Conditional Deletion of LRP1 Leads to Progressive Loss of Recombined NG2-Expressing Oligodendrocyte Precursor Cells in a Novel Mouse Model

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Generation of the New Mouse Model and Housing
2.3. Genotyping
2.4. Injection of Tamoxifen to Animals
2.5. Decapitation, Perfusion, and Dissection
2.6. Cryosections
2.7. Immunohistochemistry (IHC)
2.8. Preparation of Tissue for Reverse Transcriptase-Polymerase Chain Reaction(RT-PCR) and Western Blot Analysis
2.9. RT-PCR Analysis
2.10. Western Blot Analysis
2.11. Experimental Autoimmune Encephalomyelitis (EAE)
2.12. Imaging
2.13. Quantification
2.14. Statistics
3. Results
3.1. Induction of the NG2-Restricted LRP1-Deficient KO in the New Mouse Model
3.2. LRP1 Expression in Oligodendrocyte Development
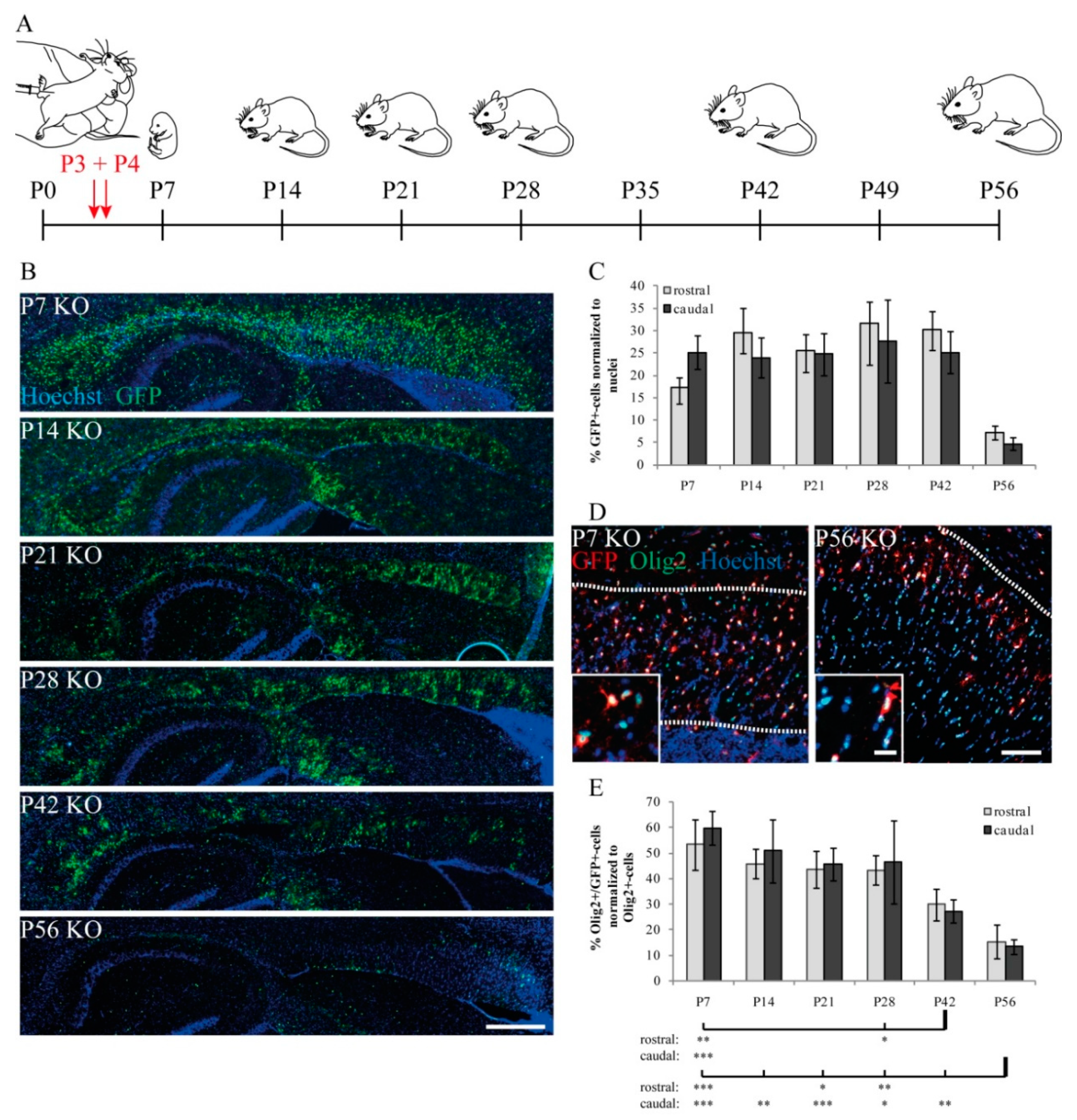
3.3. Proportion and Distribution of Control and KO Cells
3.4. Cellular Characterization of Oligodendrocyte Lineage-Specific Cells
3.5. Molecular Characterization of Oligodendrocyte Lineage-Specific Cells
3.6. Experimental Autoimmune Encephalomyelitis (EAE)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boucher, P.; Herz, J. Signaling through LRP1: Protection from atherosclerosis and beyond. Biochem. Pharmacol. 2011, 81, 1–5. [Google Scholar] [CrossRef]
- Bres, E.E.; Faissner, A. Low Density Receptor-Related Protein 1 Interactions with the Extracellular Matrix: More Than Meets the Eye. Front. Cell Dev. Biol. 2019, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Fuentealba, R.A.; Liu, Q.; Kanekiyo, T.; Zhang, J.; Bu, G. Low density lipoprotein receptor-related protein 1 promotes anti-apoptotic signaling in neurons by activating Akt survival pathway. J. Biol. Chem. 2009, 284, 34045–34053. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.M.; Kuhlmann, C.; Trossbach, S.; Jaeger, S.; Waldron, E.; Roebroek, A.; Luhmann, H.J.; Laatsch, A.; Weggen, S.; Lessmann, V.; et al. The functional role of the second NPXY motif of the LRP1 beta-chain in tissue-type plasminogen activator-mediated activation of N-methyl-D-aspartate receptors. J. Biol. Chem. 2008, 283, 12004–12013. [Google Scholar] [CrossRef] [PubMed]
- Muratoglu, S.C.; Mikhailenko, I.; Newton, C.; Migliorini, M.; Strickland, D.K. Low density lipoprotein receptor-related protein 1 (LRP1) forms a signaling complex with platelet-derived growth factor receptor-beta in endosomes and regulates activation of the MAPK pathway. J. Biol. Chem. 2010, 285, 14308–14317. [Google Scholar] [CrossRef]
- Hussain, M.M.; Strickland, D.K.; Bakillah, A. The mammalian low-density lipoprotein receptor family. Annu. Rev. Nutr. 1999, 19, 141–172. [Google Scholar] [CrossRef]
- Lorent, K.; Overbergh, L.; Delabie, J.; Van Leuven, F.; Van den Berghe, H. Distribution of mRNA coding for alpha-2-macroglobulin, the murinoglobulins, the alpha-2-macroglobulin receptor and the alpha-2-macroglobulin receptor associated protein during mouse embryogenesis and in adult tissues. Differentiation 1994, 55, 213–223. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Hennen, E.; Safina, D.; Haussmann, U.; Wörsdörfer, P.; Edenhofer, F.; Poetsch, A.; Faissner, A. A LewisX glycoprotein screen identifies the low density lipoprotein receptor-related protein. 1 (LRP1) as a modulator of oligodendrogenesis in mice. J. Biol. Chem. 2013, 288, 16538–16545. [Google Scholar] [CrossRef]
- Safina, D.; Schlitt, F.; Romeo, R.; Pflanzner, T.; Pietrzik, C.U.; Narayanaswami, V.; Edenhofer, F.; Faissner, A. Low-density lipoprotein receptor-related protein 1 is a novel modulator of radial glia stem cell proliferation, survival, and differentiation. Glia 2016, 64, 1363–1380. [Google Scholar] [CrossRef] [PubMed]
- Auderset, L.; Cullen, C.L.; Young, K.M. Low Density Lipoprotein-Receptor Related Protein 1 is Differentially Expressed by Neuronal and Glial Populations in the Developing and Mature Mouse Central Nervous System. PLoS ONE 2016, 11, e0155878. [Google Scholar] [CrossRef] [PubMed]
- Herz, J.; Clouthier, D.E.; Hammer, R.E. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell 1992, 71, 411–421. [Google Scholar] [CrossRef]
- Simpson, P.B.; Armstrong, R.C. Intracellular signals and cytoskeletal elements involved in oligodendrocyte progenitor migration. Glia 1999, 26, 22–35. [Google Scholar] [CrossRef]
- Rowitch, D.H.; Kriegstein, A.R. Developmental genetics of vertebrate glial-cell specification. Nature 2010, 468, 214–222. [Google Scholar] [CrossRef]
- Lin, J.-P.; Mironova, Y.A.; Shrager, P.; Giger, R.J. LRP1 regulates peroxisome biogenesis and cholesterol homeostasis in oligodendrocytes and is required for proper CNS myelin development and repair. eLife 2017, 6, e30498. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Pringle, N.P.; Richardson, W.D. A singularity of PDGF alpha-receptor expression in the dorsoventral axis of the neural tube may define the origin of the oligodendrocyte lineage. Development 1993, 117, 525–533. [Google Scholar]
- Horner, P.J.; Thallmair, M.; Gage, F.H. Defining the NG2-expressing cell of the adult CNS. J. Neurocytol. 2002, 31, 469–480. [Google Scholar] [CrossRef]
- Rohlmann, A.; Gotthardt, M.; Willnow, T.E.; Hammer, R.E.; Herz, J. Sustained somatic gene inactivation by viral transfer of Cre recombinase. Nat. Biotechnol. 1996, 14, 1562–1565. [Google Scholar] [CrossRef]
- Huang, W.; Zhao, N.; Bai, X.; Karram, K.; Trotter, J.; Goebbels, S.; Scheller, A.; Kirchhoff, F. Novel NG2-CreERT2 knock-in mice demonstrate heterogeneous differentiation potential of NG2 glia during development. Glia 2014, 62, 896–913. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Willet, S.G.; Bankaitis, E.D.; Xu, Y.; Wright, C.V.; Gu, G. Non-parallel recombination limits Cre-LoxP-based reporters as precise indicators of conditional genetic manipulation. Genesis 2013, 51, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Tobin, J.E.; Budde, M.D.; Chen, C.I.; Trinkaus, K.; Cross, A.H.; McDaniel, D.P.; Song, S.K.; Armstrong, R.C. Rostrocaudal analysis of corpus callosum demyelination and axon damage across disease stages refines diffusion tensor imaging correlations with pathological features. J. Neuropathol. Exp. Neurol. 2010, 69, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Haro, D.; Mora-Loyola, E.; Soria-Ortiz, B.; Garcia-Colunga, J. Regional density of glial cells in the rat corpus callosum. Biol. Res. 2013, 46, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Van Tilborg, E.; de Theije Caroline, G.M.; van Hal, M.; Wagenaar, N.; de Vries, L.S.; Benders, M.J.; Rowitch, D.H.; Nijboer, C.H. Origin and dynamics of oligodendrocytes in the developing brain: Implications for perinatal white matter injury. Glia 2018, 66, 221–238. [Google Scholar] [CrossRef] [PubMed]
- Steelman, A.J.; Thompson, J.P.; Li, J. Demyelination and remyelination in anatomically distinct regions of the corpus callosum following cuprizone intoxication. Neurosci. Res. 2012, 72, 32–42. [Google Scholar] [CrossRef]
- Wu, Q.-Z.; Yang, Q.; Cate, H.S.; Kemper, D.; Binder, M.; Wang, H.X.; Fang, K.; Quick, M.J.; Marriott, M.; Kilpatrick, T.J.; et al. MRI identification of the rostral-caudal pattern of pathology within the corpus callosum in the cuprizone mouse model. J. Magn. Reson. Imaging 2008, 27, 446–453. [Google Scholar] [CrossRef]
- Jahn, H.M.; Kasakow, C.V.; Helfer, A.; Michely, J.; Verkhratsky, A.; Maurer, H.H.; Scheller, A.; Kirchhoff, F. Refined protocols of tamoxifen injection for inducible DNA recombination in mouse astroglia. Sci. Rep. 2018, 8, 5913. [Google Scholar] [CrossRef]
- Kessaris, N.; Fogarty, M.; Iannarelli, P.; Grist, M.; Wegner, M.; Richardson, W.D. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat. Neurosci. 2006, 9, 173–179. [Google Scholar] [CrossRef]
- Huang, W.; Guo, Q.; Bai, X.; Scheller, A.; Kirchhoff, F. Early embryonic NG2 glia are exclusively gliogenic and do not generate neurons in the brain. Glia 2019, 67, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Ozerdem, U.; Grako, K.A.; Dahlin-Huppe, K.; Monosov, E.; Stallcup, W.B. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev. Dyn. 2001, 222, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Komitova, M.; Suzuki, R.; Zhu, X. Polydendrocytes (NG2 cells): Multifunctional cells with lineage plasticity. Nat. Rev. Neurosci. 2009, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Dimou, L.; Gallo, V. NG2-glia and their functions in the central nervous system. Glia 2015, 63, 1429–1451. [Google Scholar] [CrossRef] [Green Version]
- Richardson, W.D.; Young, K.M.; Tripathi, R.B.; McKenzie, I. NG2-glia as multipotent neural stem cells: Fact or fantasy? Neuron 2011, 70, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.; Gallo, V. Postnatal neurogenesis and gliogenesis in the olfactory bulb from NG2-expressing progenitors of the subventricular zone. J. Neurosci. 2004, 24, 10530–10541. [Google Scholar] [CrossRef]
- Aguirre, A.A.; Chittajallu, R.; Belachew, S.; Gallo, V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J. Cell Biol. 2004, 165, 575–589. [Google Scholar] [CrossRef]
- Liu, Q.; Trotter, J.; Zhang, J.; Peters, M.M.; Cheng, H.; Bao, J.; Han, X.; Weeber, E.J.; Bu, G. Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J. Neurosci. 2010, 30, 17068–17078. [Google Scholar] [CrossRef] [Green Version]
- Del Rio-Hortega, P. Studies on neuroglia: Glia with very few processes (oligodendroglia) by PÃ-o del RÃ-o-Hortega. 1921. Clin. Neuropathol. 2012, 31, 440–459. [Google Scholar]
- Pérez-Cerdá, F.; Sánchez-Gómez, M.V.; Matute, C. Pío del Río Hortega and the discovery of the oligodendrocytes. Front. Neuroanat. 2015, 9, 92. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Wang, S.; Anderson, D.J. Identification of a novel family of oligodendrocyte lineage-specific basic helix-loop-helix transcription factors. Neuron 2000, 25, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.C. Defining glial cells during CNS development. Nat. Rev. Neurosci. 2001, 2, 840–843. [Google Scholar] [CrossRef] [PubMed]
- Nikić, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef]
- Kipp, M.; Nyamoya, S.; Hochstrasser, T.; Amor, S. Multiple sclerosis animal models: A clinical and histopathological perspective. Brain Pathol. 2017, 27, 123–137. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- May, P. The low-density lipoprotein receptor-related protein 1 in inflammation. Curr. Opin. Lipidol. 2013, 24, 134–137. [Google Scholar] [CrossRef]
- Bechler, M.E.; Byrne, L.; Ffrench-Constant, C. CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Curr. Biol. 2015, 25, 2411–2416. [Google Scholar] [CrossRef] [Green Version]
- Ulc, A.; Zeug, A.; Bauch, J.; van Leeuwen, S.; Kuhlmann, T.; ffrench-Constant, C.; Ponimaskin, E.; Faissner, A. The guanine nucleotide exchange factor Vav3 modulates oligodendrocyte precursor differentiation and supports remyelination in white matter lesions. Glia 2019, 67, 376–392. [Google Scholar] [CrossRef]
- Zuchero, J.B.; Barres, B.A. Intrinsic and extrinsic control of oligodendrocyte development. Curr. Opin. Neurobiol. 2013, 23, 914–920. [Google Scholar] [CrossRef] [Green Version]
- Elbaz, B.; Popko, B. Molecular Control of Oligodendrocyte Development. Trends Neurosci. 2019. [Google Scholar] [CrossRef]
- Herz, J.; Strickland, D.K. LRP: A multifunctional scavenger and signaling receptor. J. Clin. Investig. 2001, 108, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Auderset, L.; Landowski, L.M.; Foa, L.; Young, K.M. Low Density Lipoprotein Receptor Related Proteins as Regulators of Neural Stem and Progenitor Cell Function. Stem Cells Int. 2016, 2016, 2108495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Sluis Bart Wijers, M.; Herz, J. News on the molecular regulation and function of hepatic low-density lipoprotein receptor and LDLR-related protein 1. Curr. Opin. Lipidol. 2017, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Krämer-Albers, E.-M.; Gehrig-Burger, K.; Thiele, C.; Trotter, J.; Nave, K.A. Perturbed interactions of mutant proteolipid protein/DM20 with cholesterol and lipid rafts in oligodendroglia: Implications for dysmyelination in spastic paraplegia. J. Neurosci. 2006, 26, 11743–11752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, E.S.; Mawdsley, D.J.; Walker, M.; Hines, J.H.; Pozzoli, M.; Appel, B. Mutation of. 3-hydroxy-3-methylglutaryl CoA synthase I reveals requirements for isoprenoid and cholesterol synthesis in oligodendrocyte migration arrest, axon wrapping, and myelin gene expression. J. Neurosci. 2014, 34, 3402–3412. [Google Scholar] [CrossRef] [PubMed]
- Saher, G.; Brügger, B.; Lappe-Siefke, C.; Mobius, W.; Tozawa, R.; Wehr, M.C.; Wieland, F.; Ishibashi, S.; Nave, K.A. High cholesterol level is essential for myelin membrane growth. Nat. Neurosci. 2005, 8, 468–475. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Product Size | Source |
|---|---|---|---|
| LRP1 | for 5′-CATACCCTCTTCAAACCCCTTCCTG rev 5′-GCAAGCTCTCCTGCTCAGACCTGGA | WT: 291 bp KO: 350 bp | Jackson Laboratory |
| NG2-Cre (NGCE) | for 5′-GGCAAACCCAGAGCCCTGCC wt rev 5′-GCTGGAGCTGACAGCGGGTG Cre-ERT2 rev 5′-GCCCGGACCGACGATGAAGC | WT: 557 bp KO: 829 bP | [21] |
| Rosa26-GCaMP3 | for wt 5′-CTCTGCTGCCTCCTGGCTTCT wt rev 5′-CGAGGCGGATCACAAGCAATA for KI 5′-CACGTGATGACAAACCTTGG rev KI 5′-GGCATTAAAGCAGCGTATCC | WT: 327 bp KO: 245 bp | [22] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schäfer, I.; Kaisler, J.; Scheller, A.; Kirchhoff, F.; Haghikia, A.; Faissner, A. Conditional Deletion of LRP1 Leads to Progressive Loss of Recombined NG2-Expressing Oligodendrocyte Precursor Cells in a Novel Mouse Model. Cells 2019, 8, 1550. https://doi.org/10.3390/cells8121550
Schäfer I, Kaisler J, Scheller A, Kirchhoff F, Haghikia A, Faissner A. Conditional Deletion of LRP1 Leads to Progressive Loss of Recombined NG2-Expressing Oligodendrocyte Precursor Cells in a Novel Mouse Model. Cells. 2019; 8(12):1550. https://doi.org/10.3390/cells8121550
Chicago/Turabian StyleSchäfer, Ina, Johannes Kaisler, Anja Scheller, Frank Kirchhoff, Aiden Haghikia, and Andreas Faissner. 2019. "Conditional Deletion of LRP1 Leads to Progressive Loss of Recombined NG2-Expressing Oligodendrocyte Precursor Cells in a Novel Mouse Model" Cells 8, no. 12: 1550. https://doi.org/10.3390/cells8121550