The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Chromosomal Instability (CIN) in Cancer
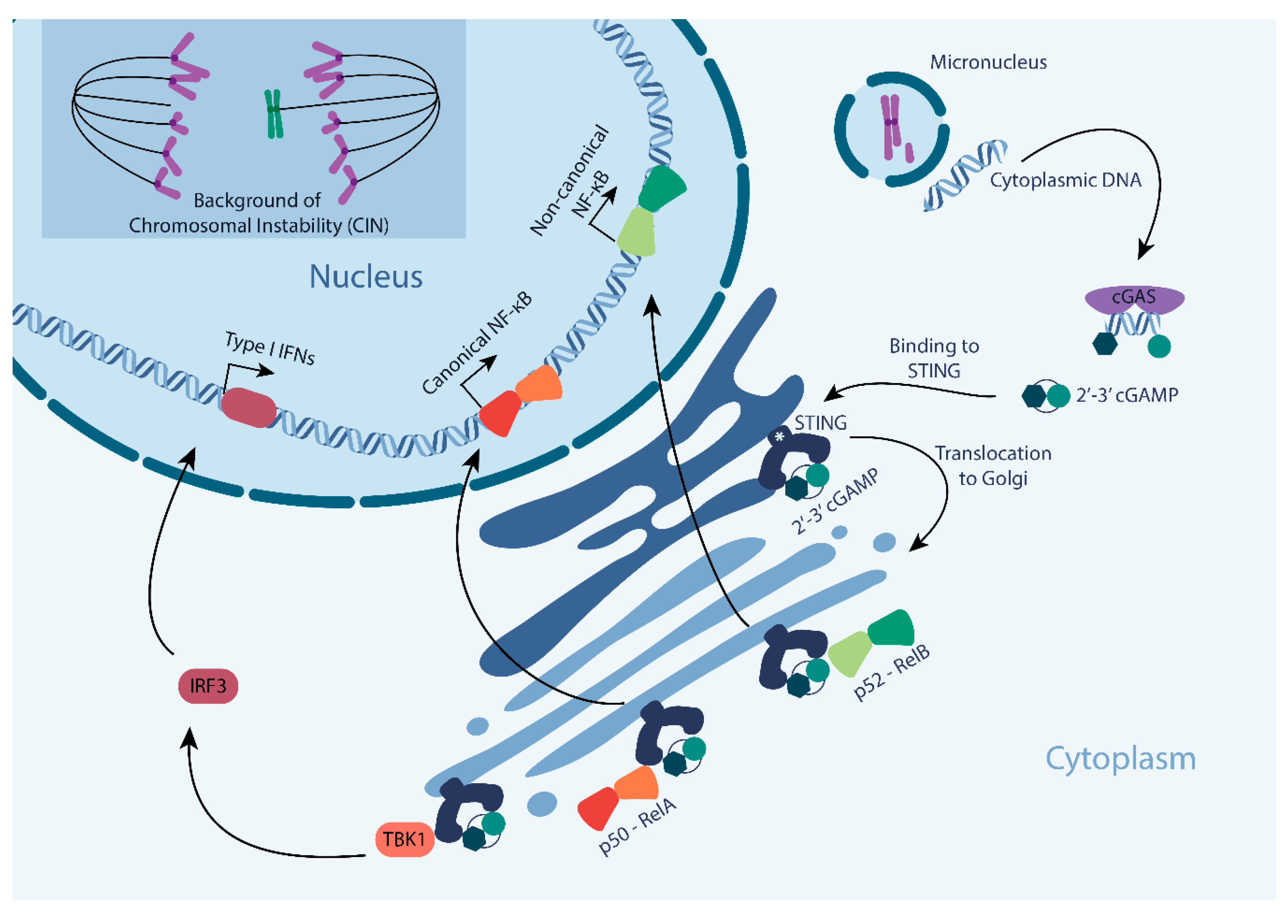
2. Upstream Triggers of the cGAS-STING Pathway
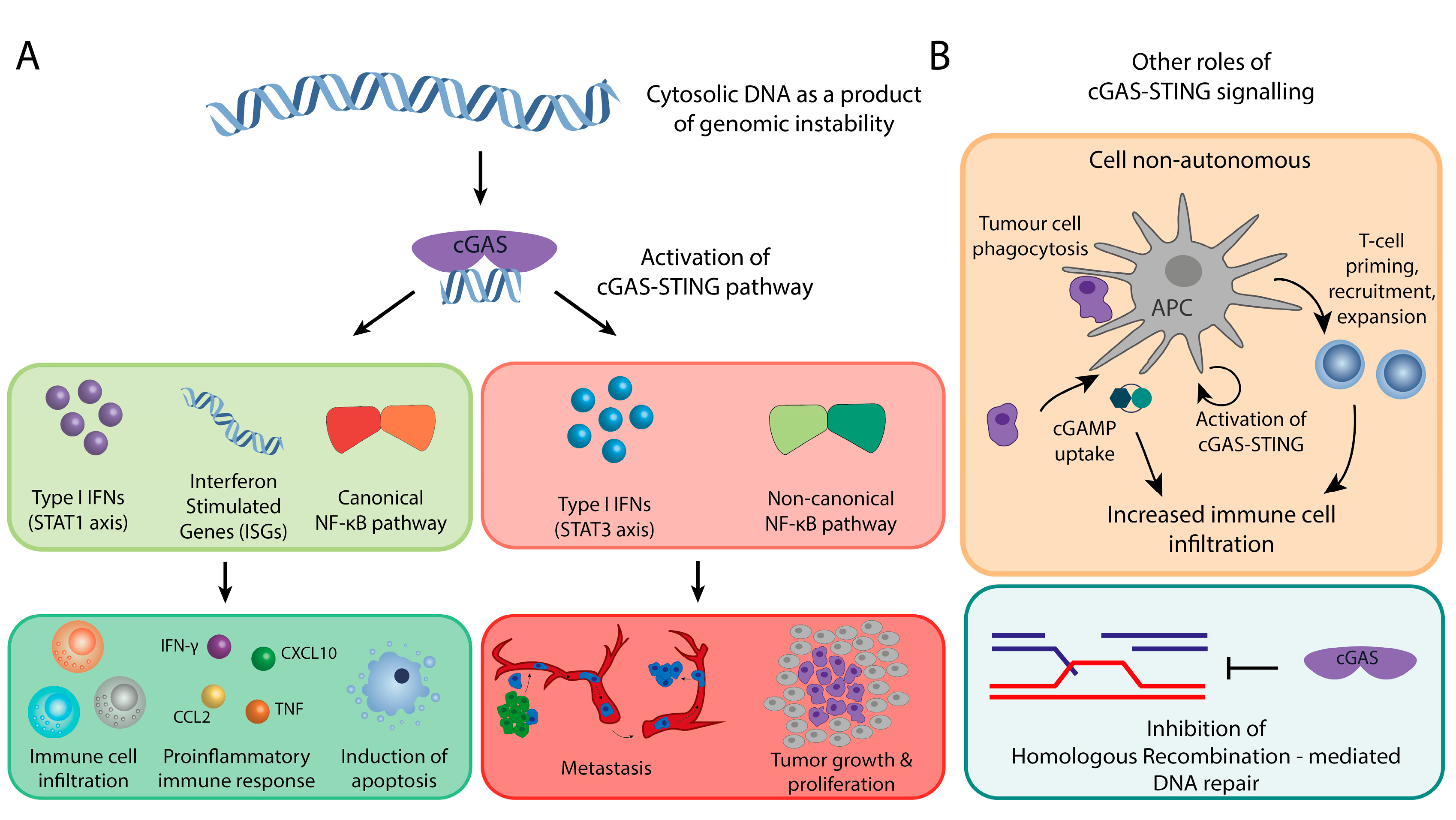
3. Downstream Effects of the cGAS-STING Pathway: The Innate Immune System Response
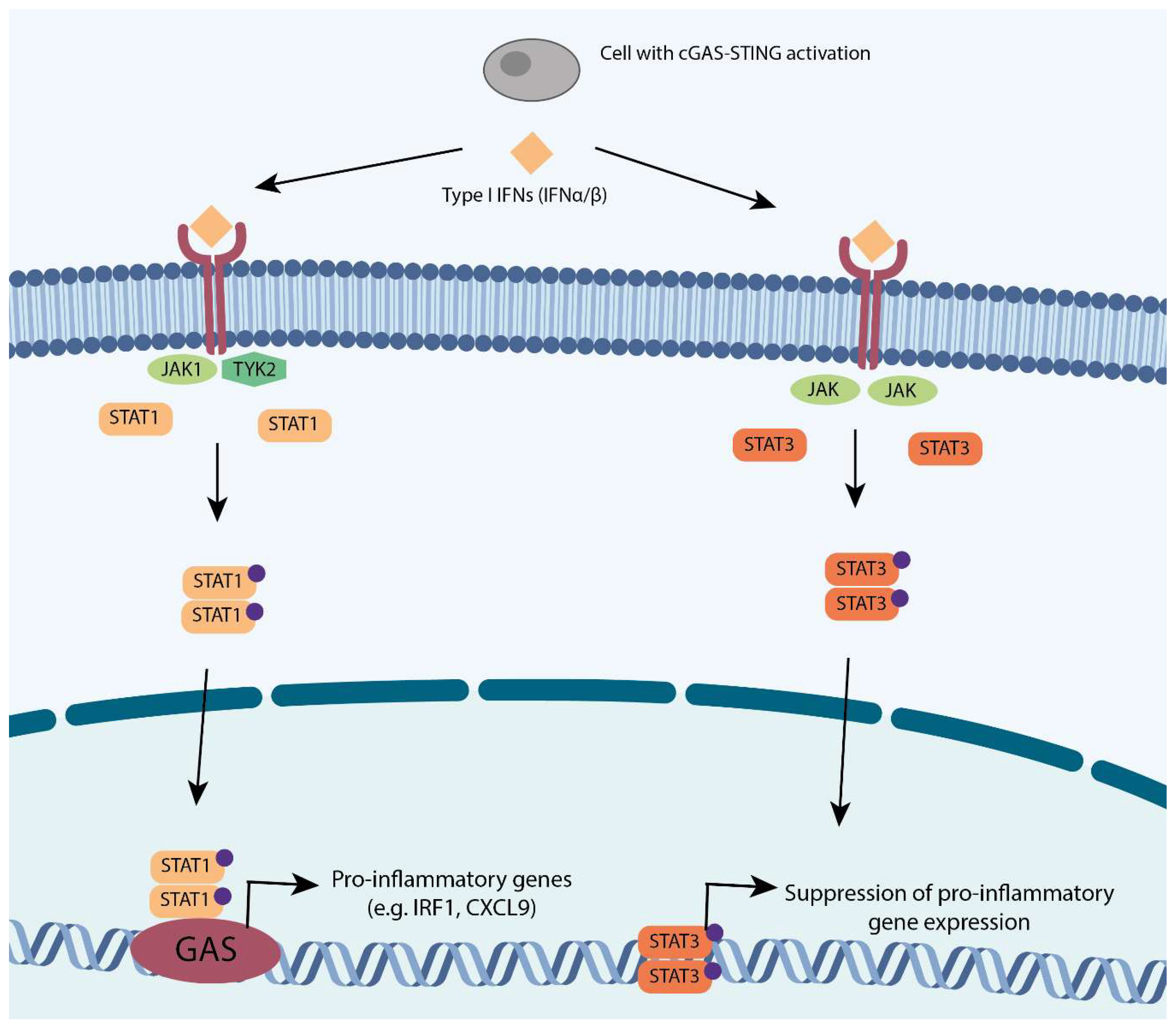
3.1. Type I Interferon (IFN) Response and Signal Transducer and Activator of Transcription (STAT) Response
3.2. Nuclear Factor kB (NF-KB) Response
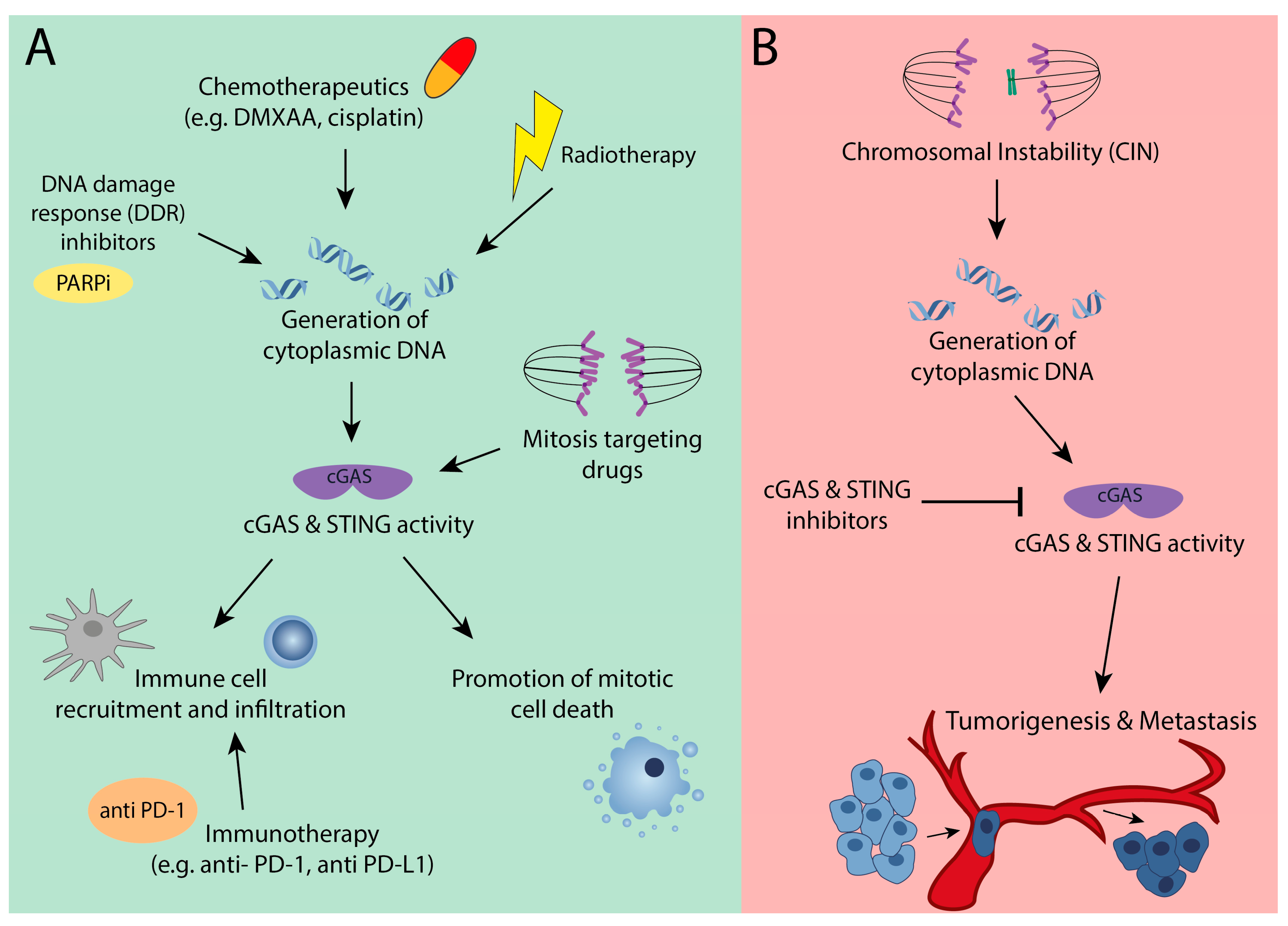
4. The Bright Side: cGAS as a Suppressor of Chromosomal Instable Tumors
5. The Dark Side: cGAS-STING Signaling is Required for CIN Tumors
6. Therapeutic Implications of Altered cGAS-STING Signaling
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Bolhaqueiro, A.C.F.; Ponsioen, B.; Bakker, B.; Klaasen, S.J.; Kucukkose, E.; van Jaarsveld, R.H.; Vivié, J.; Verlaan-Klink, I.; Hami, N.; Spierings, D.C.J.; et al. Ongoing chromosomal instability and karyotype evolution in human colorectal cancer organoids. Nat. Genet. 2019, 51, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Bakker, B.; Taudt, A.; Belderbos, M.E.; Porubsky, D.; Spierings, D.C.J.; de Jong, T.V.; Halsema, N.; Kazemier, H.G.; Hoekstra-Wakker, K.; Bradley, A.; et al. Single-cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol. 2016, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Landau, D.A. Chromosomal Instability as a Driver of Tumor Heterogeneity and Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a029611. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z. Evolution of aneuploidy: Overcoming the original CIN. Genes Dev. 2018, 32, 1459–1460. [Google Scholar] [CrossRef]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 2008, 121, 3859–3866. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Xu, H.; Litchfield, K.; Rowan, A.; Chambers, T.; Lopez, J.I.; Nicol, D.; O’Brien, T.; Larkin, J.; Horswell, S.; et al. Tracking Cancer Evolution Reveals Constrained Routes to Metastases: TRACERx Renal. Cell 2018, 173, 581–594.e12. [Google Scholar] [CrossRef] [PubMed]
- Ott, K.; Vogelsang, H.; Mueller, J.; Becker, K.; Müller, M.; Fink, U.; Siewert, J.R.; Höfler, H.; Keller, G. Chromosomal instability rather than p53 mutation is associated with response to neoadjuvant cisplatin-based chemotherapy in gastric carcinoma. Clin. Cancer Res. 2003, 9, 2307–2315. [Google Scholar] [PubMed]
- Swanton, C.; Nicke, B.; Schuett, M.; Eklund, A.C.; Ng, C.; Li, Q.; Hardcastle, T.; Lee, A.; Roy, R.; East, P.; et al. Chromosomal instability determines taxane response. Proc. Natl. Acad. Sci. USA 2009, 106, 8671–8676. [Google Scholar] [CrossRef]
- Lee, H.-S.; Lee, N.C.O.; Kouprina, N.; Kim, J.-H.; Kagansky, A.; Bates, S.; Trepel, J.B.; Pommier, Y.; Sackett, D.; Larionov, V. Therapeutics, Targets, and Chemical Biology Effects of Anticancer Drugs on Chromosome Instability and New Clinical Implications for Tumor-Suppressing Therapies. Cancer Res. 2016, 76, 902–911. [Google Scholar] [CrossRef]
- Zaki, B.I.; Suriawinata, A.A.; Eastman, A.R.; Garner, K.M.; Bakhoum, S.F. Chromosomal instability portends superior response of rectal adenocarcinoma to chemoradiation therapy. Cancer 2014, 120, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. CGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Diner, E.J.; Burdette, D.L.; Wilson, S.C.; Monroe, K.M.; Kellenberger, C.A.; Hyodo, M.; Hayakawa, Y.; Hammond, M.C.; Vance, R.E. The Innate Immune DNA Sensor cGAS Produces a Noncanonical Cyclic Dinucleotide that Activates Human STING. Cell Rep. 2013, 3, 1355–1361. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef]
- Civril, F.; Deimling, T.; de Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.-P. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef]
- Shu, C.; Li, X.; Li, P. The mechanism of double-stranded DNA sensing through the cGAS-STING pathway. Cytokine Growth Factor Rev. 2014, 25, 641–648. [Google Scholar] [CrossRef]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, S.; Woo, J.S.; Wu, B.; El-Sherbiny, Y.M.; Leung, J.; Chupradit, K.; Rice, L.; Seo, G.J.; Calmettes, G.; Ramakrishna, C.; et al. The Ca2+ sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat. Immunol. 2019, 20, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, ra20. [Google Scholar] [CrossRef] [PubMed]
- Gonugunta, V.K.; Sakai, T.; Pokatayev, V.; Yang, K.; Wu, J.; Dobbs, N.; Yan, N. Trafficking-Mediated STING Degradation Requires Sorting to Acidified Endolysosomes and Can Be Targeted to Enhance Anti-tumor Response. Cell Rep. 2017, 21, 3234–3242. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Capobianchi, M.R.; Uleri, E.; Caglioti, C.; Dolei, A. Type I IFN family members: Similarity, differences and interaction. Cytokine Growth Factor Rev. 2015, 26, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Kanda, N.; Shimizu, T.; Tada, Y.; Watanabe, S. IL-18 enhances IFN-γ-induced production of CXCL9, CXCL10, and CXCL11 in human keratinocytes. Eur. J. Immunol. 2007, 37, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 2018, 49, 754–763.e4. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, G.; Pack, M.; Thomas, D.; Paludan, C.; Schmid, D.; Strowig, T.; Bougras, G.; Muller, W.A.; Moretta, L.; Munz, C. Distinct roles of IL-12 and IL-15 in human natural killer cell activation by dendritic cells from secondary lymphoid organs. Proc. Natl. Acad. Sci. 2004, 101, 16606–16611. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; Broz, M.L.; Ranger, J.J.; Ozcelik, J.; Ahn, R.; Zuo, D.; Ursini-Siegel, J.; Hallett, M.T.; Krummel, M.; Muller, W.J. STAT3 Establishes an Immunosuppressive Microenvironment during the Early Stages of Breast Carcinogenesis to Promote Tumor Growth and Metastasis. Cancer Res. 2016, 76, 1416–1428. [Google Scholar] [CrossRef] [PubMed]
- Cacalano, N.A. Regulation of Natural Killer Cell Function by STAT3. Front. Immunol. 2016, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Grégoire, C.; Chasson, L.; Luci, C.; Tomasello, E.; Geissmann, F.; Vivier, E.; Walzer, T. The trafficking of natural killer cells. Immunol. Rev. 2007, 220, 169–182. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- D’Ignazio, L.; Batie, M.; Rocha, S. TNFSF14/LIGHT, a Non-Canonical NF-kB Stimulus, Induces the HIF Pathway. Cells 2018, 7, 102. [Google Scholar]
- House, C.D.; Grajales, V.; Ozaki, M.; Jordan, E.; Wubneh, H.; Kimble, D.C.; James, J.M.; Kim, M.K.; Annunziata, C.M. IKKε cooperates with either MEK or non-canonical NF-kB driving growth of triple-negative breast cancer cells in different contexts. BMC Cancer 2018, 18, 595. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Tegowski, M.; Baldwin, A. Noncanonical NF-κB in Cancer. Biomedicines 2018, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- WANG, J.; YI, S.; ZHOU, J.; ZHANG, Y.; GUO, F. The NF-κB subunit RelB regulates the migration and invasion abilities and the radio-sensitivity of prostate cancer cells. Int. J. Oncol. 2016, 49, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Belguise, K.; O’Neill, C.F.; Sánchez-Morgan, N.; Romagnoli, M.; Eddy, S.F.; Mineva, N.D.; Yu, Z.; Min, C.; Trinkaus-Randall, V.; et al. RelB NF-kappaB represses estrogen receptor alpha expression via induction of the zinc finger protein Blimp1. Mol. Cell. Biol. 2009, 29, 3832–3844. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.; McCann, J.L.; Starrett, G.J.; Kosyakovsky, L.; Luengas, E.M.; Molan, A.M.; Burns, M.B.; McDougle, R.M.; Parker, P.J.; Brown, W.L.; et al. The PKC/NF-κB signaling pathway induces APOBEC3B expression in multiple human cancers. Cancer Res. 2015, 75, 4538–4547. [Google Scholar] [CrossRef] [PubMed]
- Narvaiza, I.; Landry, S.; Weitzman, M.D. APOBEC3 proteins and genomic stability: the high cost of a good defense. Cell Cycle 2012, 11, 33–38. [Google Scholar] [CrossRef]
- Walker, J.-A.; Boreham, D.R.; Unrau, P.; Duncan, A.M.V. Chromosome content and ultrastructure of radiation-induced micronuclei. Mutagenesis 1996, 11, 419–424. [Google Scholar] [CrossRef]
- Santaguida, S.; Richardson, A.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651.e5. [Google Scholar] [CrossRef]
- Xu, M.M.; Pu, Y.; Han, D.; Shi, Y.; Cao, X.; Liang, H.; Chen, X.; Li, X.-D.; Deng, L.; Chen, Z.J.; et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein α Signaling. Immunity 2017, 47, 363–373.e5. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ge, M.; Lu, S.; Shi, J.; Li, X.; Wang, M.; Huang, J.; Shao, Y.; Huang, Z.; Zhang, J.; et al. Pro-inflammatory effects of the Th1 chemokine CXCL10 in acquired aplastic anaemia. Cytokine 2017, 94, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Zierhut, C.; Yamaguchi, N.; Paredes, M.; Luo, J.-D.; Carroll, T.; Funabiki, H. The Cytoplasmic DNA Sensor cGAS Promotes Mitotic Cell Death. Cell 2019, 178, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.J.; Barber, G.N. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018, 37, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Liang, D.; Xiao-Feng, H.; Guan-Jun, D.; Er-Ling, H.; Sheng, C.; Ting-Ting, W.; Qin-Gang, H.; Yan-Hong, N.; Ya-Yi, H. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim. Biophys. Acta - Mol. Basis Dis. 2015, 1852, 2494–2503. [Google Scholar] [CrossRef]
- Gaston, J.; Cheradame, L.; Yvonnet, V.; Deas, O.; Poupon, M.-F.; Judde, J.-G.; Cairo, S.; Goffin, V. Intracellular STING inactivation sensitizes breast cancer cells to genotoxic agents. Oncotarget 2016, 7, 77205. [Google Scholar] [CrossRef]
- Takashima, K.; Takeda, Y.; Oshiumi, H.; Shime, H.; Okabe, M.; Ikawa, M.; Matsumoto, M.; Seya, T. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem. Biophys. Res. Commun. 2016, 478, 1764–1771. [Google Scholar] [CrossRef]
- Tang, C.-H.A.; Zundell, J.A.; Ranatunga, S.; Lin, C.; Nefedova, Y.; Del Valle, J.R.; Hu, C.-C.A. Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res. 2016, 76, 2137–2152. [Google Scholar] [CrossRef]
- Wu, C.; Molavi, O.; Zhang, H.; Gupta, N.; Alshareef, A.; Bone, K.M.; Gopal, K.; Wu, F.; Lewis, J.T.; Douglas, D.N.; et al. STAT1 is phosphorylated and downregulated by the oncogenic tyrosine kinase NPM-ALK in ALK-positive anaplastic large-cell lymphoma. Blood 2015, 126, 336–345. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Dyer, K.F.; Kimak, M.; Zhang, Q.; Gooding, W.E.; Chaillet, J.R.; Chai, R.L.; Ferrell, R.E.; Zamboni, B.; Hunt, J.; et al. Decreased STAT1 Expression by Promoter Methylation in Squamous Cell Carcinogenesis. JNCI J. Natl. Cancer Inst. 2006, 98, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Qian, Q.; Lv, Y.; Li, P. SOCS1 is associated with clinical progression and acts as an oncogenic role in triple-negative breast cancer. IUBMB Life 2018, 70, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tahk, S.; Yee, K.M.; Yang, R.; Yang, Y.; Mackie, R.; Hsu, C.; Chernishof, V.; O’Brien, N.; Jin, Y.; et al. PIAS1 regulates breast tumorigenesis through selective epigenetic gene silencing. PLoS ONE 2014, 9, e89464. [Google Scholar] [CrossRef] [PubMed]
- Puhr, M.; Hoefer, J.; Eigentler, A.; Dietrich, D.; van Leenders, G.; Uhl, B.; Hoogland, M.; Handle, F.; Schlick, B.; Neuwirt, H.; et al. PIAS1 is a determinant of poor survival and acts as a positive feedback regulator of AR signaling through enhanced AR stabilization in prostate cancer. Oncogene 2016, 35, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Pensa, S.; Regis, G.; Novelli, F.; Poli, V. STAT1 and STAT3 in tumorigenesis: A matter of balance. JAK-STAT 2012, 1, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, L.; Jin, J.; Luan, Y.; Chen, C.; Li, Y.; Chu, H.; Wang, X.; Liao, G.; Yu, Y.; et al. p38 inhibition provides anti–DNA virus immunity by regulation of USP21 phosphorylation and STING activation. J. Exp. Med. 2017, 214, 991–1010. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Wu, X.; Ma, D.; Wu, J.; Wang, L.; Jiang, Y.; Fei, Y.; Zhu, C.; Tan, R.; et al. Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 2018, 563, 131. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Panda, S.; Xue, X.; Liang, F.; Sung, P.; Gekara, N.O. The innate immune DNA sensor cGAS is a negative regulator of DNA repair hence promotes genome instability and cell death. bioRxiv 2018, 465401. [Google Scholar] [CrossRef]
- Elsea, C.R.; Roberts, D.A.; Druker, B.J.; Wood, L.J. Inhibition of p38 MAPK Suppresses Inflammatory Cytokine Induction by Etoposide, 5-Fluorouracil, and Doxorubicin without Affecting Tumoricidal Activity. PLoS ONE 2008, 3, e2355. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yi, M.; Qin, S.; Song, Y.; Chu, Q.; Wu, K. Activating cGAS-STING pathway for the optimal effect of cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djw199. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, A.; Peterson, N.; Khalaj, K.; Vitkin, N.; Robinson, A.; Francis, J.-A.; Koti, M. STING agonist therapy in combination with PD-1 immune checkpoint blockade enhances response to carboplatin chemotherapy in high-grade serous ovarian cancer. Br. J. Cancer 2018, 119, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Heijink, A.M.; Talens, F.; Jae, L.T.; van Gijn, S.E.; Fehrmann, R.S.N.; Brummelkamp, T.R.; van Vugt, M.A.T.M. BRCA2 deficiency instigates cGAS-mediated inflammatory signaling and confers sensitivity to tumor necrosis factor-alpha-mediated cytotoxicity. Nat. Commun. 2019, 10, 100. [Google Scholar] [CrossRef]
- Vincent, J.; Adura, C.; Gao, P.; Luz, A.; Lama, L.; Asano, Y.; Okamoto, R.; Imaeda, T.; Aida, J.; Rothamel, K.; et al. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun. 2017, 8, 750. [Google Scholar] [CrossRef] [PubMed]
- Haag, S.M.; Gulen, M.F.; Reymond, L.; Gibelin, A.; Abrami, L.; Decout, A.; Heymann, M.; van der Goot, F.G.; Turcatti, G.; Behrendt, R.; et al. Targeting STING with covalent small-molecule inhibitors. Nature 2018, 559, 269–273. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, C.; Tijhuis, A.E.; Foijer, F. The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells 2019, 8, 1228. https://doi.org/10.3390/cells8101228
Hong C, Tijhuis AE, Foijer F. The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells. 2019; 8(10):1228. https://doi.org/10.3390/cells8101228
Chicago/Turabian StyleHong, Christy, Andrea E. Tijhuis, and Floris Foijer. 2019. "The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability" Cells 8, no. 10: 1228. https://doi.org/10.3390/cells8101228
APA StyleHong, C., Tijhuis, A. E., & Foijer, F. (2019). The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells, 8(10), 1228. https://doi.org/10.3390/cells8101228