1. Introduction
Exchange protein activated by cyclic AMP (EPAC) proteins are now emerging as drug targets with the potential to treat multiple disorders, including type 2 diabetes and cardiovascular diseases [
1]. EPACs are multi-domain proteins that act as a guanine nucleotide exchange factor (GEF) for the Ras-like small GTPases, Rap1 and Rap2 [
2,
3]. The EPAC protein family comprises two members, EPAC1 and EPAC2, which are encoded by two independent genes that display distinct tissue expression; with EPAC1 ubiquitously expressed in most tissues, whereas EPAC2 expression is more restricted to the liver, brain, heart, and secretory organs, including the pancreas [
4,
5]. They share the same functional domain organization and mechanisms of activation, consisting of a regulatory N-terminal region, which includes a disheveled-EGL-pleckstrin homology domain (DEP) and a cyclic nucleotide binding domain (CNBD), and a catalytic C-terminal region that includes a Ras exchange motif (REM), a Ras association domain (RA), and a CDC25 homology domain (CDC25-HD) [
4,
5]. EPAC2 includes an additional CNBD within its N-terminus (CNBD-A) that has a low affinity for cAMP and is not required for protein activation; rather it is suggested to be involved in the subcellular localization of EPAC2 [
6].
X-ray crystallography has provided conformations of the open [
7] and closed [
8] states of EPAC2 while NMR spectroscopy has been used to explore how ligand binding affects the dynamics of the conformational switch in EPAC1 [
9,
10,
11]. In addition, we have developed an in silico docking model for interactions of the known EPAC ligand, I942 (
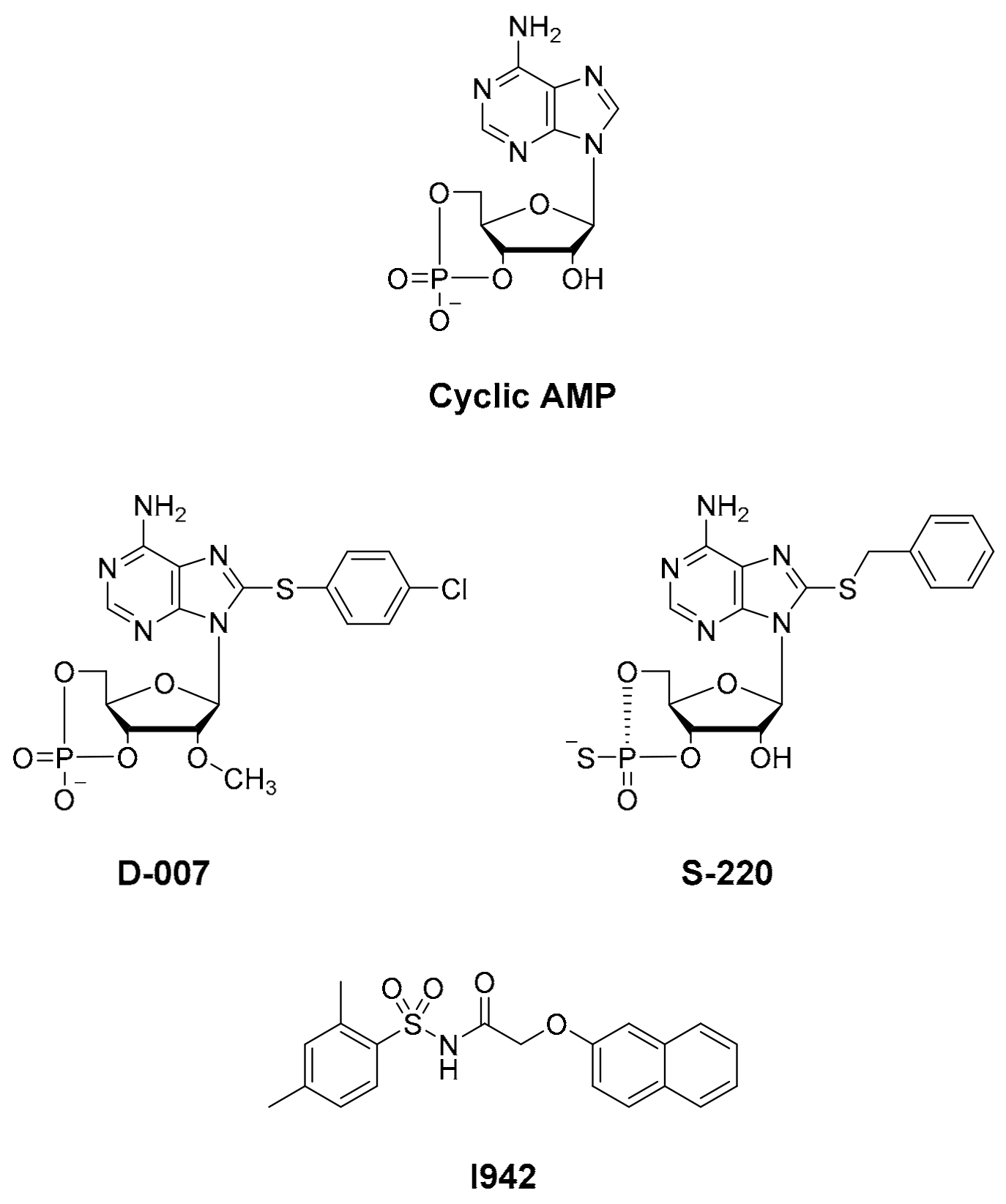
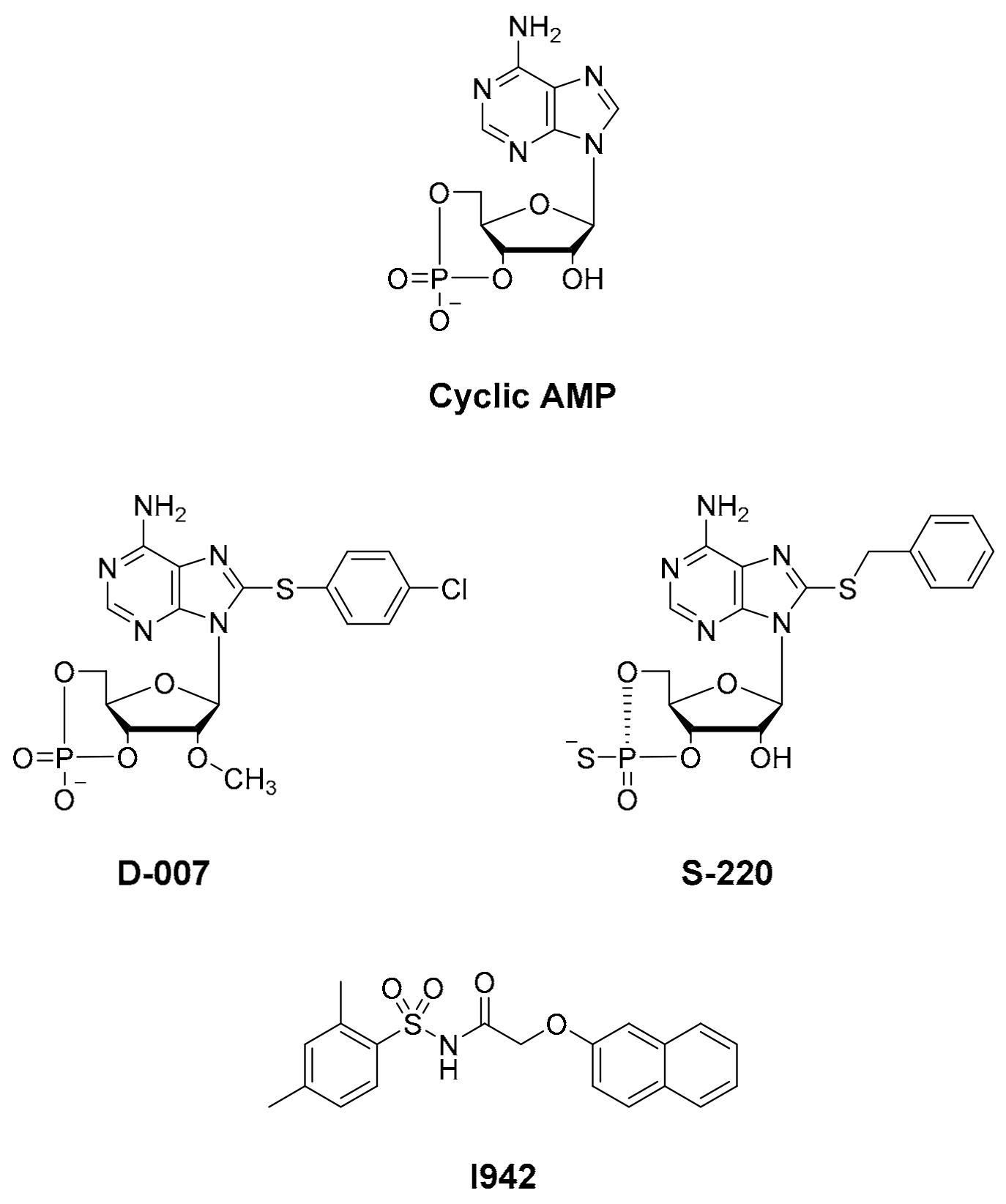
Figure 1; [
12]), with EPAC1 [
13]. From these studies, it is clear that EPAC2 exists as multiple conformations in dynamic equilibrium. These range from a closed, auto-inhibited conformation in the absence of cyclic AMP, where the regulatory region sterically blocks access to the catalytic site, to a cyclic AMP bound form in an open, catalytically active state, where the regulatory region binds to a different face of the catalytic domain. Note that an EPAC1 structure has yet to be reported and in silico models have been constructed using an EPAC2 lysine 405 to glutamine point mutation structure, which is anticipated to be a roughly analogous docking site to that of EPAC1 [
13].
A number of laboratories, including ours, have used high-throughput screening (HTS) of compound libraries to search for novel EPAC ligands that can discriminate between the active (agonists) and inactive (antagonists) conformations of EPAC. These assays principally use competition for binding of the fluorescent cyclic AMP analogue, 8-NBD-cAMP, to EPAC isoforms. Initially, 8-NBD-cAMP competition assays were applied to screens involving EPAC2 and led to the identification of the EPAC2-selective inhibitor, ESI-05 (4-methylphenyl-2,4,6-trimethylphenylsulfone) [
14]. Furthermore ESI-09 (2-(5-(
tert-butyl)isoxazol-3-yl)-
N-(3-chlorophenyl)-2-oxoacetohydrazonoyl cyanide) was proposed as a non-selective EPAC1 and EPAC2 inhibitor [
15,
16,
17], though one study showed general, unspecific protein denaturing properties of this compound at high concentrations [
18]. Both uncompetitive (CE3F4) and non-competitive EPAC1 inhibitors (5225554 and 5376753) have also been identified by HTS using an
in vitro EPAC1 guanine nucleotide exchange factor (GEF) activity assay [
19] and an EPAC-based bioluminescence resonance energy transfer-based assay [
20], respectively. Notably, none of these HTS approaches led to the identification of small molecule agonists of EPAC1 activity, and, to date, only cyclic AMP derived EPAC activators have been developed, independently of HTS, namely D-007 for EPAC1 and S-220 for EPAC2 (
Figure 1; [
21]). Recently we screened a 5195 small molecule library using HTS with competition binding of 8-NBD-cAMP to the recombinant CNBD of EPAC1 (amino acids 169-318) and identified the novel ligand, I942 [
12]. Subsequent,
in vitro EPAC1 GEF assays revealed that I942 displayed partial agonist properties towards EPAC1, leading to activation of EPAC1, in the absence of cyclic AMP, and inhibition of GEF activity in the presence of cyclic AMP, with little agonist action towards EPAC2 or protein kinase A (PKA) [
12]. This was the first observation of non-cyclic-nucleotide (NCN) small molecules with agonist properties towards EPAC1. Subsequent studies with I942 revealed that it activates EPAC1 and Rap1 GTPase in cells and exerts anti-inflammatory actions in human umbilical vascular endothelial cells (HUVECs) through the inhibition of interleukin 6 (IL-6)-promoted gene expression [
22].
Here, for the first time, we have adapted the 8-NBD-cAMP/EPAC1 CNBD competition assay for ultra HTS (uHTS) to explore further the chemical diversity of novel NCN EPAC1 agonists. By screening a chemically diverse library of approximately 350,000 compounds, we identified further NCN EPAC1 agonists that are chemically distinct from I942. We next synthesized an expanded analogue library from isolated hits and, with subsequent triage using binding assays and microscale thermophoresis (MST), we determined potency and selectivity of these analogues towards the CNBDs of EPAC1 and EPAC2. Subsequent in vitro and in cellulae EPAC1 activation assays led us to identify SY009 as an activator of EPAC1 that is chemically distinct from I942. The identification of further NCN agonists with the potential to activate EPAC1, independently of EPAC2, presents powerful experimental tools to investigate the role of EPAC1 in health and disease and, therefore, the development of future therapeutic strategies to combat diseases associated with EPAC activation.
2. Materials and Methods
2.1. Materials
Forskolin, rolipram, and cyclic AMP were purchased from Merck-Millipore (Burlington, MA, USA). Analogues of cyclic AMP, 8-NBD-cAMP, Sp-8-BnT-cAMPS (S-220), and 8-pCPT-2′-O-Me-cAMP (D-007) were purchased from Biolog Life Science Institute GmbH & Co. KG (Bremen, Germany). BL-21 cells were purchased from New England Biolabs. The test compound I942 (N-(2,4-dimethylbenzenesulfonyl)-2-(naphthalen-2-yloxy)acetamide) was sourced from MolPort (Riga, Latvia). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), GlutaMAX, and penicillin/streptomycin (5000 U/mL) were purchased from Thermo Fisher Scientific (Waltham, MA, USA) and the selective antibiotic, puromycin, and complete, EDTA-free protease inhibitor cocktail were from Sigma-Aldrich (St. Louis, Mo, USA). Rap1A/Rap1B (26B4) and vasodilator-stimulated phosphoprotein (VASP; 9A2) antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA) and HRP-conjugated anti-mouse and anti-rabbit IgG secondary antibodies from Sigma-Aldrich.
2.2. Recombinant Protein Production
EPAC1-CNBD (amino acids 169–318 of EPAC1) and EPAC2-CNBD (amino acids 304-453) cDNAs were previously sub-cloned into the multi-cloning site of the pGEX-6P-1 expression vector (GE Healthcare) by BC Bioscienes (Dundee, Scotland). Ral-GDS-RBD (amino acids 788-884) was cloned into pGEX-5X-1 as described [
23]. Recombinant EPAC1-CNBD, EPAC2-CNBD, EPAC1 (amino acids 149–881), Ral-GDS-RBD (amino acids 788-884), and Rap1B (amino acids 1–167) glutathione S-transferase (GST)-fusion proteins were then expressed and purified from BL-21
Escherichia coli, as previously described [
12,
23]. Protein concentrations were determined by the bicinchoninic acid (BCA) assay or by Nanodrop 2000/2000c (Thermo Scientific). All recombinant proteins were determined to be pure by SDS-PAGE analysis. Proteins were stored in aliquots at −80 °C until required.
For large-scale preparations of EPAC1-CNBD for uHTS, pGEX-6P-1.EPAC1-CNBD was transformed into BL21 (DE3)-R3-pRARE2 expression strain (at Structural Genomics Consortium, University of Oxford). Cells were grown at 37 °C until the O.D6oo reached between 1–1.5 and then induced with 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) at 18 °C overnight. Cells were harvested next day and re-suspended in 50 mM Tris-HCl, 300 mM NaCl, 1 mM EDTA, 1 mM TCEP pH 7.8 + 0.5 mg/mL lysozyme, 1:1000 dilution of protease inhibitor cocktail, SET III and 0.1% (v/v) Triton X-100 and benzonase. The total volume after cell resuspension was 400 mL, which was split into two fractions of 200 mL each and lysed by sonication, 35% amplitude on 750 W sonicator, 5 s ON and 5 s OFF, for 10 min (total time of 20 min). The lysate was then centrifuged at 17,500× gmax on a JA18 rotor for 1 h then the supernatant was filtered through 0.45 μm filters. Equilibrated 50% (v/v) glutathione Sepharose slurry (12 mL to 300 mL filtrate) was then added to the filtrate and then rotated at 3 rpm overnight. Next day the Sepharose was washed with 60 column volumes of the wash buffer (50 mM Tris-HCl, 300 mM NaCl, 1 mM EDTA, 1 mM TCEP pH 7.8), and then eluted with 8 × 10 mL fractions of 15 mM reduced glutathione in the elution buffer (50 mM Tris-HCl, 300 mM NaCl, 1 mM EDTA, 1 mM TCEP pH 7.8). The protein fractions were run on SDS-PAGE and the eluates were buffer-exchanged overnight with 50 mM Tris-HCl, 300 mM NaCl, 1 mM EDTA, 1 mM TCEP pH 7.8, using 3.5 kD cut off snakeskin. Protein was then aliquoted into 500 μL aliquots, at a concentration of 1.4 mg/mL, and then snap frozen.
2.3. 8-NBD-cAMP Competition Assay and Ultra-High Throughput Screening (uHTS)
8-(2-[7-nitro-4-benzofurazanyl] aminoethylthio) adenosine-3′,5′-cyclic monophosphate (8-NBD-cAMP) competition assays were performed with purified EPAC1-CNBD and EPAC2-CNBD GST-fusion proteins as previously described [
12]. Comparison of reference molecules, dose response curves (DRC) and pilot screens were done in black, low volume 384 assay plates (Greiner). Plates were incubated for 4 h before 8-NBD-cAMP fluorescence intensity at 480/535 nm (ex/em) was measured using the Envision multi-label plate reader (Perkin-Elmer; Waltham, MA, USA).
The high-throughput screen with the European Lead Factory (ELF) library (~350,000 compounds) [
24,
25] was carried out using the 8-(2-[7-nitro-4-benzofurazanyl] aminoethylthio) adenosine-3′, 5′-cyclic monophosphate (8-NBD-cAMP) competition assay to measure binding to the CNBD of EPAC1 as described [
12]. Firstly, the competition assay was optimized to run on an operational ultra-high throughput screening (uHTS) system, comprising an Echo
® 555 acoustic liquid dispenser and three EnVision multi-label plate readers (Perkin-Elmer). This involved defining dispensing volumes, plate types (384 or 1536 well formats), reagent stability and incubation times. As previously reported [
12], the assay was measured after 4 h, however, the feasibility assays showed a better signal to background ratio (S/B) for 400 nM probe (8-NBD-cAMP) with 400 nM protein (EPAC1-CNBD) and that this combination of reagents was stable for up to 20 h at 4 °C. The signal to background ratio (S/B) and Z’ values for the assays were comparable in both 384 and 1536 well formats. For uHTS, dose response curve (DRC) plates were prepared in 1536-well format at the European Screening Centre (Newhouse, Scotland) and shipped to the Pivot Park Screening Centre (Oss, Netherlands). The 1536-well source plates contained 100% DMSO in the first 6 columns followed by 6 compound DRCs consisting of 7 points with a final range of 2.00 × 10
−5 M–2.00 × 10
−8 M, resulting in a maximum of 192 compound DRCs per plate. ActivityBase High Content and Throughput Screening Software (IDBS) was used to analyze the data and to calculate quality parameters including signal to background ratio (S/B) and Z’ values. The signal to background ratio (S/B) was determined by dividing the mean maximum fluorescent signal (DMSO diluent control) by the mean minimum signal (saturating cAMP concentrations) to give the magnitude of the fluorescence change in the assay. The Z’ factor was also calculated from the minimum (
n, cAMP) and maximum (
p, DMSO) control values from the assay, together with the mean fluorescent values (
μ) and their standard deviation (
σ), using the following equation:
For all compounds, the % effect was calculated for all seven concentrations of the DRC. ActivityBase was also used to calculate the pIC
50, hill slope, and Emax (maximal inhibition of 8-NBD-cAMP binding, reported in
Figures S1 and S2). Using optimized assay conditions, the screening of all plates demonstrated an S/B of 2.8 or higher and the Z’ value above 0.6, therefore all plates were validated, resulting in the identification of 68 actives. Of these, 23 were subsequently confirmed on a re-test of the primary assay (S/B of ±4 and a Z’ value of 0.56). Assay interference compounds were removed with a deselection assay involving interference of green fluorescent protein (60 nM GFP; excitation 480; emission 535) under regular assay conditions.
2.4. Bayesian Modeling
Confirmed actives from the uHTS screen were supplemented with compounds identified from an in silico Bayesian model constructed from the original screening data to identify potential false negatives. Briefly, a threshold for activity was defined based on the Z-score and % inhibition and each molecule was placed in either an active or inactive bin. All the molecules were then broken down in silico into sub-structural features. The frequency of each sub-structural feature in the active and inactive bins was then generated. A feature enriched in the active molecules was given a positive score, with a magnitude depending on the level of enrichment. Conversely, sub-structures that occurred more frequently than chance in the inactive compounds were given a negative score. The score was further weighted by how well that feature was represented in the screening data set. A table of all the substructures represented in the screening library and their associated scores was then generated. The Bayesian score does not describe how active a molecule is predicted to be. The higher score indicates a higher confidence that the molecule will be active at the threshold used to build the model but makes no prediction about it being more or less potent.
2.5. Generation of in Silico Docking Models of EPAC1 CNBD
While there are no published EPAC1 structures in the literature, several EPAC2 structures are available in their active form in the Protein Data Bank (PDB), co-crystallized with cyclic AMP and three different analogues, including D-007. Two of the published structures contain a mutation on position 405 (EPAC2 numbering), a lysine in EPAC2 is replaced by a glutamine (as found in the analogous position of EPAC1). The difference between the two residues K ˃ Q results mainly in a change in the local structure of the hinge region. Moreover, the difference in the cAMP analogue used in 4MGY and 4MGK (both mutated K405Q) induces a change in this region as well. Crystal structures of cAMP and these analogues bound to EPAC2 and EPAC2 mutants (K405Q) were used to guide development of the EPAC1 homology model using the Schrodinger Suite molecular modeling software platform.
2.6. Chemical Synthesis—General Methods
All non-aqueous reactions were carried out under oxygen free N2 using flame-dried glassware. Tetrahydrofuran (THF) and dimethylformamide (DMF were purified by MBRAUN SPS-800 solvent purification system. Reactions using Zn(CN)2 were performed under N2 on a Schlenk line equipped with a CuSO4 bubbler to trap any HCN released. Petroleum ether refers to the fraction of petroleum ether boiling in the range of 40–60 °C and was purchased in Winchester quantities. Brine refers to a saturated solution of NaOH in water. Water was distilled water. Flash column chromatography was carried out using Matrix silica gel 60 from Fisher Chemicals. Thin layer chromatography was carried out using commercially available Merck F254 aluminum-backed silica plates visualized by UV (254 nm) or stained using either a solution of acidified ninhydrin in ethanol or aqueous acidic KMnO4. Proton (300 or 400 MHz) and carbon (75.5 or 101 MHz) NMR spectra were recorded on Bruker AV 300 or AV 400, respectively. For samples recorded in CDCl3 and (CD3)2CO, chemical shifts are quoted on parts per million, relative to CHCl3 (δH 7.26) or (CD3)2CO (δH 2.05, central line of quintet) and CDCl3 (δC 77.16, central line of triplet) or (CD3)2CO (δC 29.84 using Distortionless Enhancement by Polarization Transfer (DEPT) experiments). Coupling constants (J) are quoted in Hertz. Melting points were obtained (central line of septet). Carbon NMR spectra were recorded with broad band decoupling and assigned on a Stuart Scientific SMP 10 at ambient pressure. Infrared spectra were recorded on a Perkin-Elmer Spectrum 100 FT-IR Universal ATR Sampling Accessory deposited neat to a diamond/ZnSe plate. Mass spectra were obtained at the EPSRC UK National Mass Spectrometry Facility at Swansea University.
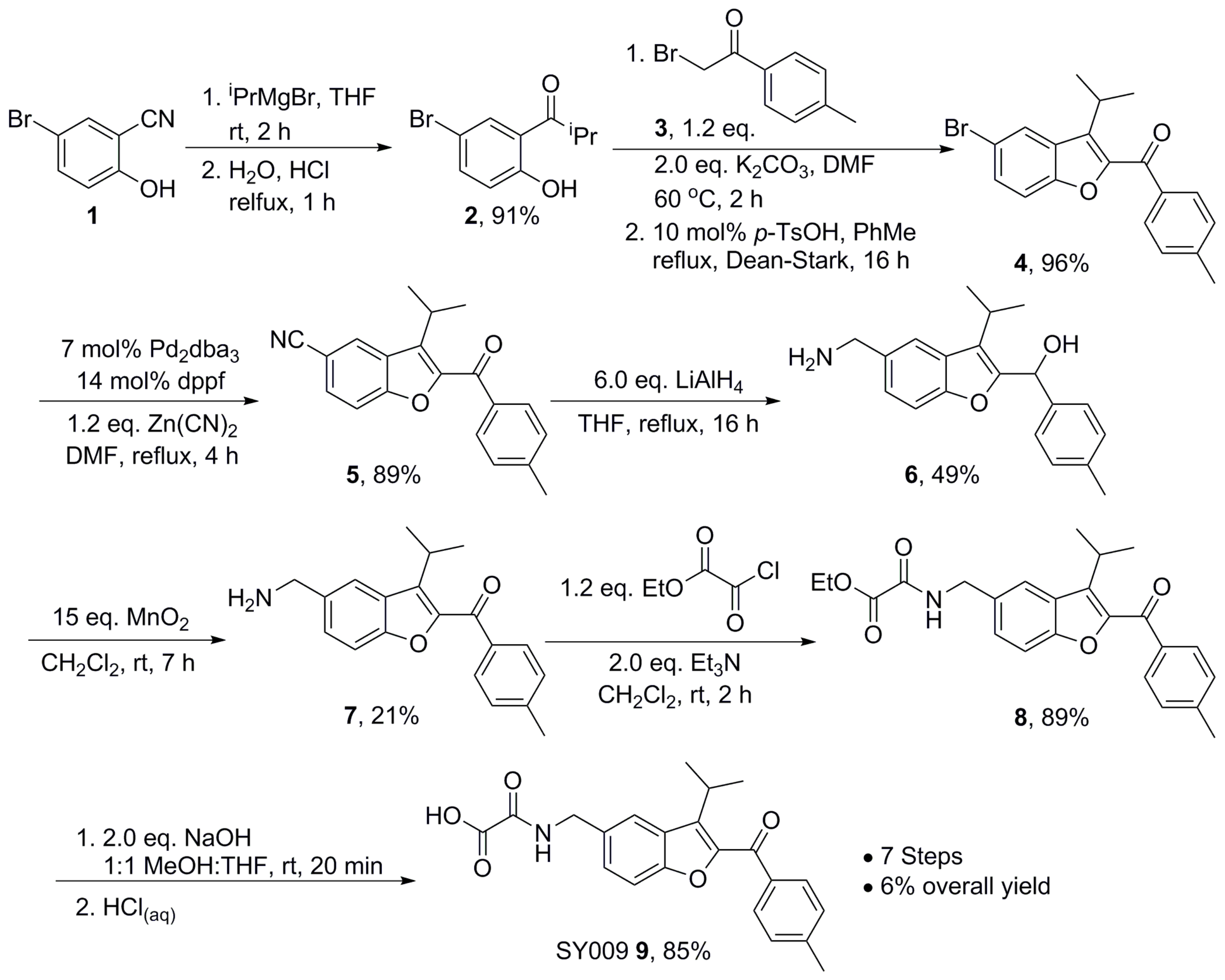
2.7. Chemical Synthesis—Representative Synthesis of SY009
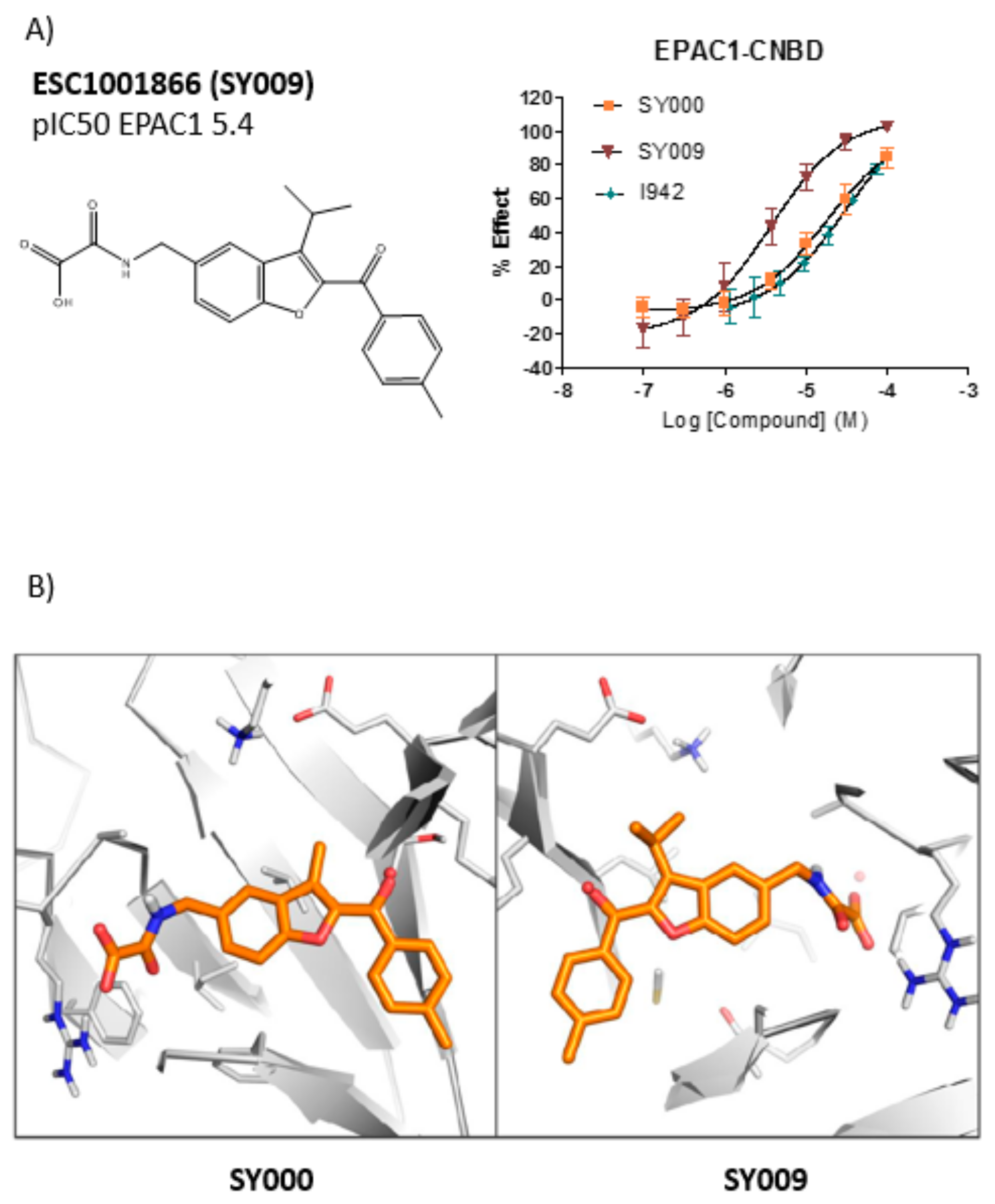
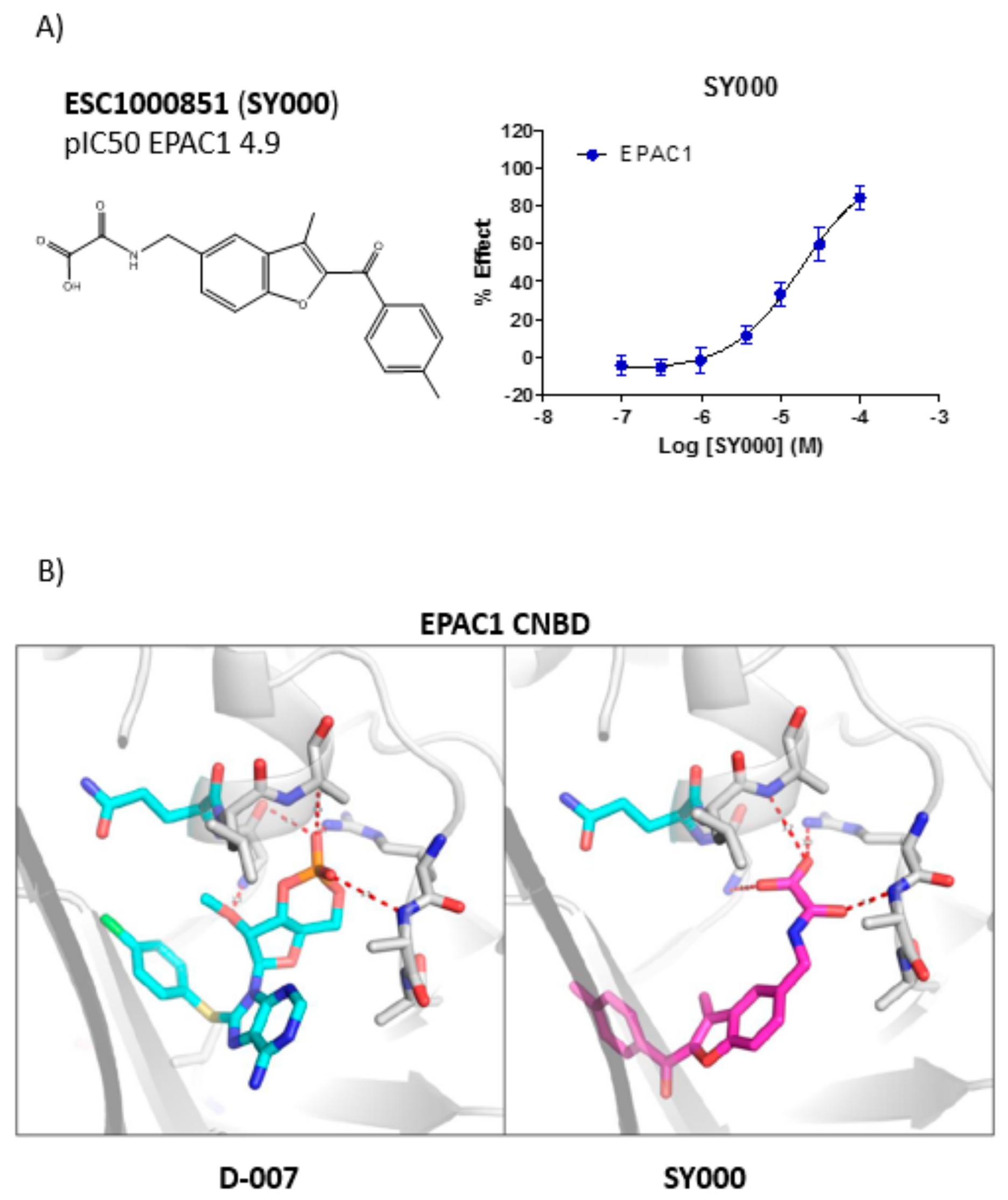
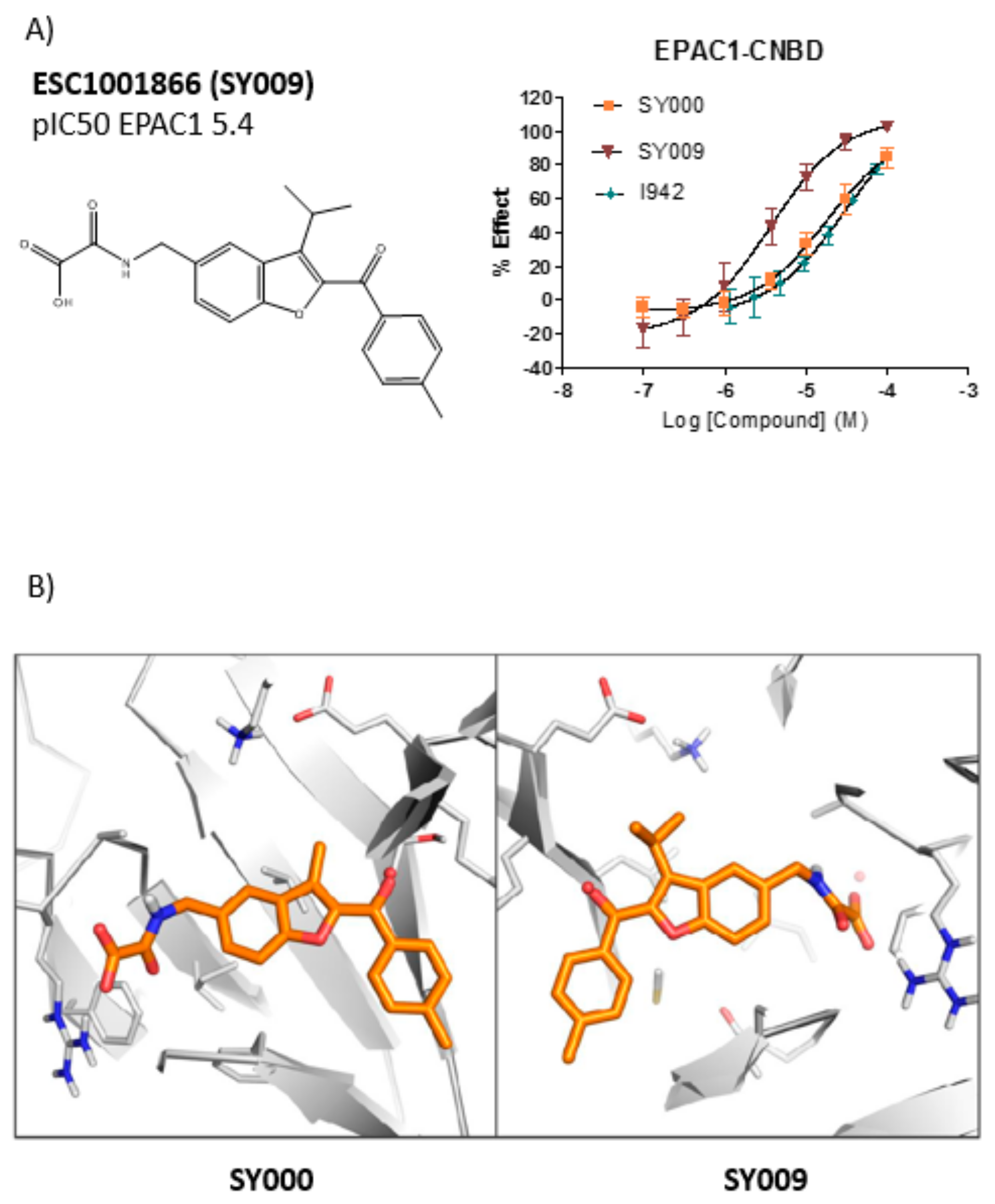
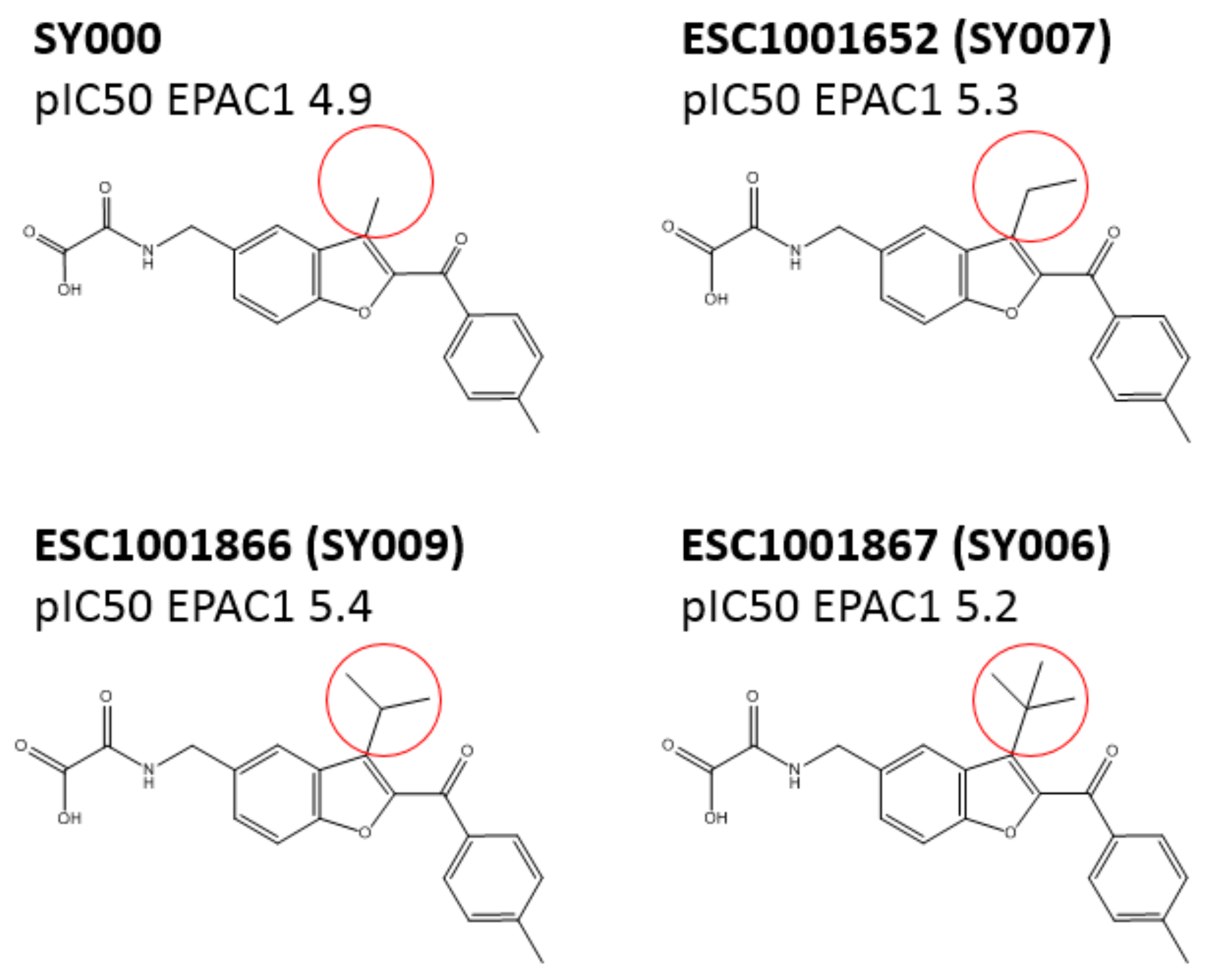
A chemical library of 69 analogues of the top hit, SY000, was generated (see
Table in Supplementary Material) following in silico modeling of the EPAC1 CNBD (described in
Section 2.5). This was supplemented with 22 additional compounds obtained from the initial screening library (shaded red in the
Supplementary Data Table). Synthesis of the improved hit, SY009 (illustrated in
Figure 2) was done as follows. Nitrile 1 was treated with an excess of
iso-propylmagnesium bromide, followed by treatment with aqueous acid to affect imine hydrolysis, giving phenol ketone 2 in 91% yield. Next, the benzofuran core was assembled: phenol 2 was treated with α-bromo ketone 3 in DMF in the presence of K
2CO
3 for 2 h at 60 °C, before a solvent exchange and refluxing in toluene for 16 h under a Dean–Stark trap in the presence of a catalytic amount of para-TsOH gave bromobenzofuran 4 in 96%. Cyanobenzofuran 5 was then obtained in 89% yield after a palladium-catalyzed cyanation using 7 mol% Pd
2(dba)
3 and 14 mol% dppf with 1.2 eq. Zn(CN)
2. The reaction was carried out over 4 h in DMF at 120 °C and run under a constant stream on N
2 on a Schlenk line equipped with a copper sulfate bubbler to capture any evolved HCN. Global reduction using LiAlH
4 gave amino alcohol 6 in 49% yield, and re-oxidation to the aminoketone 7 using MnO
2 was achieved in 21% yield, monitoring the reaction carefully using thin layer chromatography (TLC) to avoid oxidation of the amine to the corresponding aldimine. Repeated attempts to improve this yield using different conditions resulted in either inefficient oxidation of the alcohol, or complete aldimine formation. Next, amide 8 was formed in 89% yield after treatment of 7 with ethyl chlorooxoacetate, and subsequent ester hydrolysis gave SY009 9 in 85% yield. Routes analogous to the representative synthesis of SY009 described here were used to synthesize the other 69 compounds in the library. In each case, the appropriate phenol was selected to install the benzofuran core together with the relevant bromoketone or bromoamide to install the ketone or amide motif in the benzofuran 2-position. Well-established functional group transformations were then used to diversify groups at the benzofuran 5 position and at the carbonyl position.
2.8. Microscale Thermophoresis (MST)
EPAC1-CNBD and EPAC2-CNBD were labeled with the red fluorescent dye NT-647-NHS, which non-selectively couples to free amine groups in the protein, predominantly represented by exposed lysines. Briefly, 100 μL of 20 μM CNBD in PBS was incubated with 100 μL of 3-fold excess NT-647 NHS fluorescent dye for 30 min in the dark at room temperature. To remove unreacted “free” dye, the reaction was placed into a pre-equilibrated gravity flow column in MST buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM MgCl2). The labeled protein was eluted at 3 μM in MST buffer. Red fluorescence (excitation 650 nm/emission 670–690 nm) was measured for a series of labeled CNBD concentrations to confirm that labeling was successful. A concentration-dependent fluorescent signal indicated that coupling was successful. For MST a signal > 250 fluorescent units is typically considered sufficient for screening using the Monolith NT automated instrument from NanoTemper Technologies. CNBDs were therefore used at 120 nM.
2.9. Guanine Nucleotide Exchange Factor (GEF) Activity Assay
The EPAC1
in vitro GEF activities, using recombinant EPAC1 (149–881) and Rap1B (1–167), were calculated as previously described [
12]. Briefly, recombinant EPAC1 (149–881) was incubated at 100 nM with recombinant Rap1B (200 nM) preloaded with the fluorescent guanosine diphosphate (GDP) analogue 2′/3′-O-(N-Methylanthraniloyl)guanosine 5-diphosphate (MANT-GDP) in the presence of 20 μM GDP and then the fluorescence intensity was measured at 360/450 nm (ex/em) over time. Multiple reactions were performed at cyclic AMP/compound concentrations varying between 0.1 and 1000 μM and curves were fit as single exponential decay to obtain k
obs.
2.10. Active Rap1 Pull-Down Assay
U2OS cells stably transfected with EPAC1 or EPAC2 (from Holger Rehmann, University of Utrecht) were cultured in 6-well plates in DMEM, high glucose, supplemented with 10% (v/v) FBS, 1% (v/v) GlutaMAX, 1% (v/v) penicillin-streptomycin, and 2 mg/L puromycin (to ensure selection of stable transfectants). Then, 80% confluent cells were starved in culture medium with reduced FBS concentration (0.5% v/v) for 16 h and then stimulated for 10 min with either vehicle, 100 µM of test compounds, 10 µM forskolin, and 10 µM rolipram or 50 µM of D-007 in case of U2OS-EPAC1 or 100 µM of S-220 for U2OS-EPAC2. Cells were then rinsed with ice-cold PBS and lysed in 0.5 mL cell lysis buffer (from Cell Signaling Technologies) supplemented with 10 mM MgCl2 and 1 mM phenylmethylsulfonyl fluoride (PMSF), followed by clearing the lysates by centrifugation. Cell lysates were incubated for 1 h (4 °C, gentle agitation) with 40 µg GST-Ral-GDS-RBD immobilized on glutathione Sepharose 4B (GE Healthcare) to selectively capture GTP-bound Rap1. Following this, the glutathione resin was separated from supernatant by centrifugation, washed three times with cell lysis buffer, then resuspended in 2× SDS sample loading buffer and denatured for 5 min at 95 °C followed by analysis by Western blotting.
2.11. SDS-PAGE and Western Blotting
Samples were prepared by mixing equal volumes of cell lysate with 2× SDS sample loading buffer and then denaturing for 5 min at 95 °C, unless indicated otherwise. Protein samples were separated by SDS-PAGE on 10% (v/v) polyacrylamide gels, for VASP, or on 12.5% (v/v), for Rap1, and then transferred to nitrocellulose membranes. Membranes were then blocked for 1 h at room temperature in 5% (w/v) bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% (v/v) Tween 20, followed by an overnight incubation with primary antibody diluted in blocking buffer at 4 °C. Subsequently, the membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. For signal detection the SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific) was used. Images were acquired using the Fusion FX7 camera platform (Vilber; Collégien, France). Densitometry was performed with ImageJ.
2.12. Statistical Analyses
Statistical significance was determined using one-way analysis of variance (ANOVA) with Tukey’s post-tests.
4. Discussion
Screening against EPAC1 CNBD with the ELF library (~350,000 compounds) and subsequent triage and re-synthesis identified a single compound, SY000, which confirmed moderate binding activity at EPAC1. SY000 was prioritized for further work and initial SAR resulted in compound SY009 with improved activity (sub-μM). SY009 was found to display agonist properties towards EPAC1 in vitro and in cellulae and selectivity towards EPAC1, over EPAC2 and PKA, in cells. Further work will be required to determine binding to other cellular CNBDs, including cyclic nucleotide-gated (CNG) and hyperpolarization-activated cyclic nucleotide–gated (HCN) channels. Despite this, SY009 is an encouraging starting point for further work since it has been demonstrated here that it is possible to increase the binding activity of ligands with small structural alterations, as well as induce bias towards either EPAC1 or EPAC2 activity.
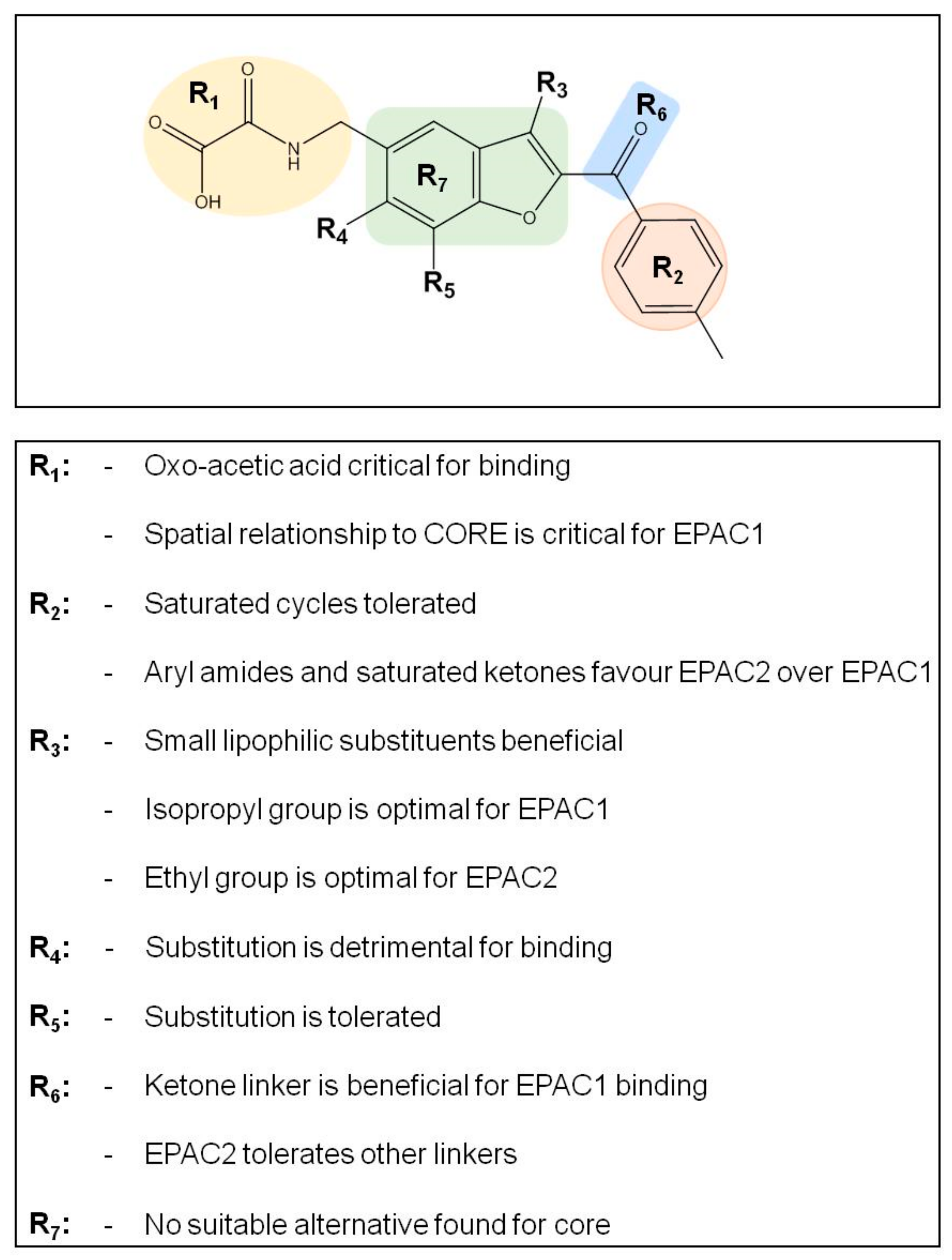
The benzofuran core is typically not a favored motif in drug discovery due to the potential susceptibility to hepatic oxidation by CYP enzymes, giving rise to reactive epoxide intermediates. In the case of this series, the C2/C3 substituents would be expected to reduce the susceptibility to epoxidation, however we were also interested in addressing whether alternative 5–6 heterocyclic systems could act as suitable replacements. We found that replacing the core benzofuran (
Supplementary Materials) resulted in a dramatic loss in EPAC1 activity. While no promising alternative cores were discovered there is scope for more extensive investigation in this area. Poor solubility, presumably due to the flat aromatic nature of the hit compounds, was identified as an important area for improvement. To address this potential limitation, analogues with reduced aromaticity were also targeted. This resulted in several analogues (e.g., ESC1001920 and ESC1001653) with equivalent potency to the original hits and significantly higher fraction sp
3 which should help to improve solubility issues with the series.
SY006, SY007, and SY009 proved to be effective activators of Rap1 in cells and therefore complement our original NCN EPAC1 agonist, I942, as chemically distinct tool molecules to probe the role of EPAC1 in cellular systems. The question remains as to the mechanisms by which the new SY series and the original I942 series promote EPAC1 activation. We know that cyclic AMP binds the CNBD leading to subtle conformational rearrangements within the core CNBD domain in the auto-inhibited closed state, this leads to “tumbling” of the CNBD, freeing EPAC to open towards the active state, and allowing the catalytic subunit to interact with Rap GTPases. In this conformation, cyclic AMP forms additional interactions, stabilizing the CNBD in the active state. As a result, EPAC exists in dynamic equilibrium between open and closed states and, with the active state being selected upon interaction with orthosteric agonists. Partial agonists, such as I942, and SY009 identified here, are less efficient than cyclic AMP in shifting the conformational equilibrium. Despite this, the work here provides further evidence that NCN EPAC1 agonists can be further developed as tool molecules with the potential for development as future therapeutic agents. In this regard SY009, being chemically distinct from I942, is predicted to adopt a distinct binding mode in the cyclic AMP binding pocket (
Figure 4). Indeed, our current model of I942/CNBD interaction involves the acidic
N-acylsulfonamide motif (pKa ~4) occupying a similar volume to the cyclic AMP phosphate, engaging a key charge-pairing arginine (Arg279) within the CNBD phosphate-binding cassette [
13]. Similarly, based on the docking model presented here the oxoacid functional group of SY006, SY007, and SY009 is also predicted to interact with the same region of the EPAC1 CNBD, in a similar fashion. However, whereas the benzofuran oxo-acetic series are predicted to orientate in the pocket in a manner analogous to cyclic AMP (
Figure 3b), I942 may form additional, hydrophobic interactions that are not accessible to cyclic AMP involving threading of the oxymethylene linker through a narrow, solvent-filled passage towards the back of the protein [
13]. Therefore, further comparative binding studies are required to fully define the binding modes to determine the molecular basis of selectivity and further develop potency of the two existing classes of NCN EPAC1 agonists.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

