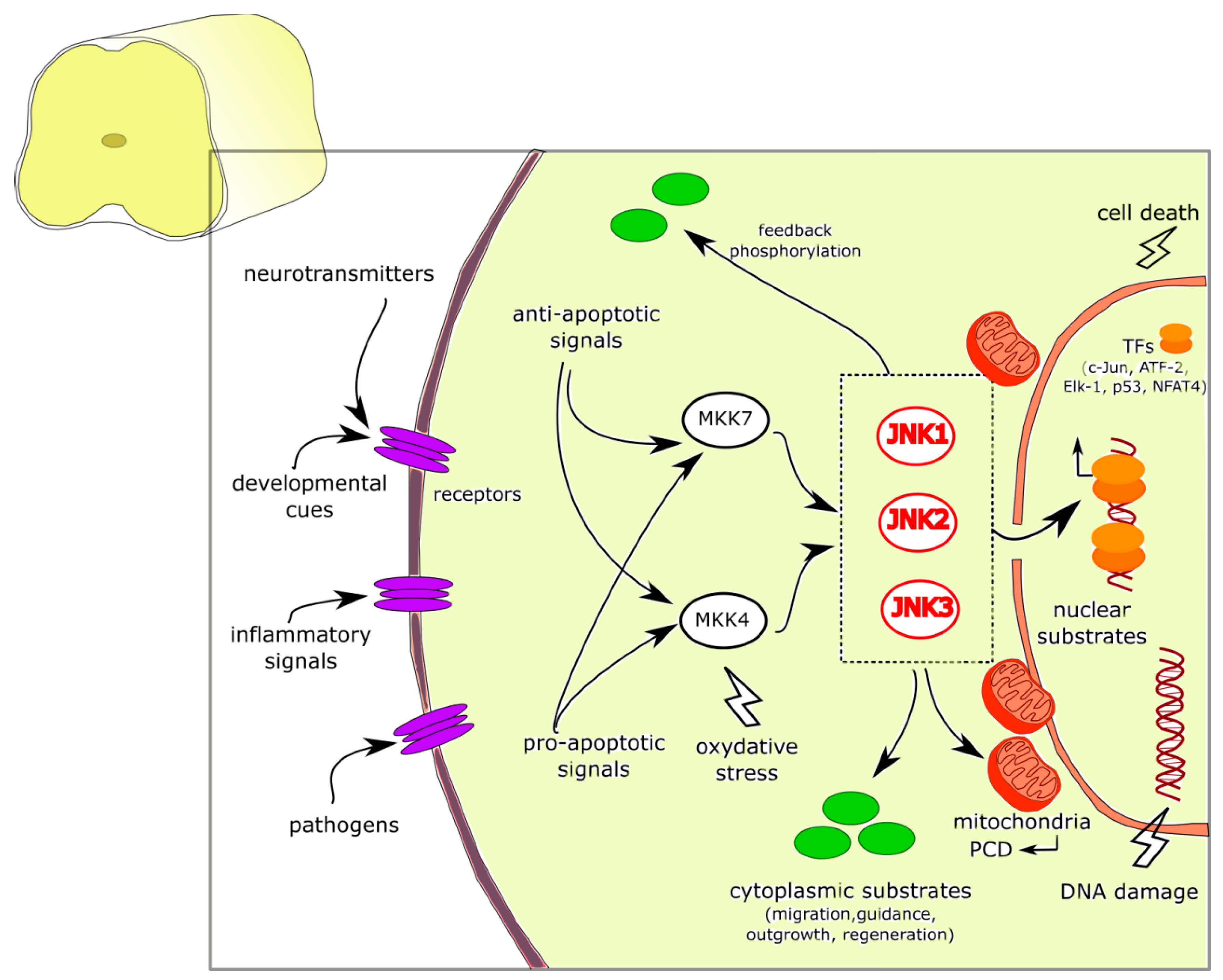
JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
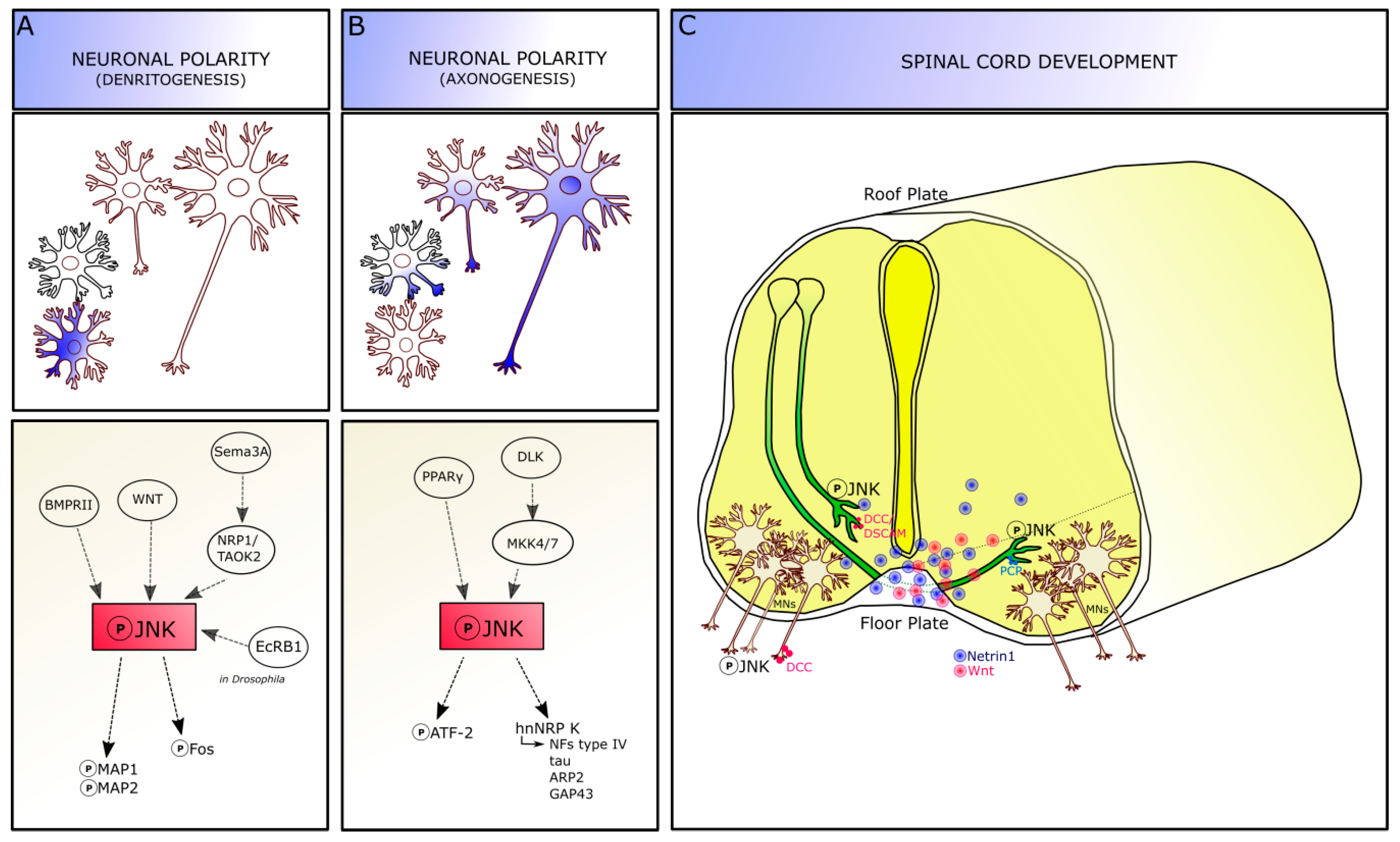
2. JNK in CNS Development
2.1. JNK Role in Dendritogenesis
2.2. JNK Role in Axonogenesis
3. JNK in Developing Spinal Cord
3.1. JNK in Commissural Axons
3.2. JNK in Motor Neuron Development
3.3. JNK in Spinal Cord Patterning
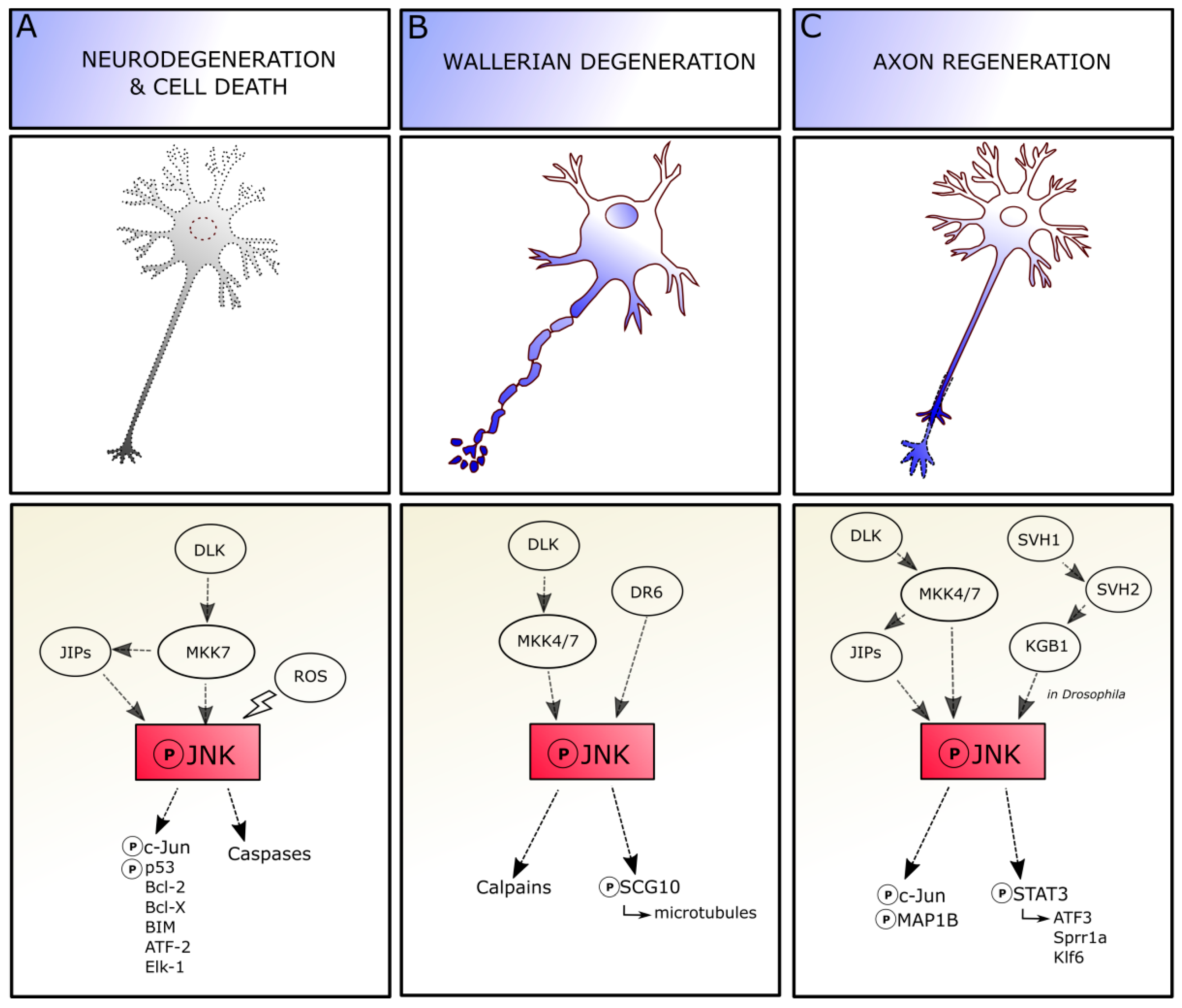
4. JNK in Adult CNS
JNK in Axonal Degeneration and Regeneration
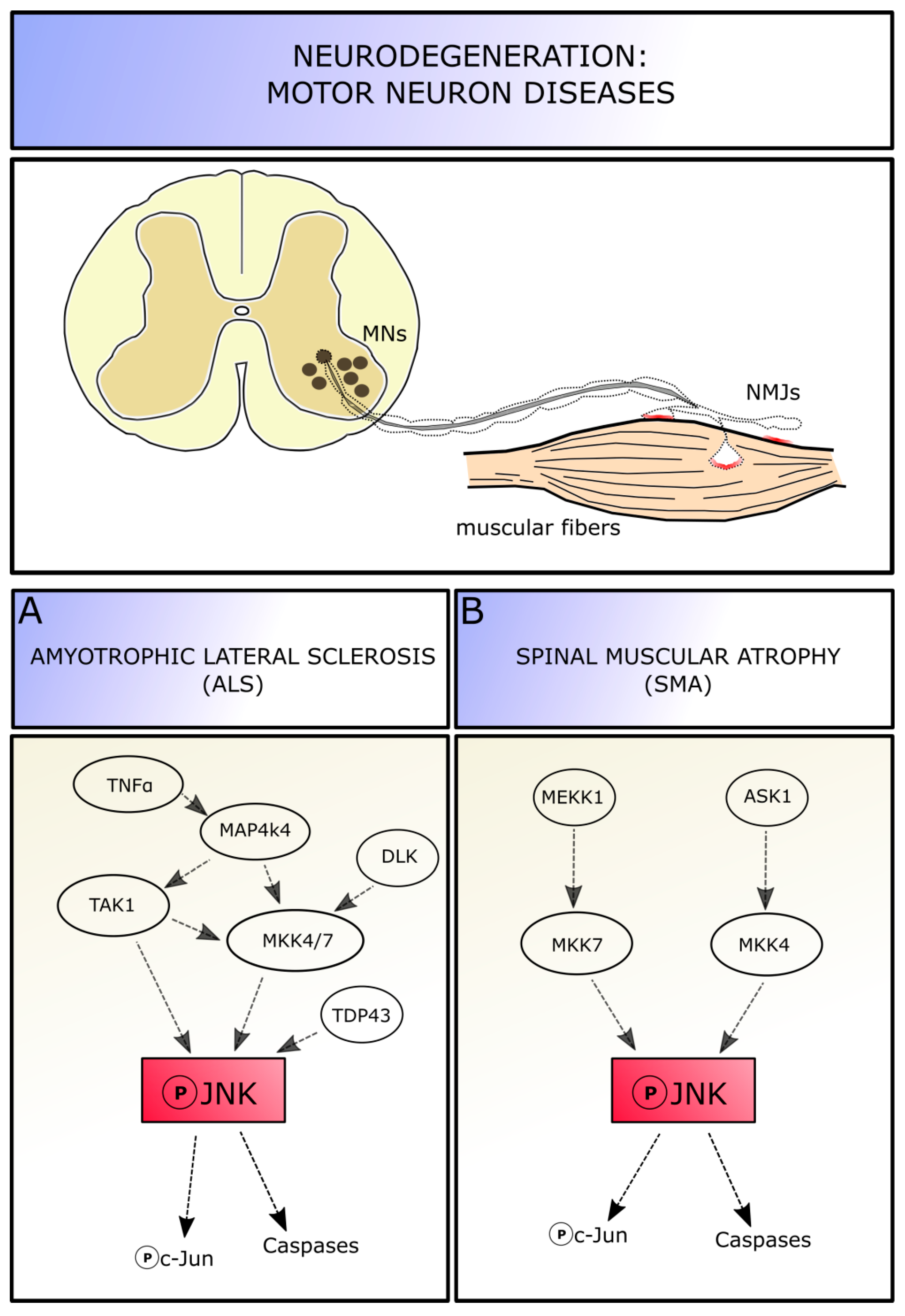
5. JNK in MN Degenerative Diseases
5.1. JNK Role in Amyotrophic Lateral Sclerosis
5.2. JNK Role in Spinal Muscular Atrophy
5.3. JNK Role in Spinal and Bulbar Muscular Atrophy
6. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Pulverer, B.J.; Kyriakis, J.M.; Avruch, J.; Nikolakaki, E.; Woodgett, J.R. Phosphorylation of c-jun mediated by MAP kinases. Nature 1991, 353, 670–674. [Google Scholar] [CrossRef]
- Smeal, T.; Binetruy, B.; Mercola, D.A.; Birrer, M.; Karin, M. Oncogenic and transcriptional cooperation with Ha-Ras requires phosphorylation of c-Jun on serines 63 and 73. Nature 1991, 354, 494–496. [Google Scholar] [CrossRef] [PubMed]
- Dérijard, B.; Raingeaud, J.; Barrett, T.; Wu, I.H.; Han, J.; Ulevitch, R.J.; Davis, R.J. Independent human +MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 1995, 267, 682–685, Erratum in: Science 1995, 269, 17. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C. The 2 Faces of JNK Signaling in Cancer. Genes Cancer 2013, 4, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Destrument, A.; Tournier, C. Physiological roles of MKK4 and MKK7: Insights from animal models. Biochim. Biophys. Acta 2007, 1773, 1349–1357. [Google Scholar] [CrossRef]
- Dickens, M.; Rogers, J.S.; Cavanagh, J.; Raitano, A.; Xia, Z.; Halpern, J.R.; Greenberg, M.E.; Sawyers, C.L.; Davis, R.J. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science 1997, 277, 693–696. [Google Scholar] [CrossRef]
- Yasuda, J.; Whitmarsh, A.J.; Cavanagh, J.; Sharma, M.; Davis, R.J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 1999, 19, 7245–7254. [Google Scholar] [CrossRef]
- Ito, M.; Yoshioka, K.; Akechi, M.; Yamashita, S.; Takamatsu, N.; Sugiyama, K.; Hibi, M.; Nakabeppu, Y.; Shiba, T.; Yamamoto, K.I. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signalling pathway. Mol. Cell. Biol. 1999, 19, 7539–7548. [Google Scholar] [CrossRef]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Derijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef]
- Yamasaki, T.; Kawasaki, H.; Nishina, H. Diverse roles of JNK and MKK pathways in the brain. J. Signal. Transduct. 2012, 2012, 459265. [Google Scholar] [CrossRef]
- Coffey, E.T.; Courtney, M.J. Regulation of SAPKs in CNS neurons. Biochem. Soc. Trans. 1997, 25, S568. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Metzler, B.; Xu, Q. Discordant activation of stress-activated protein kinases or c-Jun NH2-terminal protein kinases in tissues of heat-stressed mice. J. Biol. Chem. 1997, 272, 9113–9119. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T.; Hongisto, V.; Dickens, M.; Davis, R.J.; Courtney, M.J. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J. Neurosci. 2000, 20, 7602–7613. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Miller, C.A. A splicing variant of a death domain protein that is regulated by a mitogenactivated kinase is a substrate for c-Jun N-terminal kinase in the human central nervous system. Proc. Natl Acad. Sci. USA 1998, 95, 2586–2591. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yang, T.; Xu, Z. The JNK pathway and neuronal migration. J. Genet. Genom. 2007, 34, 957–965. [Google Scholar] [CrossRef]
- Coffey, E.T. Nuclear and cytosolic JNK signaling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Waetzig, V.; Zhao, Y.; Herdegen, T. The bright side of JNKs-multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog. Neurobiol. 2006, 80, 84–97. [Google Scholar] [CrossRef]
- Zhao, Y.; Spigolon, G.; Bonny, C.; Culman, J.; Vercelli, A.; Herdegen, T. The JNK inhibitor D-JNKI-1 blocks apoptotic JNK signaling in brain mitochondria. Mol. Cell. Neurosci. 2012, 49, 300–310. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bimrelated members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Dong, C.; Turner, T.K.; Jones, S.N.; Flavell, R.A.; Davis, R.J. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001, 15, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Winchester, C.L.; Ohzeki, H.; Vouyiouklis, D.A.; Thompson, R.; Penninger, J.M.; Yamagami, K.; Norrie, J.D.; Hunter, R.; Pratt, J.A.; Morris, B.J. Converging evidence that sequence variations in the novel candidate gene MAP2K7 (MKK7) are functionally associated with schizophrenia. Hum. Mol. Genet. 2012, 21, 4910–4921. [Google Scholar] [CrossRef] [PubMed]
- Kunde, S.A.; Rademacher, N.; Tzschach, A.; Wiedersberg, E.; Ullmann, R.; Kalscheuer, V.M.; Shoichet, S.A. Characterisation of de novo MAPK10/JNK3 truncation mutations associated with cognitive disorders in two unrelated patients. Hum. Genet. 2013, 132, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H. When signaling kinases meet histones and histone modifiers in the nucleus. Mol. Cell. 2011, 42, 274–284. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef]
- Björkblom, B.; Ostman, N.; Hongisto, V.; Komarovski, V.; Filén, J.J.; Nyman, T.A.; Kallunki, T.; Courtney, M.J.; Coffey, E.T. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: Role of microtubule-associated protein 2 as an effector. J. Neurosci. 2005, 25, 6350–6361. [Google Scholar] [CrossRef]
- Tararuk, T.; Ostman, N.; Li, W.; Bjorkblom, B.; Padzik, A.; Zdrojewska, J.; Hongisto, V.; Herdegen, T.; Konopka, W.; Courtney, M.J.; et al. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J. Cell Biol. 2006, 173, 265–277. [Google Scholar] [CrossRef]
- Tönges, L.; Planchamp, V.; Koch, J.C.; Herdegen, T.; Bähr, M.; Lingor, P. JNK isoforms differentially regulate neurite growth and regeneration in dopaminergic neurons in vitro. J. Mol. Neurosci. 2011, 45, 284–293. [Google Scholar] [CrossRef]
- Chang, L.; Jones, Y.; Ellisman, M.H.; Goldstein, L.S.; Karin, M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev. Cell. 2003, 4, 521–533. [Google Scholar] [CrossRef]
- Podkowa, M.; Zhao, X.; Chow, C.W.; Coffey, E.; Davis, R.J.; Attisano, L. Microtubule stabilization by bone morphogenetic protein receptor-mediated scaffolding of c-Jun N-terminal kinase promotes dendrite formation. Mol. Cell. Biol. 2010, 30, 2241–2250. [Google Scholar] [CrossRef] [PubMed]
- Rosso, S.B.; Sussman, D.; Wynshaw-Boris, A.; Salinas, P.C. Wnt signaling through dishevelled, Rac and JNK regulates dendritic development. Nat.Neurosci. 2005, 8, 34–42. [Google Scholar] [CrossRef] [PubMed]
- de Anda, F.C.; Rosario, A.L.; Durak, O.; Tran, T.; Gräff, J.; Meletis, K.; Rei, D.; Soda, T.; Madabhushi, R.; Ginty, D.D.; et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat. Neurosci. 2012, 15, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Chen, R.; Soba, P.; Jan, Y.N. JNK signaling coordinates with ecdysone signaling to promote pruning of Drosophila sensory neuron dendrites. Development 2019, 146, dev163592. [Google Scholar] [CrossRef] [PubMed]
- Oliva, A.A., Jr.; Atkins, C.M.; Copenagle, L.; Banker, G.A. Activated c-Jun N-terminal kinase is required for axon formation. J. Neurosci. 2006, 26, 9462–9470. [Google Scholar] [CrossRef]
- Esch, T.; Lemmon, V.; Banker, G. Local presentation of substrate molecules directs axon specification by cultured hippocampal neurons. J. Neurosci. 1999, 19, 6417–6426. [Google Scholar] [CrossRef]
- Hao, Y.; Waller, T.J.; Nye, D.M.; Li, J.; Zhang, Y.; Hume, R.I.; Rolls, M.M.; Collins, C.A. Degeneration of injured axons and dendrites requires restraint of a protective JNK signaling pathway by the transmembrane protein raw. J. Neurosci. 2019, 39, 8457–8470. [Google Scholar] [CrossRef]
- Hutchins, E.J.; Szaro, B.G. c-jun N-terminal kinase phosphorylation of heterogeneous nuclear ribonucleoprotein K regulates vertebrate axon outgrowth via a posttranscriptional mechanism. J. Neurosci. 2013, 33, 14666–14680. [Google Scholar] [CrossRef]
- Abe, K.; Aoyagi, A.; Saito, H. Sustained phosphorylation of mitogen-activated protein kinase is required for basic fibroblast growth factor-mediated axonal branch formation in cultured rat hippocampal neurons. Neurochem. Int. 2001, 38, 309–315. [Google Scholar] [CrossRef]
- Herdegen, T.; Leah, J.D. Inducible and constitutive transcription factors in the mammalian nervous system: Control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res. Rev. 1998, 28, 370–490. [Google Scholar] [CrossRef]
- Kreutz, M.R.; Bien, A.; Vorwerk, C.K.; Bockers, T.M.; Seidenbecher, C.I.; Tischmeyer, W.; Sabel, B.A. Co-expression of c-jun and ATF-2 characterizes the surviving retinal ganglion cells which maintain axonal connections after partial optic nerve injury. Brain Res. Mol. Brain Res. 1999, 69, 232–241. [Google Scholar] [CrossRef]
- Quintanilla, R.A.; Godoy, J.A.; Alfaro, I.; Cabezas, D.; von Bernhardi, R.; Bronfman, M.; Inestrosa, N.C. Thiazolidinediones promote axonal growth through the activation of the JNK pathway. PLoS ONE 2013, 8, e65140. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Li, W.; Shao, Q.; Dwyer, T.; Huang, H.; Yang, T.; Liu, G. c-jun N-terminal kinase 1 (JNK1) is required for coordination of netrin signaling in axon guidance. J. Biol. Chem. 2013, 288, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Chédotal, A. Roles of axon guidance molecules in neuronal wiring in the developing spinal cord. Nat. Rev. Neurosci. 2019, 20, 380–396. [Google Scholar] [CrossRef]
- Ducuing, H.; Gardette, T.; Pignata, A.; Tauszig-Delamasure, S.; Castellani, V. Commissural axon navigation in the spinal cord: A repertoire of repulsive forces is in command. Semin. Cell Dev. Biol. 2019, 85, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Shafer, B.; Onishi, K.; Lo, C.; Colakoglu, G.; Zou, Y. Vangl2 promotes Wnt/planar cell polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev. Cell. 2011, 20, 177–191. [Google Scholar] [CrossRef]
- Serafini, T.; Colamarino, S.A.; Leonardo, E.D.; Wang, H.; Beddington, R.; Skarnes, W.C.; Tessier-Lavigne, M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 1996, 87, 1001–1014. [Google Scholar] [CrossRef]
- Fazeli, A.; Dickinson, S.L.; Hermiston, M.L.; Tighe, R.V.; Steen, R.G.; Small, C.G.; Stoeckli, E.T.; Keino-Masu, K.; Masu, M.; Rayburn, H.; et al. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature 1997, 386, 796–804. [Google Scholar] [CrossRef]
- Hirai, S.; Banba, Y.; Satake, T.; Ohno, S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-jun N-terminal kinase pathway. J. Neurosci. 2011, 31, 6468–6480. [Google Scholar] [CrossRef]
- Zou, Y. Wnt signaling in axon guidance. Trends Neurosci. 2004, 27, 528–532. [Google Scholar] [CrossRef]
- Yamanaka, H.; Moriguchi, T.; Masuyama, N.; Kusakabe, M.; Hanafusa, H.; Takada, R.; Takada, S.; Nishida, E. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements invertebrates. EMBO Rep. 2002, 3, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T. Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science 1992, 256, 1555–1560. [Google Scholar]
- Fraher, J.P.; Dockery, P.; O’Donoghue, O.; Riedewald, B.; O’Leary, D. Initial motor axon outgrowth from the developing central nervous system. J. Anat. 2007, 211, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Bonanomi, D. Axon pathfinding for locomotion. Semin. Cell Dev. Biol. 2019, 85, 26–35. [Google Scholar] [CrossRef]
- Kim, M.; Fontelonga, T.M.; Lee, C.H.; Barnum, S.J.; Mastick, G.S. Motor axons are guided to exit points in the spinal cord by Slit and Netrin signals. Dev. Biol. 2017, 432, 178–191. [Google Scholar] [CrossRef]
- Kuan, C.Y.; Yang, D.D.; Samanta Roy, D.R.; Davis, R.J.; Rakic, P.; Flavell, R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999, 22, 667–676. [Google Scholar] [CrossRef]
- Messina, A.; Jaworowski, A.; Bell, C. Detection of jun but not fos protein during developmental cell death in sympathetic neurons. J. Comp. Neurol. 1996, 372, 544–550. [Google Scholar] [CrossRef]
- Raivich, G.; Behrens, A. Role of the AP-1 transcription factor c-jun in developing, adult and injured brain. Prog. Neurobiol. 2006, 78, 347–363. [Google Scholar] [CrossRef]
- Sun, W.; Gould, T.W.; Newbern, J.; Milligan, C.; Choi, S.Y.; Kim, H.; Oppenheim, R.W. Phosphorylation of c-jun in avian and mammalian motoneurons in vivo during programmed cell death: An early reversible event in the apoptotic cascade. J. Neurosci. 2005, 25, 5595–5603. [Google Scholar] [CrossRef]
- Ham, J.; Babij, C.; Whitfield, J.; Pfarr, C.M.; Lallemand, D.; Yaniv, M.; Rubin, L.L. A c-jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron 1995, 14, 927–939. [Google Scholar] [CrossRef]
- Borasio, G.D.; Horstmann, S.; Anneser, J.M.; Neff, N.T.; Glicksman, M.A. CEP-1347/KT7515, a JNK pathway inhibitor, supports the in vitro survival of chick embryonic neurons. Neuroreport 1998, 9, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Geden, M.J.; Deshmukh, M. Axon degeneration: Context defines distinct pathways. Curr. Opin. Neurobiol. 2016, 39, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Centeno, C.; Tomasi, S.; Forloni, G.; Bonny, C.; Vercelli, A.; Borsello, T. Time-course of c-jun N-terminal kinase activation after cerebral ischemia and effect of D-JNKI1 on c-jun and caspase-3 activation. Neuroscience 2007, 150, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Spigolon, G.; Veronesi, C.; Bonny, C.; Vercelli, A. c-jun N-terminal kinase signaling pathway in excitotoxic cell death following kainic acid-induced status epilepticus. Eur. J. Neurosci. 2010, 31, 1261–1272. [Google Scholar] [CrossRef]
- Sclip, A.; Tozzi, A.; Abaza, A.; Cardinetti, D.; Colombo, I.; Calabresi, P.; Salmona, M.; Welker, E.; Borsello, T. c-jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis. 2014, 5, e1019. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal kinase (JNK) signaling as a therapeutic target for Alzheimer’s disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Medress, Z.A.; Barres, B.A. Axon degeneration: Molecular mechanism of a self-destruction pathway. J. Cell Biol. 2012, 196, 7–18. [Google Scholar] [CrossRef]
- Becker, E.B.; Howell, J.; Kodama, Y.; Barker, P.A.; Bonni, A. Characterization of the c-jun N-terminal kinase-BimEL signaling pathway in neuronal apoptosis. J. Neurosci. 2004, 24, 8762–8770. [Google Scholar] [CrossRef]
- Newbern, J.; Taylor, A.; Robinson, M.; Lively, M.O.; Milligan, C.E. c-jun N-terminal kinase signaling regulates events associated with both health and degeneration in motoneurons. Neuroscience 2007, 147, 680–692. [Google Scholar] [CrossRef]
- Brecht, S.; Kirchhof, R.; Chromik, A.; Willesen, M.; Nicolaus, T.; Raivich, G.; Wessig, J.; Waetzig, V.; Goetz, M.; Claussen, M.; et al. Specific pathophysiological functions of JNK isoforms in the brain. Eur. J. Neurosci. 2005, 21, 363–377. [Google Scholar] [CrossRef]
- Keramaris, E.; Vanderluit, J.L.; Bahadori, M.; Mousavi, K.; Davis, R.J.; Flavell, R.; Slack, R.S.; Park, D.S. c-jun N-terminal kinase 3 deficiency protects neurons from axotomy-induced death in vivo through mechanisms independent of c-jun phosphorylation. J. Biol. Chem. 2005, 280, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Carulli, D.; Buffo, A.; Botta, C.; Altruda, F.; Strata, P. Regenerative and survival capabilities of Purkinje cells overexpressing c-jun. Eur. J. Neurosci. 2002, 16, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, S.; Yone, K.; Sakou, T.; Wada, S.; Nagamine, T.; Niiyama, T.; Ichijo, H. Induction of apoptosis signal regulating kinase 1 (ASK1) after spinal cord injury in rats: Possible involvement of ASK1-JNK and -p38 pathways in neuronal apoptosis. J. Neuropathol. Exp. Neurol. 1999, 58, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Kim, G.M.; Lee, J.M.; He, Y.Y.; Xu, J.; Hsu, C.Y. JNK activation contributes to DP5 induction and apoptosis following traumatic spinal cord injury. Neurobiol. Dis. 2005, 20, 881–889. [Google Scholar] [CrossRef]
- Repici, M.; Chen, X.; Morel, M.P.; Doulazmi, M.; Sclip, A.; Cannaya, V.; Veglianese, P.; Kraftsik, R.; Mariani, J.; Borsello, T.; et al. Specific inhibition of the JNK pathway promotes locomotor recovery and neuroprotection after mouse spinal cord injury. Neurobiol. Dis. 2012, 46, 710–721. [Google Scholar] [CrossRef]
- Lee, J.Y.; Maeng, S.; Kang, S.R.; Choi, H.Y.; Oh, T.H.; Ju, B.G.; Yune, T.Y. Valproic acid protects motor neuron death by inhibiting oxidative stress and endoplasmic reticulum stress-mediated cytochrome C release after spinal cord injury. J. Neurotrauma. 2014, 31, 582–594. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target. Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef]
- Li, G.Z.; Tao, H.L.; Zhou, C.; Wang, D.D.; Peng, C.B. Midazolam prevents motor neuronal death from oxidative stress attack mediated by JNK-ERK pathway. Hum. Cell. 2018, 31, 64–71. [Google Scholar] [CrossRef]
- Waller, A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos. Trans. R. Soc. Lond. 1850, 140, 423–429. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3. [Google Scholar] [CrossRef]
- Miller, B.R.; Press, C.; Daniels, R.W.; Sasaki, Y.; Milbrandt, J.; DiAntonio, A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat. Neurosci. 2009, 12, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Gamage, K.K.; Cheng, I.; Park, R.E.; Karim, M.S.; Edamura, K.; Hughes, C.; Spano, A.J.; Erisir, A.; Deppmann, C.D. Death receptor 6 promotes wallerian degeneration in peripheral axons. Curr. Biol. 2017, 27, 890–896. [Google Scholar] [CrossRef] [PubMed]
- Antonsson, B.; Kassel, D.B.; Di Paolo, G.; Lutjens, R.; Riederer, B.M.; Grenningloh, G. Identification of in vitro phosphorylation sites in the growth cone protein SCG10. Effect Of phosphorylation site mutants on microtubule-destabilizing activity. J. Biol. Chem. 1998, 273, 8439–8446. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.E.; Miller, B.R.; Babetto, E.; Cho, Y.; Sasaki, Y.; Qayum, S.; Russler, E.V.; Cavalli, V.; Milbrandt, J.; DiAntonio, A. SCG10 is a JNK target in the axonal degeneration pathway. Proc. Natl. Acad. Sci. USA 2012, 109, E3696–E3705. [Google Scholar] [CrossRef] [PubMed]
- Rallis, A.; Lu, B.; Ng, J. Molecular chaperones protect against JNK- and Nmnat-regulated axon degeneration in Drosophila. J. Cell Sci. 2013, 126, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Lindwall, C.; Dahlin, L.; Lundborg, G.; Kanje, M. Inhibition of c-jun phosphorylation reduces axonal outgrowth of adult rat nodose ganglia and dorsal root ganglia sensory neurons. Mol. Cell Neurosci. 2004, 27, 267–279. [Google Scholar] [CrossRef]
- Bouquet, C.; Soares, S.; von Boxberg, Y.; Ravaille-Veron, M.; Propst, F.; Nothias, F. Microtubule-associated protein 1B controls directionality of growth cone migration and axonal branching in regeneration of adult dorsal root ganglia neurons. J. Neurosci. 2004, 24, 7204–7213. [Google Scholar] [CrossRef]
- Barnat, M.; Enslen, H.; Propst, F.; Davis, R.J.; Soares, S.; Nothias, F. Distinct roles of c-jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J. Neurosci. 2010, 30, 7804–7816. [Google Scholar] [CrossRef]
- Broude, E.; McAtee, M.; Kelley, M.S.; Bregman, B.S. Fetal spinal cord transplants and exogenous neurotrophic support enhance c-jun expression in mature axotomized neurons after spinal cord injury. Exp. Neurol. 1999, 155, 65–78. [Google Scholar] [CrossRef]
- Herdegen, T.; Skene, P.; Bähr, M. The c-jun transcription factor—bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997, 20, 227–231. [Google Scholar] [CrossRef]
- Li, C.; Hisamoto, N.; Nix, P.; Kanao, S.; Mizuno, T.; Bastiani, M.; Matsumoto, K. The growth factor SVH-1 regulates axon regeneration in C. elegans via the JNK MAPK cascade. Nat. Neurosci. 2012, 15, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Brownlees, J.; Yates, A.; Bajaj, N.P.; Davis, D.; Anderton, B.H.; Leigh, P.N.; Shaw, C.E.; Miller, C.C. Phosphorylation of neurofilament heavy chain side-arms by stress activated protein kinase-1b/Jun N-terminal kinase-3. J. Cell Sci. 2000, 113, 401–407. [Google Scholar] [PubMed]
- Levy, D.E.; Lee, C.K. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar] [CrossRef]
- Schweizer, U.; Gunnersen, J.; Karch, C.; Wiese, S.; Holtmann, B.; Takeda, K.; Akira, S.; Sendtner, M. Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J. Cell Biol. 2002, 156, 287–297. [Google Scholar] [CrossRef]
- Sheu, J.Y.; Kulhanek, D.J.; Eckenstein, F.P. Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp. Neurol. 2000, 166, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Herdegen, T.; Waetzig, V. The JNK and p38 signal transduction following axotomy. Restor. Neurol. Neurosci. 2001, 19, 29–39. [Google Scholar] [PubMed]
- Nix, P.; Hisamoto, N.; Matsumoto, K.; Bastiani, M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 10738–10743. [Google Scholar] [CrossRef]
- Tedeschi, A.; Bradke, F. The DLK signaling pathway—A double-edged sword in neural development and regeneration. EMBO Rep. 2013, 14, 605–614. [Google Scholar] [CrossRef]
- Watkins, T.A.; Wang, B.; Huntwork-Rodriguez, S.; Yang, J.; Jiang, Z.; Eastham-Anderson, J.; Modrusa, Z.; Kaminker, J.S.; Tessier-Lavigne, M.; Lewcock, J.W. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc. Natl. Acad. Sci. USA 2013, 110, 4039–4044. [Google Scholar] [CrossRef]
- Leigh, P.N.; Dodson, A.; Swash, M.; Brion, J.P.; Anderton, B.H. Cytoskeletal abnormalities in motor neuron disease. An immunocytochemical study. Brain 1989, 112, 521–535. [Google Scholar] [CrossRef]
- Robberecht, W. Oxidative stress in amyotrophic lateral sclerosis. J. Neurol. 2000, 247, I1. [Google Scholar] [CrossRef] [PubMed]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The “dying-back” phenomenon of motor neurons in ALS. J. Mol. Neurosci. 2011, 43, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Piotrkiewicz, M.; Hausmanowa-Petrusewicz, I. Amyotrophic lateral sclerosis: A dying motor unit? Front. Aging Neurosci. 2013, 5, 7. [Google Scholar] [CrossRef]
- Boille’e, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their non-neuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef]
- Wu, C.; Watts, M.E.; Rubin, L.L. MAP4K4 activation mediates motor neuron degeneration in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 26, 1143–1156. [Google Scholar] [CrossRef]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signaling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef]
- Xu, P.; Das, M.; Reilly, J.; Davis, R.J. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011, 25, 310–322. [Google Scholar] [CrossRef]
- Suzuki, H.; Matsuoka, M. The JNK/c-jun signaling axis contributes to the TDP-43-induced cell death. Mol. Cell Biochem. 2013, 372, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Le Pichon, C.E.; Meilandt, W.J.; Dominguez, S.; Solanoy, H.; Lin, H.; Ngu, H.; Gogineni, A.; Sengupta Ghosh, A.; Jiang, Z.; Lee, S.H.; et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 2017, 9, eaag0394. [Google Scholar] [CrossRef] [PubMed]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic correction of SOD1 mutant iPSCs reveals ERK and JNK activated AP1 as a driver of neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wen, D.; Duan, W.; Yin, J.; Cui, C.; Wang, Y.; Li, Z.; Liu, Y.; Li, C. Systemic administration of scAAV9-IGF1 extends survival in SOD1(G93A) ALS mice via inhibiting p38 MAPK and the JNK-mediated apoptosis pathway. Brain Res. Bull. 2018, 139, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Lorson, C.L.; Hahnen, E.; Androphy, E.J.; Wirth, B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 1999, 96, 6307–6311. [Google Scholar] [CrossRef] [PubMed]
- Rossoll, W.; Jablonka, S.; Andreassi, C.; Kroning, A.K.; Karle, K.; Monani, U.R.; Sendtner, M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of {beta}-actin mRNA in growth cones of motoneurons. J. Cell Biol. 2003, 163, 801–812. [Google Scholar] [CrossRef]
- Ahmad, S.; Wang, Y.; Shaik, G.M.; Burghes, A.H.; Gangwani, L. The zinc finger protein ZPR1 is a potential modifier of spinal muscular atrophy. Hum. Mol. Genet. 2012, 21, 2745–2758. [Google Scholar] [CrossRef]
- Genabai, N.K.; Ahmad, S.; Zhang, Z.; Jiang, X.; Gabaldon, C.A.; Gangwani, L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Hum. Mol. Genet. 2015, 24, 6986–7004. [Google Scholar] [CrossRef]
- Ahmad, S.; Bhatia, K.; Kannan, A.; Gangwani, L. Molecular mechanisms of neurodegeneration in spinal muscular atrophy. J. Exp. Neurosci. 2016, 10, 39–49. [Google Scholar] [CrossRef]
- Simone, C.; Nizzardo, M.; Rizzo, F.; Ruggieri, M.; Riboldi, G.; Salani, S.; Bucchia, M.; Bresolin, N.; Comi, G.P.; Corti, S. iPSC-Derived neural stem cells act via kinase inhibition to exert neuroprotective effects in spinal muscular atrophy with respiratory distress type 1. Stem Cell Rep. 2014, 3, 297–311. [Google Scholar] [CrossRef]
- Borsello, T.; Clarke, P.G.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- Sclip, A.; Antoniou, X.; Colombo, A.; Camici, G.G.; Pozzi, L.; Cardinetti, D.; Feligioni, M.; Veglianese, P.; Bahlmann, F.H.; Cervo, L.; et al. c-jun N-terminal kinase regulates soluble Ab oligomers and cognitive impairment in AD mouse model. J. Biol. Chem. 2011, 286, 43871–43880. [Google Scholar] [CrossRef] [PubMed]
- Sclip, A.; Arnaboldi, A.; Colombo, I.; Veglianese, P.; Colombo, L.; Messa, M.; Mancini, S.; Cimini, S.; Morelli, F.; Antoniou, X.; et al. Soluble Ab oligomer-induced synaptopathy: C-jun N-terminal kinase’s role. J. Mol. Cell Biol. 2013, 5, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Manassero, G.; Repetto, I.E.; Cobianchi, S.; Valsecchi, V.; Bonny, C.; Rossi, F.; Vercelli, A. Role of JNK isoforms in the development of neuropathic pain following sciatic nerve transection in the mouse. Mol. Pain 2012, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Schellino, R.; Boido, M.; Borsello, T.; Vercelli, A. Pharmacological c-jun NH(2)-terminal kinase (JNK) pathway inhibition reduces severity of spinal muscular atrophy disease in mice. Front. Mol. Neurosci. 2018, 11, 308. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, M.; Beauvais, A.; Anderson, C.L.; Kothary, R. Rho-kinase inactivation prolongs survival of an intermediate SMA mouse model. Hum. Mol. Genet. 2010, 19, 1468–1478. [Google Scholar] [CrossRef]
- Gangwani, L.; Flavell, R.A.; Davis, R.J. ZPR1 is essential for survival and is required for localization of the survival motor neurons (SMN) protein to Cajal bodies. Mol. Cell Biol. 2005, 25, 2744–2756. [Google Scholar] [CrossRef]
- Jiang, X.; Kannan, A.; Gangwani, L. ZPR1-dependent neurodegeneration is mediated by the JNK signaling pathway. J. Exp. Neurosci. 2019, 13, 1179069519867915. [Google Scholar] [CrossRef]
- Adams, L. Motor neuron disease: Nusinersen potentially effective in SMA. Nat. Rev. Neurol. 2017, 13, 66. [Google Scholar] [CrossRef]
- Goyal, N.; Narayanaswami, P. Making sense of antisense oligonucleotides: A narrative review. Muscle Nerve 2018, 57, 356–370. [Google Scholar] [CrossRef]
- Pilato, C.M.; Park, J.H.; Kong, L.; d’Ydewalle, C.; Valdivia, D.; Chen, K.S.; Griswold-Prenner, I.; Sumner, C.J. Motor neuron loss in SMA is not associated with somal stress-activated JNK/c-jun signaling. Hum. Mol. Genet. 2019, 28, 3282–3292. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.M.; Dai, Y.; Van Alstyne, M.; Koutsioumpa, C.; Pagiazitis, J.G.; Chalif, J.I.; Wang, X.; Rabinowitz, J.E.; Henderson, C.E.; Pellizzoni, L.; et al. Converging mechanisms of p53 activation drive motor neuron degeneration in spinal muscular atrophy. Cell Rep. 2017, 21, 3767–3780. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.P.; Fischbeck, K.H. Spinal and bulbar muscular atrophy: A trinucleotide-repeat expansion neurodegenerative disease. Trends Neurosci. 1995, 18, 459–461. [Google Scholar] [CrossRef]
- Morfini, G.; Pigino, G.; Szebenyi, G.; You, Y.; Pollema, S.; Brady, S.T. JNK mediates pathogenic effects of polyglutamine-expanded androgen receptor on fast axonal transport. Nat. Neurosci. 2006, 9, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Young, J.E.; Garden, G.A.; Martinez, R.A.; Tanaka, F.; Sandoval, C.M.; Smith, A.C.; Sopher, B.L.; Lin, A.; Fischbeck, K.H.; Ellerby, L.M.; et al. Polyglutamine-expanded androgen receptor truncation fragments activate a Bax-dependent apoptotic cascade mediated by DP5/Hrk. J. Neurosci. 2009, 29, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Beitel, L.K.; Alvarado, C.; Mokhtar, S.; Paliouras, M.; Trifiro, M. Mechanisms mediating spinal and bulbar muscular atrophy: Investigations into polyglutamine-expanded androgen receptor function and dysfunction. Front. Neurol. 2013, 4, 53. [Google Scholar] [CrossRef]
- Arnold, F.J.; Pluciennik, A.; Merry, D.E. Impaired nuclear export of polyglutamine-expanded androgen receptor in spinal and bulbar muscular atrophy. Sci. Rep. 2019, 9, 119. [Google Scholar] [CrossRef]
- Graczyk, P.P. JNK inhibitors as anti-inflammatory and neuroprotective agents. Future Med. Chem. 2013, 5, 539–551. [Google Scholar] [CrossRef]
- Björkblom, B.; Vainio, J.C.; Hongisto, V.; Herdegen, T.; Courtney, M.J.; Coffey, E.T. All JNKs can kill, but nuclear localization is critical for neuronal death. J. Biol. Chem. 2008, 283, 19704–19713. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schellino, R.; Boido, M.; Vercelli, A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells 2019, 8, 1576. https://doi.org/10.3390/cells8121576
Schellino R, Boido M, Vercelli A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells. 2019; 8(12):1576. https://doi.org/10.3390/cells8121576
Chicago/Turabian StyleSchellino, Roberta, Marina Boido, and Alessandro Vercelli. 2019. "JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death" Cells 8, no. 12: 1576. https://doi.org/10.3390/cells8121576
APA StyleSchellino, R., Boido, M., & Vercelli, A. (2019). JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells, 8(12), 1576. https://doi.org/10.3390/cells8121576