jeong, J.H.; Lee, S.H.; Park, K.H.; Park, J.B.; Suh, S.W. Combined treatment with dichloroacetic acid and pyruvate reduces hippocampal neuronal death after transient cerebral ischemia. Front Neurol. 2018, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 2001, 14, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Cuajungco, M.P.; Lees, G.J. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiol. Dis. 1997, 4, 137–169. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Hernandez, M.D.; McGinty, J.F. Translocation of zinc may contribute to seizure-induced death of neurons. Brain Res. 1989, 480, 317–321. [Google Scholar] [CrossRef]
- Inoue, K.; Branigan, D.; Xiong, Z.G. Zinc-induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J. Biol. Chem. 2010, 285, 7430–7439. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Lee, B.E.; Kim, J.H.; Kim, H.J.; Sohn, M.; Song, H.K.; Chung, T.N.; Suh, S.W. Colchicine induced intraneuronal free zinc accumulation and dentate granule cell degeneration. Metallomics 2014, 6, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Calcium-mediated neurotoxicity: Relationship to specific channel types and role in ischemic damage. Trends Neurosci. 1988, 11, 465–469. [Google Scholar] [CrossRef]
- Chuah, M.I.; Tennent, R.; Jacobs, I. Response of olfactory Schwann cells to intranasal zinc sulfate irrigation. J. Neurosci. Res. 1995, 42, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of protocatechuic acid (PCA) on global cerebral ischemia-induced hippocampal neuronal death. Int. J. Mol. Sci. 2018, 19, 1420. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kwon, L.M.; Sohn, M.; Song, H.K.; Suh, S.W. Zinc chelation reduces hippocampal neurogenesis after pilocarpine-induced seizure. PLoS ONE 2012, 7, e48543. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Hamby, A.M.; Gum, E.T.; Shin, B.S.; Won, S.J.; Sheline, C.T.; Chan, P.H.; Swanson, R.A. Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death. J. Cereb. Blood Flow Metab. 2008, 28, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Desai, B.N.; Navarro, B.; Donovan, A.; Andrews, N.C.; Clapham, D.E. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 2008, 322, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; Iihara, K.; Wei, W.L.; Xiong, Z.G.; Arundine, M.; Cerwinski, W.; MacDonald, J.F.; Tymianski, M. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003, 115, 863–877. [Google Scholar] [CrossRef]
- Jiang, H.; Tian, S.L.; Zeng, Y.; Li, L.L.; Shi, J. TrkA pathway(s) is involved in regulation of TRPM7 expression in hippocampal neurons subjected to ischemic-reperfusion and oxygen-glucose deprivation. Brain Res. Bull. 2008, 76, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.S.; Jackson, M.F.; Martin, L.J.; Jansen, K.; Teves, L.; Cui, H.; Kiyonaka, S.; Mori, Y.; Jones, M.; Forder, J.P.; et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat. Neurosci. 2009, 12, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, W.T.; Zhang, S.Y.; Zhou, Y.F.; Zhang, B.F.; Liang, Z.Q.; Liu, Y.H.; Wei, Y.; Li, C.K.; Meng, X.J.; Xia, M.; et al. Carvacrol attenuates traumatic neuronal injury through store-operated Ca2+ entry-independent regulation of intracellular Ca2+ homeostasis. Neurochem. Int. 2015, 90, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Kovac, S.; Morris, G.; Walker, M.C. Carvacrol after status epilepticus (SE) prevents recurrent SE, early seizures, cell death, and cognitive decline. Epilepsia 2017, 58, 263–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dati, L.M.; Ulrich, H.; Real, C.C.; Feng, Z.P.; Sun, H.S.; Britto, L.R. Carvacrol promotes neuroprotection in the mouse hemiparkinsonian model. Neuroscience 2017, 356, 176–181. [Google Scholar] [CrossRef] [PubMed]
- da Silva Lima, M.; Quintans-Junior, L.J.; de Santana, W.A.; Kaneto, C.M.; Soares, M.B.P.; Villarreal, C.F. Anti-inflammatory effects of carvacrol: Evidence for a key role of interleukin-10. Eur. J. Pharmacol. 2013, 699, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Parnas, M.; Peters, M.; Dadon, D.; Lev, S.; Vertkin, I.; Slutsky, I.; Minke, B. Carvacrol is a novel inhibitor of Drosophila TRPL and mammalian TRPM7 channels. Cell Calcium 2009, 45, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, J.C.; Mattsson, B.; Andreasen, A.; Johansson, B.B. Rapid disappearance of zinc positive terminals in focal brain ischemia. Brain Res. 1998, 812, 265–269. [Google Scholar] [CrossRef]
- Shuttleworth, C.W.; Weiss, J.H. Zinc: New clues to diverse roles in brain ischemia. Trends Pharmacol. Sci. 2011, 32, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.; Samman, S. Zinc and redox signaling: Perturbations associated with cardiovascular disease and diabetes mellitus. Antioxid. Redox Signal. 2010, 13, 1549–1573. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Onorato, J.M.; Thorpe, S.R.; Baynes, J.W. Immunohistochemical and ELISA assays for biomarkers of oxidative stress in aging and disease. Ann. NY Acad. Sci. 1998, 854, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.S. Role of TRPM7 in cerebral ischaemia and hypoxia. J. Physiol. 2017, 595, 3077–3083. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.W. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988, 1, 623–634. [Google Scholar] [CrossRef]
- Lai, T.W.; Shyu, W.C.; Wang, Y.T. Stroke intervention pathways: NMDA receptors and beyond. Trends Mol. Med. 2011, 17, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Lee, B.E.; Lee, S.H.; Kho, A.R.; Jung, H.J.; Sohn, M.; Song, H.K.; Suh, S.W. Decreased cysteine uptake by EAAC1 gene deletion exacerbates neuronal oxidative stress and neuronal death after traumatic brain injury. Amino Acids 2016, 48, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Garnier, P.; Aoyama, K.; Chen, Y.; Swanson, R.A. Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol. Dis. 2004, 16, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xu, B.; Xiao, A.; Liu, L.; Fang, X.; Liu, R.; Turlova, E.; Barszczyk, A.; Zhong, X.; Sun, C.L.; et al. TRPM7 inhibitor carvacrol protects brain from neonatal hypoxic-ischemic injury. Mol Brain 2015, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Kozai, D.; Ogawa, N.; Mori, Y. Redox regulation of transient receptor potential channels. Antioxid Redox Signal 2014, 21, 971–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, Y.; Wakamori, M.; Ishii, M.; Maeno, E.; Nishida, M.; Yoshida, T.; Yamada, H.; Shimizu, S.; Mori, E.; Kudoh, J.; et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol. Cell 2002, 9, 163–173. [Google Scholar] [CrossRef]
- Takahashi, N.; Kozai, D.; Kobayashi, R.; Ebert, M.; Mori, Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011, 50, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Dantas, B.P.; Alves, Q.L.; de Assis, K.S.; Ribeiro, T.P.; de Almeida, M.M.; de Vasconcelos, A.P.; de Araujo, D.A.; de Andrade Braga, V.; de Medeiros, I.A.; Alencar, J.L.; et al. Participation of the TRP channel in the cardiovascular effects induced by carvacrol in normotensive rat. Vascul. Pharmacol. 2015, 67–69, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Vogt-Eisele, A.K.; Weber, K.; Sherkheli, M.A.; Vielhaber, G.; Panten, J.; Gisselmann, G.; Hatt, H. Monoterpenoid agonists of TRPV3. Br. J. Pharmacol. 2007, 151, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Macianskiene, R.; Martisiene, I.; Zablockaite, D.; Gendviliene, V. Characterization of Mg2+-regulated TRPM7-like current in human atrial myocytes. J. Biomed. Sci. 2012, 19, 75. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.N.; Fiskum, G.; Beal, M.F. Mitochondria in neurodegeneration: Bioenergetic function in cell life and death. J. Cereb. Blood Flow Metab. 1999, 19, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Auer, R.N.; Siesjo, B.K. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol. 1984, 64, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Kasarskis, E.J.; Ringo, D.; Frederickson, R.E. A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 1987, 20, 91–103. [Google Scholar] [CrossRef]
- Choi, B.Y.; Hong, D.K.; Suh, S.W. ZnT3 gene deletion reduces colchicine-induced dentate granule cell degeneration. Int. J. Mol. Sci. 2017, 18, 2189. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, D.K.; Choi, B.Y.; Kho, A.R.; Lee, S.H.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.-H.; Suh, S.W. Carvacrol Attenuates Hippocampal Neuronal Death after Global Cerebral Ischemia via Inhibition of Transient Receptor Potential Melastatin 7. Cells 2018, 7, 231. https://doi.org/10.3390/cells7120231
Hong DK, Choi BY, Kho AR, Lee SH, Jeong JH, Kang BS, Kang DH, Park K-H, Suh SW. Carvacrol Attenuates Hippocampal Neuronal Death after Global Cerebral Ischemia via Inhibition of Transient Receptor Potential Melastatin 7. Cells. 2018; 7(12):231. https://doi.org/10.3390/cells7120231
Chicago/Turabian StyleHong, Dae Ki, Bo Young Choi, A Ra Kho, Song Hee Lee, Jeong Hyun Jeong, Beom Seok Kang, Dong Hyeon Kang, Kyoung-Ha Park, and Sang Won Suh. 2018. "Carvacrol Attenuates Hippocampal Neuronal Death after Global Cerebral Ischemia via Inhibition of Transient Receptor Potential Melastatin 7" Cells 7, no. 12: 231. https://doi.org/10.3390/cells7120231
APA StyleHong, D. K., Choi, B. Y., Kho, A. R., Lee, S. H., Jeong, J. H., Kang, B. S., Kang, D. H., Park, K.-H., & Suh, S. W. (2018). Carvacrol Attenuates Hippocampal Neuronal Death after Global Cerebral Ischemia via Inhibition of Transient Receptor Potential Melastatin 7. Cells, 7(12), 231. https://doi.org/10.3390/cells7120231