Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Patient Samples
2.2. Annexin V/Propidum Iodide Staining for Apoptotic Cells
2.3. Protein Isolation/Quantification and Western Blot Analysis
2.4. SAM/SAH Reverse Competition ELISA
2.5. Histone Isolation
2.6. Chromatin Immunopreciptation (ChIP)
2.7. RT-PCR
2.8. Clinical Chemotherapeutics
2.9. In Vivo Xenograft Studies
2.10. Statistical Analysis
3. Results
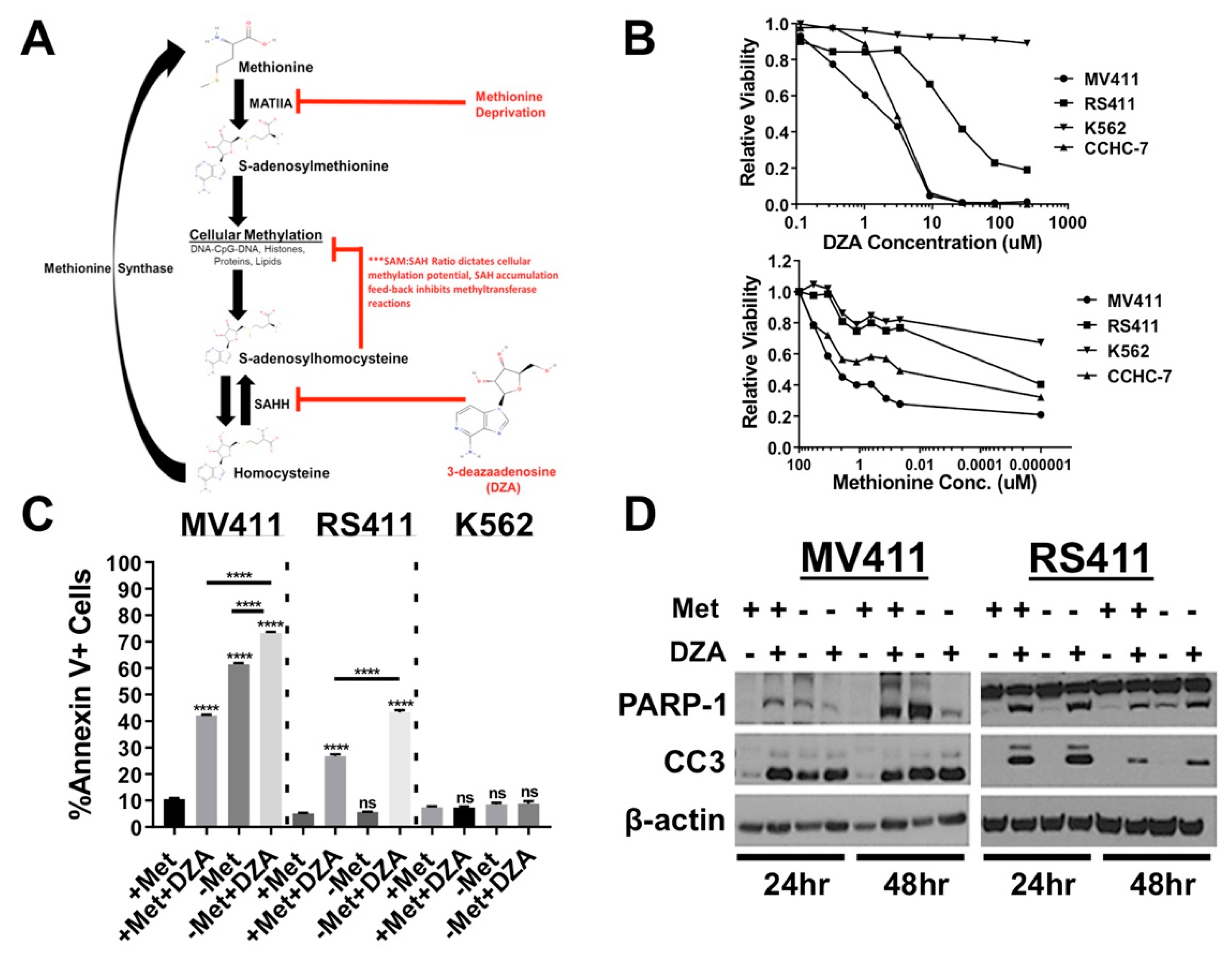
3.1. Alteration of Met/SAM Metabolism Impairs Cellular Viability and Induces Apoptosis in MLL-R Cell Lines
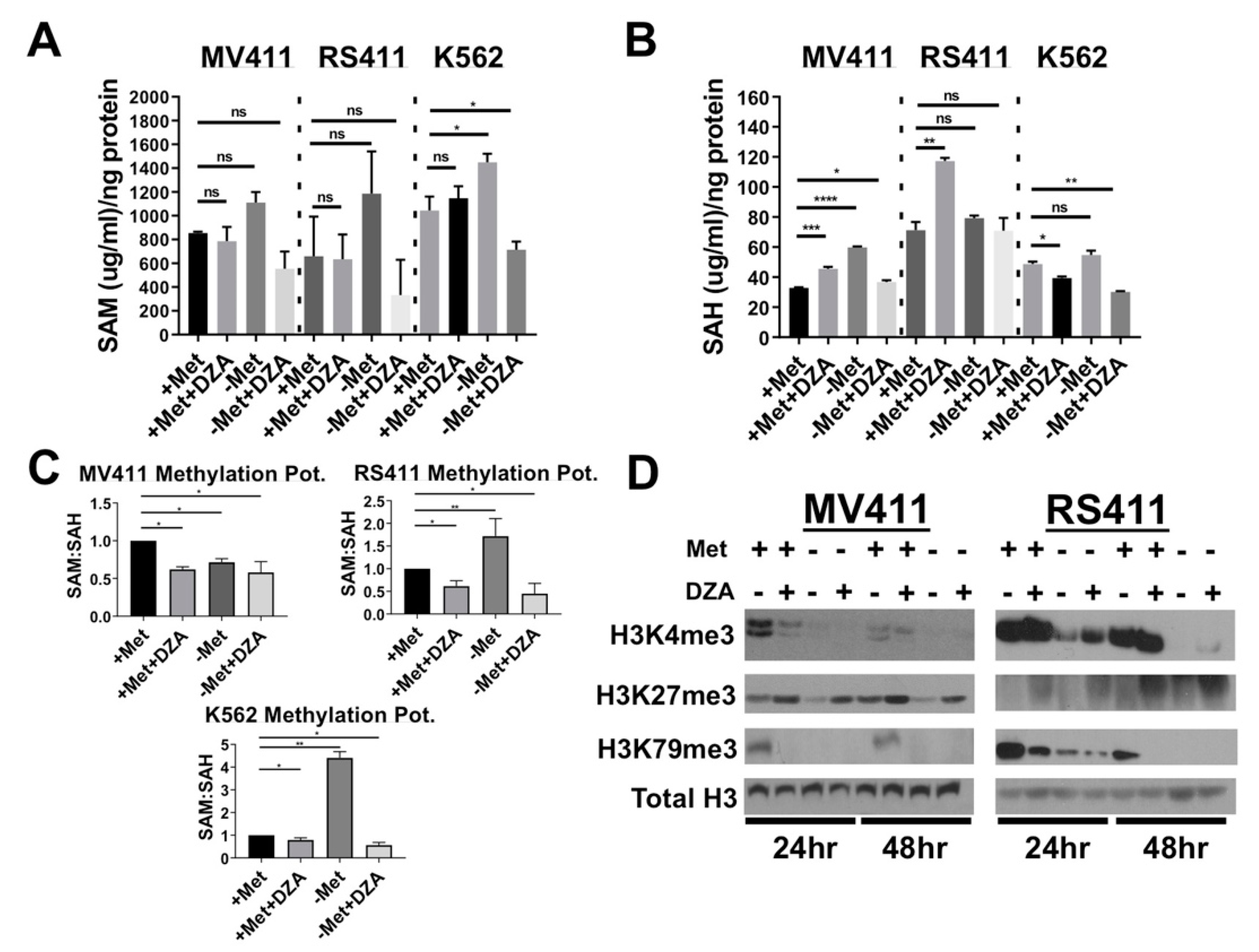
3.2. Perturbation of Met/SAM Metabolism Increases Intracellular SAH, Decreases Overall Methylation Potential, and Alters Global Histone Methylation Dynamics in MLL-R Cell Lines
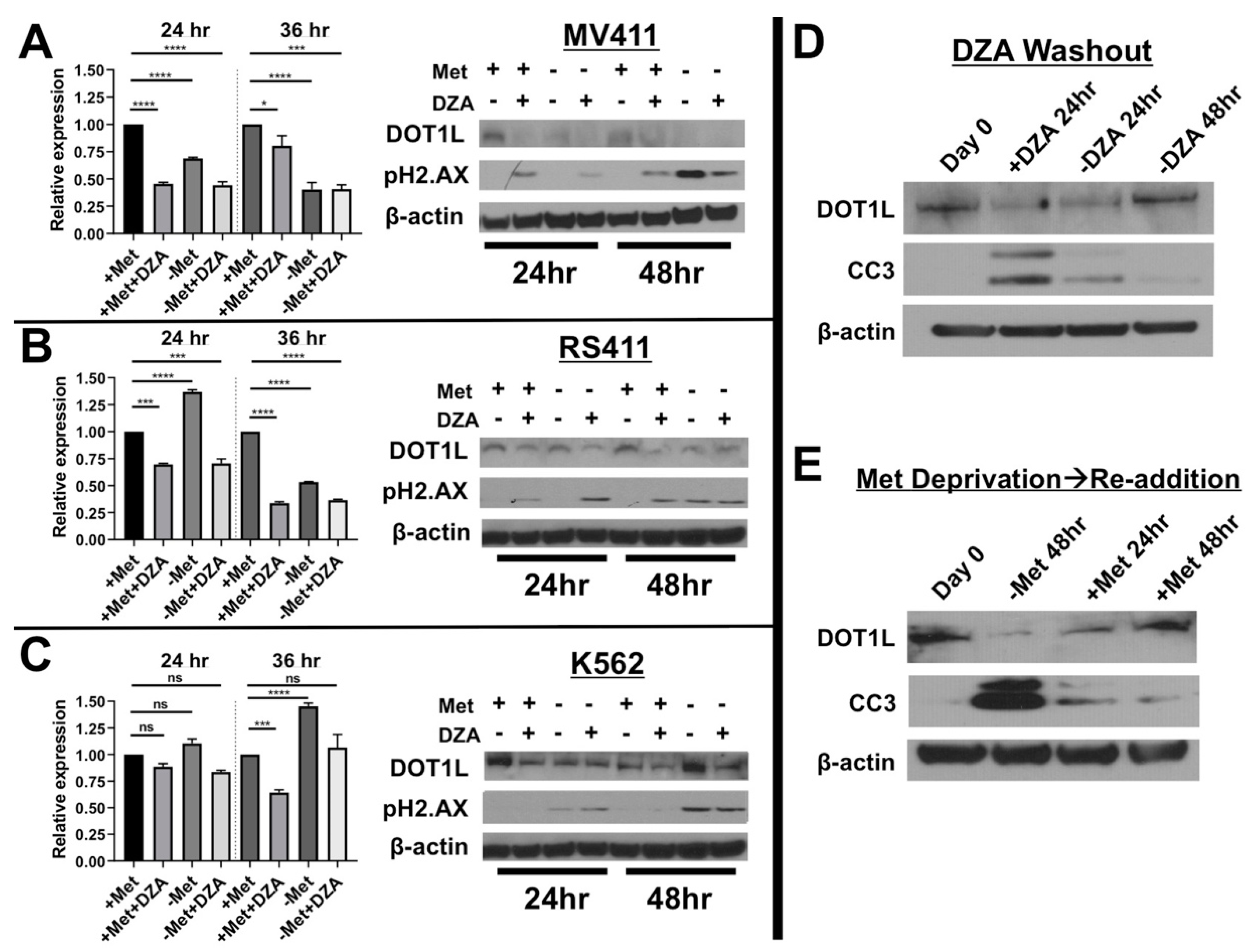
3.3. Disruption of Met/SAM Metabolism Reduces mRNA Expression and Protein Levels of the H3K79 Methyltransferase DOT1L and Induces DNA Damage
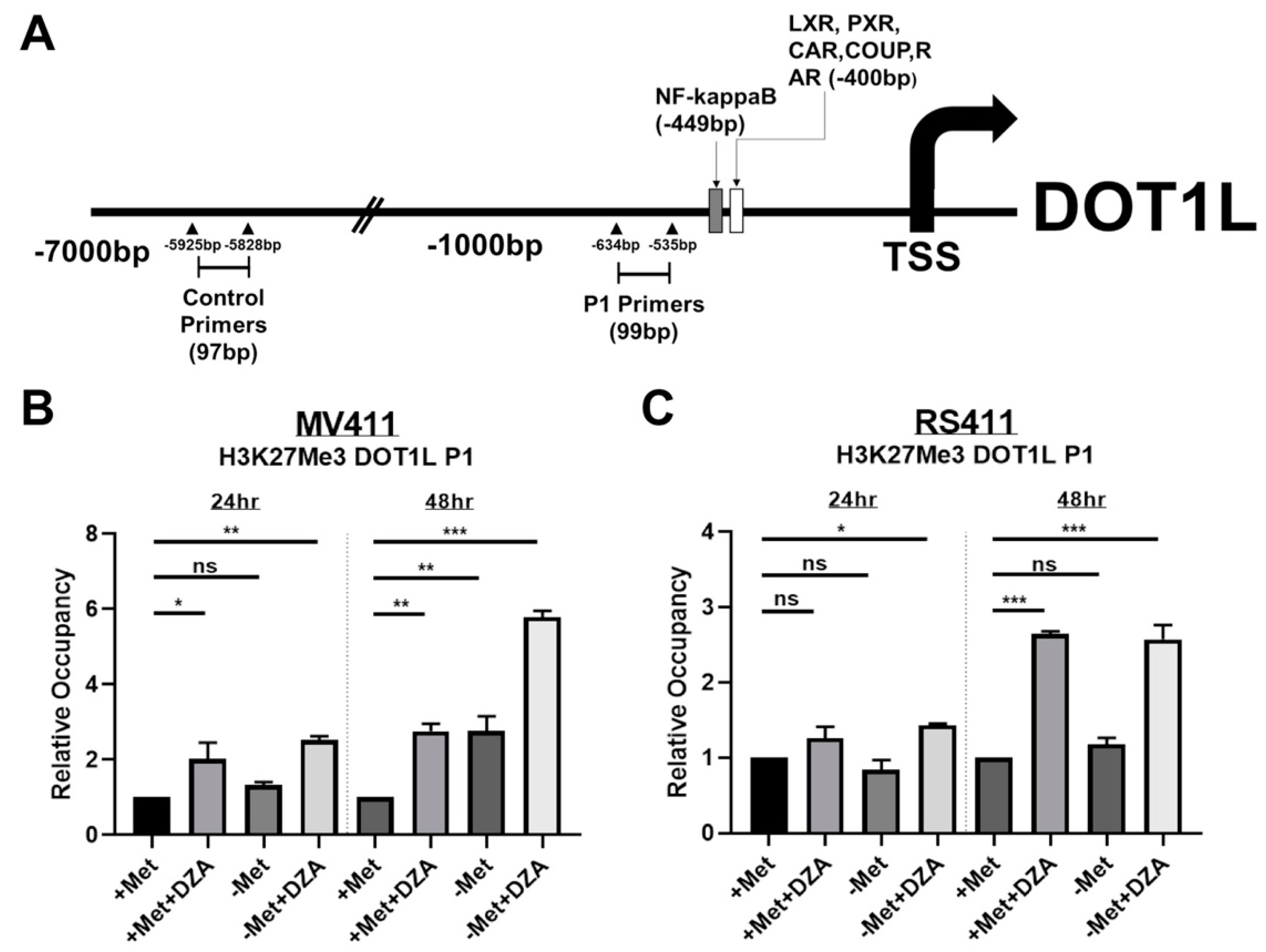
3.4. Decreased DOT1L Expression is Correlated to Changes in Histone Methylation Dynamics at the DOT1L Promoter
3.5. Patient-Derived MLL-R Leukemic Blasts are Sensitive to Alterations in Met/SAM Metabolism and Show Corresponding Changes in Global and Promoter-Specific Histone Methylation
3.6. Pharmacologic Inhibition of SAH Metabolism Significantly Prolongs the Survival of MLL-R Xenograft–Bearing Mice, in Combination with SOC Induction Therapy
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Biondi, A.; Cimino, G.; Pieters, R.; Pui, C.H. Biological and therapeutic aspects of infant leukemia. Blood 2000, 96, 24–33. [Google Scholar] [CrossRef]
- Bernt, K.M.; Armstrong, S.A. Targeting epigenetic programs in MLL-rearranged leukemias. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Jiang, Q.; Lemieux, M.; Jeannotte, L.; Su, L.; Zhang, Y. Leukaemic transformation by CALM-AF10 involves upregulation of Hoxa5 by hDOT1L. Nat. Cell Biol. 2006, 8, 1017–1024. [Google Scholar] [CrossRef]
- Chen, L.; Deshpande, A.J.; Banka, D.; Bernt, K.M.; Dias, S.; Buske, C.; Olhava, E.J.; Daigle, S.R.; Richon, V.M.; Pollock, R.M.; et al. Abrogation of MLL-AF10 and CALM-AF10-mediated transformation through genetic inactivation or pharmacological inhibition of the H3K79 methyltransferase Dot1l. Leukemia 2013, 27, 813–822. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, A.J.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.T.; Taranova, O.; He, J.; Zhang, Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 2011, 117, 6912–6922. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, B.R.; Deshpande, A. Epigenetic Regulators in the Development, Maintenance, and Therapeutic Targeting of Acute Myeloid Leukemia. Front. Oncol. 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Bottiglieri, T. S-Adenosyl-L-methionine (SAMe): From the bench to the bedside--molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 2002, 76, 1151S–1157S. [Google Scholar] [CrossRef] [PubMed]
- Catoni, G.L. S-Adenosylmethionine; a new intermediate formed enzymatically from L-methionine and adenosinetriphosphate. J. Biol. Chem. 1953, 204, 403–416. [Google Scholar]
- Banerjee, R.V.; Matthews, R.G. Cobalamin-dependent methionine synthase. Faseb J 1990, 4, 1450–1459. [Google Scholar] [CrossRef]
- Dominguez-Salas, P.; Moore, S.E.; Cole, D.; da Costa, K.A.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Innis, S.M.; Waterland, R.A.; Zeisel, S.H.; et al. DNA methylation potential: Dietary intake and blood concentrations of one-carbon metabolites and cofactors in rural African women. Am. J. Clin. Nutr. 2013, 97, 1217–1227. [Google Scholar] [CrossRef]
- Jani, T.S.; Gobejishvili, L.; Hote, P.T.; Barve, A.S.; Joshi-Barve, S.; Kharebava, G.; Suttles, J.; Chen, T.; McClain, C.J.; Barve, S. Inhibition of methionine adenosyltransferase II induces FasL expression, Fas-DISC formation and caspase-8-dependent apoptotic death in T leukemic cells. Cell Res. 2009, 19, 358–369. [Google Scholar] [CrossRef]
- Saforo, D.; Omer, L.; Smolenkov, A.; Barve, A.; Casson, L.; Boyd, N.; Clark, G.; Siskind, L.; Beverly, L. Primary lung cancer samples cultured under microenvironment-mimetic conditions enrich for mesenchymal stem-like cells that promote metastasis. Sci. Rep. 2019, 9, 4177. [Google Scholar] [CrossRef] [PubMed]
- Ghare, S.S.; Joshi-Barve, S.; Moghe, A.; Patil, M.; Barker, D.F.; Gobejishvili, L.; Brock, G.N.; Cave, M.; McClain, C.J.; Barve, S.S. Coordinated histone H3 methylation and acetylation regulate physiologic and pathologic fas ligand gene expression in human CD4+ T cells. J. Immunol. 2014, 193, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Cashman, J.D.; Hogge, D.E.; Humphries, R.K.; Eaves, C.J. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia 2004, 18, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Barve, A.; Casson, L.; Krem, M.; Wunderlich, M.; Mulloy, J.C.; Beverly, L.J. Comparative utility of NRG and NRGS mice for the study of normal hematopoiesis, leukemogenesis, and therapeutic response. Exp. Hematol. 2018, 67, 18–31. [Google Scholar] [CrossRef]
- Lee, C.Y.; Su, G.C.; Huang, W.Y.; Ko, M.Y.; Yeh, H.Y.; Chang, G.D.; Lin, S.J.; Chi, P. Promotion of homology-directed DNA repair by polyamines. Nat. Commun. 2019, 10, 65. [Google Scholar] [CrossRef]
- Snyder, R.D. Inhibition of X-ray-induced DNA strand break repair in polyamine-depleted HeLa cells. Int. J. Radiat. Biol. 1989, 55, 773–782. [Google Scholar] [CrossRef]
- Snyder, R.D.; Sunkara, P.S. Effect of polyamine depletion on DNA damage and repair following UV irradiation of HeLa cells. Photochem. Photobiol. 1990, 52, 525–532. [Google Scholar] [CrossRef]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [Green Version]
- Cavuoto, P.; Fenech, M.F. A review of methionine dependency and the role of methionine restriction in cancer growth control and life-span extension. Cancer Treat. Rev. 2012, 38, 726–736. [Google Scholar] [CrossRef]
- Erener, S.; Petrilli, V.; Kassner, I.; Minotti, R.; Castillo, R.; Santoro, R.; Hassa, P.O.; Tschopp, J.; Hottiger, M.O. Inflammasome-activated caspase 7 cleaves PARP1 to enhance the expression of a subset of NF-kappaB target genes. Mol. Cell 2012, 46, 200–211. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, S.; Song, J. Caspase-dependent apoptosis and -independent poly(ADP-ribose) polymerase cleavage induced by transforming growth factor beta1. Int. J. Biochem. Cell Biol. 2004, 36, 223–234. [Google Scholar] [CrossRef]
- Chaitanya, G.V.; Steven, A.J.; Babu, P.P. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun Signal 2010, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Lieber, C.S.; Packer, L. S-Adenosylmethionine: Molecular, biological, and clinical aspects--an introduction. Am. J. Clin. Nutr. 2002, 76, 1148S–1150S. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Lorin, S.; Blommaart, E.F.; Codogno, P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015, 47, 2037–2063. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; Van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutter, B.M.; Wu, X.; Laxman, S.; Tu, B.P. Methionine inhibits autophagy and promotes growth by inducing the SAM-responsive methylation of PP2A. Cell 2013, 154, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Horowitz, S.; Trievel, R.C. Structure and function of histone H3 lysine 9 methyltransferases and demethylases. Chembiochem. 2011, 12, 254–263. [Google Scholar] [CrossRef]
- Horiuchi, K.Y.; Eason, M.M.; Ferry, J.J.; Planck, J.L.; Walsh, C.P.; Smith, R.F.; Howitz, K.T.; Ma, H. Assay development for histone methyltransferases. Assay Drug Dev. Technol. 2013, 11, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Zielinski, T.; Lowery, R.G. Biochemical Assay Development for Histone Methyltransferases Using a Transcreener-Based Assay for S-Adenosylhomocysteine. Assay Drug Dev. Technol. 2015, 13, 200–209. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barve, A.; Vega, A.; Shah, P.P.; Ghare, S.; Casson, L.; Wunderlich, M.; Siskind, L.J.; Beverly, L.J. Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia. Cells 2019, 8, 1322. https://doi.org/10.3390/cells8111322
Barve A, Vega A, Shah PP, Ghare S, Casson L, Wunderlich M, Siskind LJ, Beverly LJ. Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia. Cells. 2019; 8(11):1322. https://doi.org/10.3390/cells8111322
Chicago/Turabian StyleBarve, Aditya, Alexis Vega, Parag P. Shah, Smita Ghare, Lavona Casson, Mark Wunderlich, Leah J. Siskind, and Levi J. Beverly. 2019. "Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia" Cells 8, no. 11: 1322. https://doi.org/10.3390/cells8111322
APA StyleBarve, A., Vega, A., Shah, P. P., Ghare, S., Casson, L., Wunderlich, M., Siskind, L. J., & Beverly, L. J. (2019). Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia. Cells, 8(11), 1322. https://doi.org/10.3390/cells8111322