Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges



Abstract
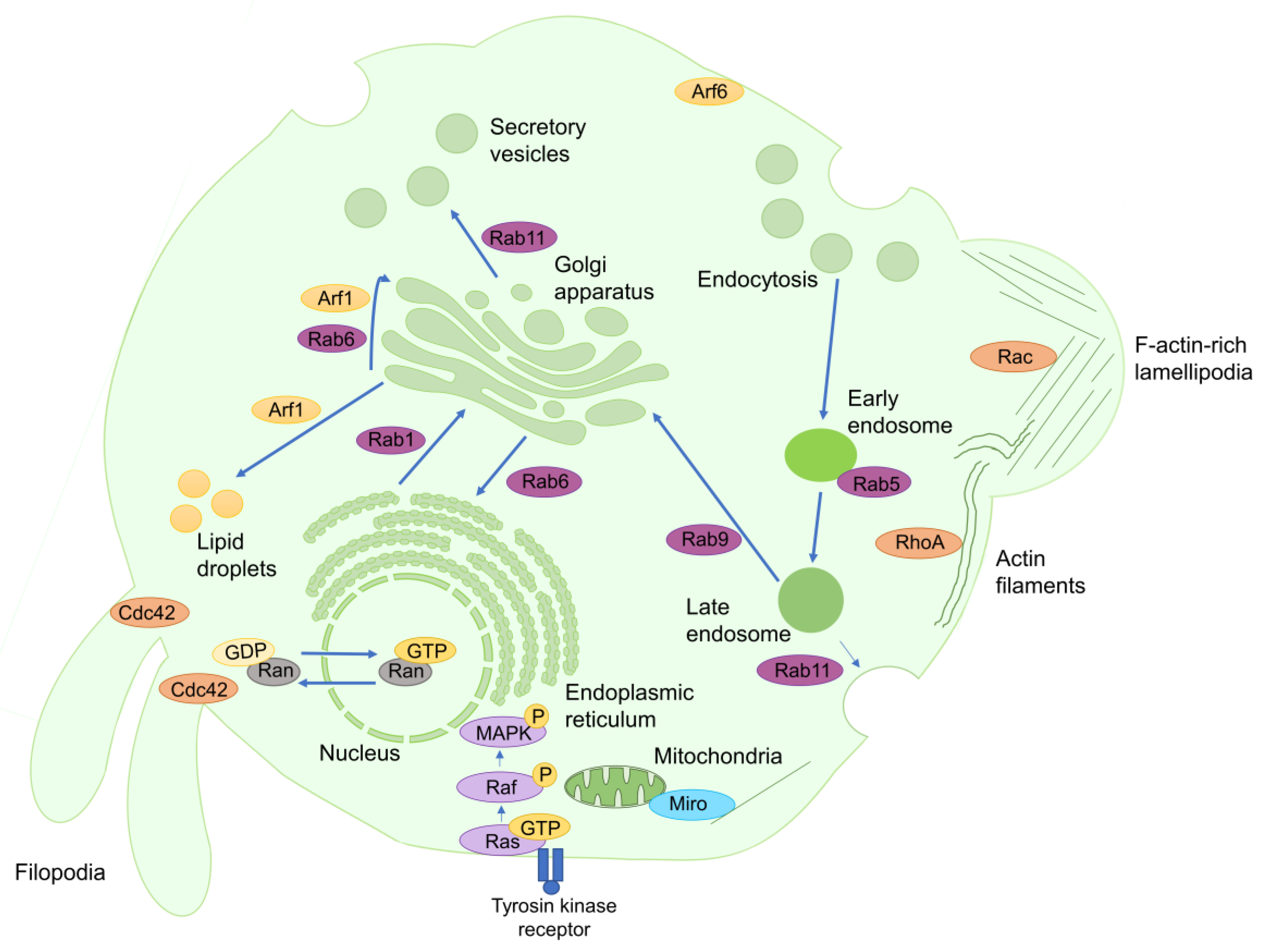
:1. Introduction
2. Arf1 and Its Inhibitors in Cancer Therapy
3. Ras and Its Inhibitors in Cancer Therapy
3.1. Chemical Strategies for Suppressing Ras Activity
3.2. Non-Chemical Mechanisms for Suppressing Ras Activity
4. Rac and Its Inhibitors in Cancer Therapy
5. Cdc42 and Its Inhibitors in Cancer Therapy
6. Targeting other Small GTPases in Cancer Therapy
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK | Anaplastic lymphoma kinase |
| ARL | Arf-like proteins |
| ARNO | Arf nucleotide-binding site opener |
| AZA1 | N*2*,N*4*-Bis-(2-methyl-1H-indol-5-yl)-pyrimidine-2,4-diamine |
| BART | Binder of Arl Two |
| BFA | Brefeldin A |
| CD4 | Cluster of differentiation 4 |
| Cdc42 | Cell division control protein 42 homolog |
| CERT | Ceramide transfer protein |
| EGFR | Epithelial growth factor receptor |
| EMT | Epithelial to mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| ERα | Estrogen receptor-alpha |
| Exo84 | Exocyst complex component 84 |
| FAK | Focal adhesion kinase |
| FAPP1 | Phosphatidylinositol-four-phosphate adapter protein 1 |
| GAP | GTPase-activating proteins |
| GATA1 | GATA-binding factor 1 |
| GEF | Guanine-nucleotide exchange factors |
| GRE | Rad and Gem-related proteins |
| H2 | Second α-helix of Ras |
| HIV | Human immunodeficiency virus |
| HNSCC | Head and neck squamous cell carcinoma |
| IMCT | Protein-S-isoprenylcysteine O-methyltransferase |
| ITSN | Intersectin |
| LARG | Leukemia-associated RhoGEF |
| M2+-cyclens | Divalent metal-cyclens |
| M6PR | Mannose-6-phosphate receptor |
| MAPK | Mitogen-activated protein kinases |
| MET | Mesenchymal to epithelial transition |
| Miro | Mitochondrial Rho GTPase |
| MMP | Matrix metalloproteinase |
| mTORC1 | mechanistic target of rapamycin complex 1 |
| NF-κB | nuclear factor-κB |
| NSAID | Non-steroidal anti-inflammatory drug |
| NSCLC | Non-small cell lung cancer |
| p70S6K | Ribosomal protein S6 kinase beta-1 |
| PAK | p21-activated kinase |
| PI3K | Phosphatidylinositol 3-kinase |
| PLCε | Phospholipase C epsilon |
| PLD1 | Phospholipase D1 |
| PREX1 | Phosphatidylinositol-3,4,5-trisphosphate dependent Rac exchange factor |
| Rab | Ras-related in brain |
| RabGGTase | Rab geranylgeranyltransferase |
| Rac | Ras-related C3 botulinum toxin substrate |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| Rad | Ras-related associated with diabetes |
| Ral | Ras-like proteins |
| Ran | Ras-related nuclear protein |
| Rap | Ras-proximal proteins |
| RCE1 | Prenyl protein-specific endoprotease 2 |
| RGMA | Repulsive guidance molecule A |
| Rheb | Ras homolog enriched in brain |
| Rho | Ras-homolog |
| RhoA | Rho-related proteins |
| RhoBTB3 | Rho-related BTB domain-containing protein 3 |
| RhoGDI1 | Rho guanine dissociation inhibitor 1 |
| Rit | Ras-like in all tissues proteins |
| Rnd | Rho-related GTP-binding protein Rho6 precursor |
| RRAS | Ras-related proteins |
| SecinH3 | Sec7 inhibitor H3 |
| Sema4D | Semaphorin 4D |
| SNARE | NSF-attachment protein receptor |
| SOS1 | Son of sevenless homolog 1 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TCL | TC10 and T-cell leukemia/lymphoma protein 1A |
| Tiam1 | T-lymphoma invasion and metastasis-inducing protein 1 |
| VEGFR | VEGF receptor |
| VEGF | Vascular endothelial growth factor |
| Zn2+-BPA | Zn2+-bis (2-picolyl) amine |
References
- Citi, S.; Spadaro, D.; Schneider, Y.; Stutz, J.; Pulimeno, P. Regulation of small GTPases at epithelial cell-cell junctions. Mol. Membr. Biol. 2011, 28, 427–444. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Nassar, N.; Wang, J. A mechanism of catalyzed GTP hydrolysis by Ras protein through magnesium ion. Chem. Phys. Lett. 2011, 516, 233–238. [Google Scholar] [CrossRef]
- Shi, G.X.; Andres, D.A.; Cai, W. Ras family small GTPase-mediated neuroprotective signaling in stroke. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 114–137. [Google Scholar] [CrossRef]
- Johnson, D.S.; Chen, Y.H. Ras family of small GTPases in immunity and inflammation. Curr. Opin. Pharmacol. 2012, 12, 458–463. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [Green Version]
- Heider, D.; Hauke, S.; Pyka, M.; Kessler, D. Insights into the classification of small GTPases. Adv. Appl. Bioinforma. Chem. 2010, 3, 15–24. [Google Scholar] [CrossRef]
- Colicelli, J. Human RAS superfamily proteins and related GTPases. Sci. STKE 2004, 2004, RE13. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Jackson, C.L. Arf family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef]
- Roth, M.G. Snapshots of ARF1: Implications for mechanisms of activation and inactivation. Cell 1999, 97, 149–152. [Google Scholar] [CrossRef]
- Casalou, C.; Faustino, A.; Barral, D.C. Arf proteins in cancer cell migration. Small GTPases 2016, 7, 270–282. [Google Scholar] [CrossRef] [Green Version]
- Kannangai, R.; Vivekanandan, P.; Martinez-Murillo, F.; Choti, M.; Torbenson, M. Fibrolamellar carcinomas show overexpression of genes in the RAS, MAPK, PIK3, and xenobiotic degradation pathways. Hum. Pathol. 2007, 38, 639–644. [Google Scholar] [CrossRef]
- Davis, J.E.; Xie, X.; Guo, J.; Huang, W.; Chu, W.M.; Huang, S.; Teng, Y.; Wu, G. ARF1 promotes prostate tumorigenesis via targeting oncogenic MAPK signaling. Oncotarget 2016, 7, 39834–39845. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, S.; Ramirez, R.A.; Claing, A. ARF1 regulates adhesion of MDA-MB-231 invasive breast cancer cells through formation of focal adhesions. Cell. Signal. 2015, 27, 403–415. [Google Scholar] [CrossRef]
- Li, R.; Peng, C.; Zhang, X.; Wu, Y.; Pan, S.; Xiao, Y. Roles of Arf6 in cancer cell invasion, metastasis and proliferation. Life Sci. 2017, 182, 80–84. [Google Scholar] [CrossRef]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras superfamily of small GTPases: The unlocked secrets. In Ras Signaling: Methods and Protocols; Trabalzini, L., Retta, S.F., Eds.; Springer: Berlin, Germany, 2014; Volume 1120, pp. 1–18. ISBN 9781627037914. [Google Scholar]
- Shirakawa, R.; Horiuchi, H. Ral GTPases: Crucial mediators of exocytosis and tumourigenesis. J. Biochem. 2015, 157, 285–299. [Google Scholar] [CrossRef]
- Di, J.; Huang, H.; Qu, D.; Tang, J.; Cao, W.; Lu, Z.; Cheng, Q.; Yang, J.; Bai, J.; Zhang, Y.; et al. Rap2B promotes proliferation, migration, and invasion of human breast cancer through calcium-related ERK1/2 signaling pathway. Sci. Rep. 2015, 5, 12363. [Google Scholar] [CrossRef] [Green Version]
- Gloerich, M.; Bos, J.L. Regulating Rap small G-proteins in time and space. Trends Cell Biol. 2011, 21, 615–623. [Google Scholar] [CrossRef]
- Armijo, M.E.; Campos, T.; Fuentes-Villalobos, F.; Palma, M.E.; Pincheira, R.; Castro, A.F. Rheb signaling and tumorigenesis: mTORC1 and new horizons. Int. J. Cancer 2016, 138, 1815–1823. [Google Scholar] [CrossRef]
- Shi, G.X.; Cai, W.; Andres, D.A. Rit subfamily small GTPases: Regulators in neuronal differentiation and survival. Cell Signal. 2013, 25, 2060–2068. [Google Scholar] [CrossRef]
- Karlsson, R.; Pedersen, E.D.; Wang, Z.; Brakebusch, C. Rho GTPase function in tumorigenesis. Biochim. Biophys. Acta 2009, 1796, 91–98. [Google Scholar] [CrossRef]
- Olson, M.F. Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases 2018, 9, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Sadok, A.; Marshall, C.J. Rho GTPases: Masters of cell migration. Small GTPases 2014, 5, e29710. [Google Scholar] [CrossRef]
- Subramani, D.; Alahari, S.K. Integrin-mediated function of Rab GTPases in cancer progression. Mol. Cancer 2010, 9, 312. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Hu, C.; Wu, F.; He, S. Rab25 GTPase: Functional roles in cancer. Oncotarget 2017, 8, 64591–64599. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M. Molecular mechanism of the nuclear protein import cycle. Nat. Rev. Mol. Cell Biol. 2007, 8, 195–208. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed]
- Dworak, N.; Makosa, D.; Chatterjee, M.; Jividen, K.; Yang, C.S.; Snow, C.; Simke, W.C.; Johnson, I.G.; Kelley, J.B.; Paschal, B.M. A nuclear lamina-chromatin-Ran GTPase axis modulates nuclear import and DNA damage signaling. Aging Cell 2019, 18, e12851. [Google Scholar] [CrossRef]
- Yudin, D.; Fainzilber, M. Ran on tracks-cytoplasmic roles for a nuclear regulator. J. Cell Sci. 2009, 122, 587–593. [Google Scholar] [CrossRef]
- Sheng, C.; Qiu, J.; Wang, Y.; He, Z.; Wang, H.; Wang, Q.; Huang, Y.; Zhu, L.; Shi, F.; Chen, Y.; et al. Knockdown of Ran GTPase expression inhibits the proliferation and migration of breast cancer cells. Mol. Med. Rep. 2018, 18, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Klosowiak, J.L.; Focia, P.J.; Chakravarthy, S.; Landahl, E.C.; Freymann, D.M.; Rice, S.E. Structural coupling of the EF hand and C-terminal GTPase domains in the mitochondrial protein Miro. EMBO Rep. 2013, 14, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Lu, B. The myriad roles of Miro in the nervous system: Axonal transport of mitochondria and beyond. Front. Cell. Neurosci. 2014, 8, 330. [Google Scholar] [CrossRef] [PubMed]
- Cromm, P.M.; Spiegel, J.; Grossmann, T.N.; Waldmann, H. Direct modulation of small GTPase activity and function. Angew. Chem. Int. Ed. 2015, 54, 13516–13537. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin. Drug Discov. 2015, 10, 991–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flisiak, S.; Zeeh, J.C.; Guibert, B.; Cherfils, J.; Zeghouf, M. An Arf1 GTPase mutant with different responses to GEF inhibitors. Biochem. Biophys. Res. Commun. 2008, 377, 156–160. [Google Scholar] [CrossRef]
- Xie, X.; Tang, S.C.; Cai, Y.; Pi, W.; Deng, L.; Wu, G.; Chavanieu, A.; Teng, Y. Suppression of breast cancer metastasis through the inactivation of ADP-ribosylation factor 1. Oncotarget 2016, 7, 58111–58120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, C.E. Paxillin interactions. J. Cell Sci. 2000, 113, 4139–4140. [Google Scholar] [PubMed]
- Bourgoin, S.G.; El Azreq, M.A. Small inhibitors of ADP-ribosylation factor activation and function in mammalian cells. World J. Pharmacol. 2012, 1, 55–64. [Google Scholar] [CrossRef]
- Spooner, R.A.; Watson, P.; Smith, D.C.; Boal, F.; Amessou, M.; Johannes, L.; Clarkson, G.J.; Lord, J.M.; Stephens, D.J.; Roberts, L.M. The secretion inhibitor Exo2 perturbs trafficking of Shiga toxin between endosomes and the trans-Golgi network. Biochem. J. 2008, 414, 471–484. [Google Scholar] [CrossRef]
- Sorieul, M.; Langhans, M.; Guetzoyan, L.; Hillmer, S.; Clarkson, G.; Lord, J.M.; Roberts, L.M.; Robinson, D.G.; Spooner, R.A.; Frigerio, L. An Exo2 derivative affects ER and Golgi morphology and vacuolar sorting in a tissue-specific manner in Arabidopsis. Traffic 2011, 12, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Pauloin, A.; Adenot, P.; Hue-Beauvais, C.; Chanat, E. The perilipin-2 (adipophilin) coat of cytosolic lipid droplets is regulated by an Arf1-dependent mechanism in HC11 mouse mammary epithelial cells. Cell Biol. Int. 2016, 40, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Lang, L.; Shay, C.; Zhao, X.; Teng, Y. Combined targeting of Arf1 and Ras potentiates anticancer activity for prostate cancer therapeutics. J. Exp. Clin. Cancer Res. 2017, 36, 112. [Google Scholar] [CrossRef]
- He, L.; Gao, L.; Shay, C.; Lang, L.; Lv, F.; Teng, Y. Histone deacetylase inhibitors suppress aggressiveness of head and neck squamous cell carcinoma via histone acetylation-independent blockade of the EGFR-Arf1 axis. J. Exp. Clin. Cancer Res. 2019, 38, 84. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Corpina, R.A.; Goldberg, J. Crystal structure of ARF1•Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 2003, 12, 1403–1411. [Google Scholar] [CrossRef]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Zeeh, J.C.; Zeghouf, M.; Grauffel, C.; Guibert, B.; Martin, E.; Dejaegere, A.; Cherfils, J. Dual specificity of the interfacial inhibitor Brefeldin A for Arf proteins and Sec7 domains. J. Biol. Chem. 2006, 281, 11805–11814. [Google Scholar] [CrossRef]
- Toda, T.; Watanabe, M.; Kawato, J.; Kadin, M.E.; Higashihara, M.; Kunisada, T.; Umezawa, K.; Horie, R. Brefeldin A exerts differential effects on anaplastic lymphoma kinase positive anaplastic large cell lymphoma and classical Hodgkin lymphoma cell lines. Br. J. Haematol. 2015, 170, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Anadu, N.O.; Davisson, V.J.; Cushman, M. Synthesis and anticancer activity of Brefeldin A ester derivatives. J. Med. Chem. 2006, 49, 3897–3905. [Google Scholar] [CrossRef]
- He, B.; Wang, Y.; Zheng, Y.; Chen, W.; Zhu, Q. Synthesis and cytotoxic evaluation of acylated Brefeldin A derivatives as potential anticancer agents. Chem. Biol. Drug Des. 2013, 82, 307–316. [Google Scholar] [CrossRef]
- Seehafer, K.; Rominger, F.; Helmchen, G.; Langhans, M.; Robinson, D.G.; Özata, B.; Brügger, B.; Strating, J.R.P.M.; Van Kuppeveld, F.J.M.; Klein, C.D. Synthesis and biological properties of novel Brefeldin A analogues. J. Med. Chem. 2013, 56, 5872–5884. [Google Scholar] [CrossRef] [PubMed]
- Shiina, I.; Umezaki, Y.; Ohashi, Y.; Yamazaki, Y.; Dan, S.; Yamori, T. Total synthesis of AMF-26, an antitumor agent for inhibition of the Golgi system, targeting ADP-ribosylation factor 1. J. Med. Chem. 2013, 56, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, Y.; Iijima, H.; Yamaotsu, N.; Yamazaki, K.; Sato, S.; Okamura, M.; Sugimoto, K.; Dan, S.; Hirono, S.; Yamori, T. AMF-26, a novel inhibitor of the Golgi system, targeting ADP-ribosylation factor 1 (Arf1) with potential for cancer therapy. J. Biol. Chem. 2012, 287, 3885–3897. [Google Scholar] [CrossRef]
- Ohashi, Y.; Okamura, M.; Hirosawa, A.; Tamaki, N.; Akatsuka, A.; Wu, K.M.; Choi, H.W.; Yoshimatsu, K.; Shiina, I.; Yamori, T.; et al. M-COPA, a Golgi disruptor, inhibits cell surface expression of MET protein and exhibits antitumor activity against MET-addicted gastric cancers. Cancer Res. 2016, 76, 3895–3903. [Google Scholar] [CrossRef] [PubMed]
- Watari, K.; Nakamura, M.; Fukunaga, Y.; Furuno, A.; Shibata, T.; Kawahara, A.; Hosoi, F.; Kuwano, T.; Kuwano, M.; Ono, M. The antitumor effect of a novel angiogenesis inhibitor (an octahydronaphthalene derivative) targeting both VEGF receptor and NF-κB pathway. Int. J. Cancer 2012, 131, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Hara, Y.; Obata, Y.; Horikawa, K.; Tasaki, Y.; Suzuki, K.; Murata, T.; Shiina, I.; Abe, R. M-COPA suppresses endolysosomal Kit-Akt oncogenic signalling through inhibiting the secretory pathway in neoplastic mast cells. PLoS ONE 2017, 12, e0175514. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Watanabe-Takahashi, M.; Shiina, I.; Ohashi, Y.; Dan, S.; Nishikawa, K.; Yamori, T.; Naito, M. M-COPA, a novel Golgi system disruptor, suppresses apoptosis induced by Shiga toxin. Genes Cells 2016, 21, 901–906. [Google Scholar] [CrossRef] [Green Version]
- Bi, X.; Schmitz, A.; Hayallah, A.M.; Song, J.N.; Famulok, M. Affinity-based labeling of cytohesins with a bifunctional SecinH3 photoaffinity probe. Angew. Chem. Int. Ed. 2008, 47, 9565–9568. [Google Scholar] [CrossRef]
- Hafner, M.; Schmitz, A.; Grüne, I.; Srivatsan, S.G.; Paul, B.; Kolanus, W.; Quast, T.; Kremmer, E.; Bauer, I.; Famulok, M. Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature 2006, 444, 941–944. [Google Scholar] [CrossRef]
- Jayaram, B.; Syed, I.; Kyathanahalli, C.N.; Rhodes, C.J.; Kowluru, A. Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 β-cells and rat islets. Biochem. Pharmacol. 2011, 81, 1016–1027. [Google Scholar] [CrossRef] [Green Version]
- Davidson, A.C.; Humphreys, D.; Brooks, A.B.; Hume, P.J.; Koronakis, V. The Arf GTPase-activating protein family is exploited by Salmonella enterica serovar Typhimurium to invade nonphagocytic host cells. MBio 2015, 6, e02253-14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ahirwar, D.K.; Oghumu, S.; Wilkie, T.; Powell, C.A.; Nasser, M.W.; Satoskar, A.R.; Li, D.Y.; Ganju, R.K. Endothelial Robo4 suppresses breast cancer growth and metastasis through regulation of tumor angiogenesis. Mol. Oncol. 2016, 10, 272–281. [Google Scholar] [CrossRef]
- Bill, A.; Schmitz, A.; König, K.; Heukamp, L.C.; Hannam, J.S.; Famulok, M. Anti-proliferative effect of cytohesin inhibition in gefitinib-resistant lung cancer cells. PLoS ONE 2012, 7, e41179. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Sun, J.; Hu, J.; Hu, Y.; Zhou, J.; Chen, Z.; Xu, D.; Xu, W.; Zheng, S.; Zhang, S. Cytohesins/ARNO: The function in colorectal cancer cells. PLoS ONE 2014, 9, e90997. [Google Scholar] [CrossRef] [PubMed]
- Germer, K.; Leonard, M.; Zhang, X. RNA aptamers and their therapeutic and diagnostic applications. Int. J. Biochem. Mol. Biol. 2013, 4, 27–40. [Google Scholar]
- Mayer, G.; Blind, M.; Nagel, W.; Böhm, T.; Knorr, T.; Jackson, C.L.; Kolanus, W.; Famulok, M. Controlling small guanine-nucleotide-exchange factor function through cytoplasmic RNA intramers. Proc. Natl. Acad. Sci. USA 2001, 98, 4961–4965. [Google Scholar] [CrossRef] [Green Version]
- Van Hattum, H.; Waldmann, H. Chemical biology tools for regulating RAS signaling complexity in space and time. Chem. Biol. 2014, 21, 1185–1195. [Google Scholar] [CrossRef]
- Wang, W.; Fang, G.; Rudolph, J. Ras inhibition via direct Ras binding-is there a path forward? Bioorg. Med. Chem. Lett. 2012, 22, 5766–5776. [Google Scholar] [CrossRef]
- Spencer-Smith, R.; Li, L.; Prasad, S.; Koide, A.; Koide, S.; O’Bryan, J.P. Targeting the α4-α5 interface of RAS results in multiple levels of inhibition. Small GTPases 2018, in press. [Google Scholar] [CrossRef]
- Patgiri, A.; Yadav, K.K.; Arora, P.S.; Bar-Sagi, D. An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol. 2011, 7, 585–587. [Google Scholar] [CrossRef]
- Lu, S.; Jang, H.; Zhang, J.; Nussinov, R. Inhibitors of Ras-SOS interactions. ChemMedChem 2016, 11, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Schöpel, M.; Jockers, K.F.; Düppe, P.M.; Autzen, J.; Potheraveedu, V.N.; Ince, S.; Yip, K.T.; Heumann, R.; Herrmann, C.; Scherkenbeck, J.; et al. Bisphenol A binds to Ras proteins and competes with guanine nucleotide exchange: Implications for GTPase-selective antagonists. J. Med. Chem. 2013, 56, 9664–9672. [Google Scholar] [CrossRef]
- Peri, F.; Airoldi, C.; Colombo, S.; Martegani, E.; van Neuren, A.S.; Stein, M.; Marinzi, C.; Nicotra, F. Design, synthesis and biological evaluation of sugar-derived Ras inhibitors. ChemBioChem 2005, 6, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Leshchiner, E.S.; Parkhitko, A.; Bird, G.H.; Luccarelli, J.; Bellairs, J.A.; Escudero, S.; Opoku-Nsiah, K.; Godes, M.; Perrimon, N.; Walensky, L.D. Direct inhibition of oncogenic KRAS by hydrocarbon-stapled SOS1 helices. Proc. Natl. Acad. Sci. USA 2015, 112, 1761–1766. [Google Scholar] [CrossRef] [PubMed]
- Taveras, A.G.; Remiszewski, S.W.; Doll, R.J.; Cesarz, D.; Huang, E.C.; Kirschmeier, P.; Pramanik, B.N.; Snow, M.E.; Wang, Y.S.; del Rosario, J.D.; et al. Ras oncoprotein inhibitors: The discovery of potent, Ras nucleotide exchange inhibitors and the structural determination of a drug-protein complex. Bioorg. Med. Chem. 1997, 5, 125–133. [Google Scholar] [CrossRef]
- Ganguly, A.K.; Wang, Y.S.; Pramanik, B.N.; Doll, R.J.; Snow, M.E.; Taveras, A.G.; Remiszewski, S.; Cesarz, D.; del Rosario, J.; Vibulbhan, B.; et al. Interaction of a novel GDP exchange inhibitor with the Ras protein. Biochemistry 1998, 37, 15631–15637. [Google Scholar] [CrossRef]
- Palmioli, A.; Sacco, E.; Abraham, S.; Thomas, C.J.; Di Domizio, A.; De Gioia, L.; Gaponenko, V.; Vanoni, M.; Peri, F. First experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand. Bioorg. Med. Chem. Lett. 2009, 19, 4217–4222. [Google Scholar] [CrossRef] [PubMed]
- Kalbitzer, H.R.; Spoerner, M. State 1(T) inhibitors of activated Ras. In Inhibitors of the Ras Superfamily G-Proteins, Part A; Tamanoi, F., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 33, pp. 69–94. ISBN 9780124167490. [Google Scholar]
- Spoerner, M.; Graf, T.; König, B.; Kalbitzer, H.R. A novel mechanism for the modulation of the Ras-effector interaction by small molecules. Biochem. Biophys. Res. Commun. 2005, 334, 709–713. [Google Scholar] [CrossRef] [PubMed]
- Rosnizeck, I.C.; Graf, T.; Spoerner, M.; Tränkle, J.; Filchtinski, D.; Herrmann, C.; Gremer, L.; Vetter, I.R.; Wittinghofer, A.; König, B.; et al. Stabilizing a weak binding state for effectors in the human Ras protein by cyclen complexes. Angew. Chem. Int. Ed. 2010, 49, 3830–3833. [Google Scholar] [CrossRef]
- Liu, L.J.; Wang, W.; Huang, S.Y.; Hong, Y.; Li, G.; Lin, S.; Tian, J.; Cai, Z.; Wang, H.M.D.; Ma, D.L.; et al. Inhibition of the Ras/Raf interaction and repression of renal cancer xenografts in vivo by an enantiomeric iridium(III) metal-based compound. Chem. Sci. 2017, 8, 4756–4763. [Google Scholar] [CrossRef]
- Kauke, M.J.; Traxlmayr, M.W.; Parker, J.A.; Kiefer, J.D.; Knihtila, R.; McGee, J.; Verdine, G.; Mattos, C.; Wittrup, K.D. An engineered protein antagonist of K-Ras/B-Raf interaction. Sci. Rep. 2017, 7, 5831. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.; Block, C.; Geisen, C.; Haas, K.; Weber, C.; Winde, G.; Möröy, T.; Müller, O. Sulindac sulfide inhibits Ras signaling. Oncogene 1998, 17, 1769–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilad, L.A.; Bresler, T.; Gnainsky, J.; Smirnoff, P.; Schwartz, B. Regulation of vitamin D receptor expression via estrogen-induced activation of the ERK 1/2 signaling pathway in colon and breast cancer cells. J. Endocrinol. 2005, 185, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, M.R.; Chang, H.C.; Hung, W.C. Non-steroidal anti-inflammatory drugs suppress the ERK signaling pathway via block of Ras/c-Raf interaction and activation of MAP kinase phosphatases. Cell. Signal. 2008, 20, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Khazak, V.; Eyrisch, S.; Kato, J.; Tamanoi, F.; Golemis, E.A. A two-hybrid approach to identify inhibitors of the RAS-RAF interaction. In Inhibitors of the Ras Superfamily G-Proteins, Part A; Tamanoi, F., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; Volume 33, pp. 213–248. ISBN 9780124167490. [Google Scholar]
- Skobeleva, N.; Menon, S.; Weber, L.; Golemis, E.A.; Khazak, V. In vitro and in vivo synergy of MCP compounds with mitogen-activated protein kinase pathway- and microtubule-targeting inhibitors. Mol. Cancer Ther. 2007, 6, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Oinuma, I.; Katoh, H.; Negishi, M. Semaphorin 4D/Plexin-B1–mediated R-Ras GAP activity inhibits cell migration by regulating β1 integrin activity. J. Cell Biol. 2006, 173, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, M.; Yamashita, T. Inactivation of Ras by p120GAP via focal adhesion kinase dephosphorylation mediates RGMa-induced growth cone collapse. J. Neurosci. 2009, 29, 6649–6662. [Google Scholar] [CrossRef]
- Oinuma, I.; Ishikawa, Y.; Katoh, H.; Negishi, M. The semaphorin 4D receptor plexin-B1 is a GTPase activating protein for R-Ras. Science 2004, 305, 862–865. [Google Scholar] [CrossRef]
- Quah, S.Y.; Tan, M.S.; Teh, Y.H.; Stanslas, J. Pharmacological modulation of oncogenic Ras by natural products and their derivatives: Renewed hope in the discovery of novel anti-Ras drugs. Pharmacol. Ther. 2016, 162, 35–57. [Google Scholar] [CrossRef]
- Laheru, D.; Shah, P.; Rajeshkumar, N.V.; McAllister, F.; Taylor, G.; Goldsweig, H.; Le, D.T.; Donehower, R.; Jimeno, A.; Linden, S.; et al. Integrated preclinical and clinical development of S-trans, trans-farnesylthiosalicylic acid (FTS, Salirasib) in pancreatic cancer. Investig. New Drugs 2012, 30, 2391–2399. [Google Scholar] [CrossRef] [Green Version]
- Haklai, R.; Elad-Sfadia, G.; Egozi, Y.; Kloog, Y. Orally administered FTS (salirasib) inhibits human pancreatic tumor growth in nude mice. Cancer Chemother. Pharmacol. 2008, 61, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Zundelevich, A.; Elad-Sfadia, G.; Haklai, R.; Kloog, Y. Suppression of lung cancer tumor growth in a nude mouse model by the Ras inhibitor salirasib (farnesylthiosalicylic acid). Mol. Cancer Ther. 2007, 6, 1765–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charette, N.; De Saeger, C.; Lannoy, V.; Horsmans, Y.; Leclercq, I.; Stärkel, P. Salirasib inhibits the growth of hepatocarcinoma cell lines in vitro and tumor growth in vivo through Ras and mTOR inhibition. Mol. Cancer 2010, 9, 256. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.A.; Conaway, M.C.; Gregory, C.W.; Yue, W.; Santen, R.J. The novel Ras antagonist, farnesylthiosalicylate, suppresses growth of prostate cancer in vitro. Prostate 2004, 58, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Riely, G.J.; Johnson, M.L.; Medina, C.; Rizvi, N.A.; Miller, V.A.; Kris, M.G.; Pietanza, M.C.; Azzoli, C.G.; Krug, L.M.; Pao, W.; et al. A phase II trial of Salirasib in patients with lung adenocarcinomas with KRAS mutations. J. Thorac. Oncol. 2011, 6, 1435–1437. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Yu, H.; Hughes, N.W.; Liu, B.; Kendirli, A.; Klein, K.; Chen, W.W.; Lander, E.S.; Sabatini, D.M. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 2017, 168, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Heid, I.; Lubeseder-Martellato, C.; Sipos, B.; Mazur, P.K.; Lesina, M.; Schmid, R.M.; Siveke, J.T. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 2011, 141, 719–730. [Google Scholar] [CrossRef]
- Kumar, M.S.; Hancock, D.C.; Molina-Arcas, M.; Steckel, M.; East, P.; Diefenbacher, M.; Armenteros-Monterroso, E.; Lassailly, F.; Matthews, N.; Nye, E.; et al. The GATA2 transcriptional network is requisite for RAS oncogene-driven non-small cell lung cancer. Cell 2012, 149, 642–655. [Google Scholar] [CrossRef]
- Kawada, K.; Toda, K.; Sakai, Y. Targeting metabolic reprogramming in KRAS-driven cancers. Int. J. Clin. Oncol. 2017, 22, 651–659. [Google Scholar] [CrossRef]
- Chang, H.H.; Moro, A.; Takakura, K.; Su, H.Y.; Mo, A.; Nakanishi, M.; Waldron, R.T.; French, S.W.; Dawson, D.W.; Hines, J.; et al. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS ONE 2017, 12, e0184455. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mack, N.A.; Whalley, H.J.; Castillo-Lluva, S.; Malliri, A. The diverse roles of Rac signaling in tumorigenesis. Cell Cycle 2011, 10, 1571–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef] [Green Version]
- Akbar, H.; Cancelas, J.; Williams, D.A.; Zheng, J.; Zheng, Y. Rational design and applications of a Rac GTPase-specific small molecule inhibitor. In Regulators and Effectors of Small GTPases: Rho Family; Balch, W.E., Der, C.J., Hall, A., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2006; Volume 406, pp. 554–565. [Google Scholar]
- Kaneto, N.; Yokoyama, S.; Hayakawa, Y.; Kato, S.; Sakurai, H.; Saiki, I. RAC1 inhibition as a therapeutic target for gefitinib-resistant non-small-cell lung cancer. Cancer Sci. 2014, 105, 788–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Hein, A.L.; Etekpo, A.; Burchett, K.M.; Lin, C.; Enke, C.A.; Batra, S.K.; Cowan, K.H.; Ouellette, M.M. Inhibition of RAC1 GTPase sensitizes pancreatic cancer cells to γ-irradiation. Oncotarget 2014, 5, 10251–10270. [Google Scholar] [CrossRef] [Green Version]
- Hernández, E.; de la Mota-Peynado, A.; Dharmawardhane, S.; Vlaar, C.P. Novel inhibitors of Rac1 in metastatic breast cancer. P. R. Health Sci. J. 2010, 29, 348–356. [Google Scholar]
- Montalvo-Ortiz, B.L.; Castillo-Pichardo, L.; Hernández, E.; Humphries-Bickley, T.; de la Mota-Peynado, A.; Cubano, L.A.; Vlaar, C.P.; Dharmawardhane, S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012, 287, 13228–13238. [Google Scholar] [CrossRef]
- Castillo-Pichardo, L.; Humphries-Bickley, T.; de la Parra, C.; Forestier-Roman, I.; Martinez-Ferrer, M.; Hernandez, E.; Vlaar, C.; Ferrer-Acosta, Y.; Washington, A.V.; Cubano, L.A.; et al. The Rac inhibitor EHop-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl. Oncol. 2014, 7, 546–555. [Google Scholar] [CrossRef]
- Martin, H.; Mali, R.S.; Ma, P.; Chatterjee, A.; Ramdas, B.; Sims, E.; Munugalavadla, V.; Ghosh, J.; Mattingly, R.R.; Visconte, V.; et al. Pak and Rac GTPases promote oncogenic KIT-induced neoplasms. J. Clin. Investig. 2013, 123, 4449–4463. [Google Scholar] [CrossRef]
- Okada, T.; Lee, A.Y.; Qin, L.X.; Agaram, N.; Mimae, T.; Shen, Y.; O’Connor, R.; López-Lago, M.A.; Craig, A.; Miller, M.L.; et al. Integrin-α10 dependency identifies RAC and RICTOR as therapeutic targets in high-grade myxofibrosarcoma. Cancer Discov. 2016, 6, 1148–1165. [Google Scholar] [CrossRef] [Green Version]
- Veluthakal, R.; Tunduguru, R.; Arora, D.K.; Sidarala, V.; Syeda, K.; Vlaar, C.P.; Turmond, D.C.; Kowluru, A. VAV2, a guanine nucleotide exchange factor for Rac1, regulates glucose-stimulated insulin secretion in pancreatic beta cells. Diabetologia 2015, 58, 2573–2581. [Google Scholar] [CrossRef] [Green Version]
- Shutes, A.; Onesto, C.; Picard, V.; Leblond, B.; Schweighoffer, F.; Der, C.J. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J. Biol. Chem. 2007, 282, 35666–35678. [Google Scholar] [CrossRef]
- Onesto, C.; Shutes, A.; Picard, V.; Schweighoffer, F.; Der, C.J. Characterization of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. In Small GTPases in Disease, Part B; Balch, W.E., Der, C.J., Hall, A., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2008; Volume 439, pp. 111–129. [Google Scholar]
- Rosenblatt, A.E.; Garcia, M.I.; Lyons, L.; Xie, Y.; Maiorino, C.; Désiré, L.; Slingerland, J.; Burnstein, K.L. Inhibition of the Rho GTPase, Rac1, decreases estrogen receptor levels and is a novel therapeutic strategy in breast cancer. Endocr. Relat. Cancer 2011, 18, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Katz, E.; Sims, A.H.; Sproul, D.; Caldwell, H.; Dixon, J.M.; Meehan, R.R.; Harrison, D.J. Targeting of Rac GTPases blocks the spread of intact human breast cancer. Oncotarget 2012, 3, 608–619. [Google Scholar] [CrossRef] [Green Version]
- Jim Leu, S.J.; Sung, J.S.; Huang, M.L.; Chen, M.Y.; Tsai, T.W. A novel anti-CCN1 monoclonal antibody suppresses Rac-dependent cytoskeletal reorganization and migratory activities in breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 885–891. [Google Scholar] [CrossRef]
- Niebel, B.; Wosnitza, C.I.; Famulok, M. RNA-aptamers that modulate the RhoGEF activity of Tiam1. Bioorg. Med. Chem. 2013, 21, 6239–6246. [Google Scholar] [CrossRef]
- Taniuchi, K.; Yokotani, K.; Saibara, T. BART inhibits pancreatic cancer cell invasion by Rac1 inactivation through direct binding to active Rac1. Neoplasia 2012, 14, 440–450. [Google Scholar] [CrossRef]
- Shan, D.; Chen, L.; Njardarson, J.T.; Gaul, C.; Ma, X.; Danishefsky, S.J.; Huang, X.Y. Synthetic analogues of migrastatin that inhibit mammary tumor metastasis in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 3772–3776. [Google Scholar] [CrossRef] [Green Version]
- Dütting, S.; Heidenreich, J.; Cherpokova, D.; Amin, E.; Zhang, S.C.; Ahmadian, M.R.; Brakebusch, C.; Nieswandt, B. Critical off-target effects of the widely used Rac1 inhibitors NSC23766 and EHT1864 in mouse platelets. J. Thromb. Haemost. 2015, 13, 827–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Bai, L.; Nan, Q.Z. Activation of Rho GTPase Cdc42 promotes adhesion and invasion in colorectal cancer cells. Med. Sci. Monit. Basic Res. 2013, 19, 201–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias-Romero, L.E.; Chernoff, J. Targeting Cdc42 in cancer. Expert Opin. Ther. Targets 2013, 17, 1263–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stengel, K.R.; Zheng, Y. Essential role of Cdc42 in Ras-induced transformation revealed by gene targeting. PLoS ONE 2012, 7, e37317. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.J.; Tu, S.; Cerione, R.A. Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell 2003, 114, 715–725. [Google Scholar] [CrossRef]
- Qadir, M.I.; Parveen, A.; Ali, M. Cdc42: Role in cancer management. Chem. Biol. Drug Des. 2015, 86, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Pelish, H.E.; Peterson, J.R.; Salvarezza, S.B.; Rodriguez-Boulan, E.; Chen, J.L.; Stamnes, M.; Macia, E.; Feng, Y.; Shair, M.D.; Kirchhausen, T. Secramine inhibits Cdc42-dependent functions in cells and Cdc42 activation in vitro. Nat. Chem. Biol. 2006, 2, 39–46. [Google Scholar] [CrossRef]
- Xu, B.; Pelish, H.; Kirchhausen, T.; Hammond, G.B. Large scale synthesis of the Cdc42 inhibitor Secramine A and its inhibition of cell spreading. Org. Biomol. Chem. 2006, 4, 4149–4157. [Google Scholar] [CrossRef] [PubMed]
- Ambrogio, C.; Voena, C.; Manazza, A.D.; Martinengo, C.; Costa, C.; Kirchhausen, T.; Hirsch, E.; Inghirami, G.; Chiarle, R. The anaplastic lymphoma kinase controls cell shape and growth of anaplastic large cell lymphoma through Cdc42 activation. Cancer Res. 2008, 68, 8899–8907. [Google Scholar] [CrossRef]
- Baltiérrez-Hoyos, R.; Roa-Espitia, A.L.; Hernández-González, E.O. The association between CDC42 and caveolin-1 is involved in the regulation of capacitation and acrosome reaction of guinea pig and mouse sperm. Reproduction 2012, 144, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, D.S.; Lehmann, M.; Felts, R.; Garcia, E.; Blanchet, F.P.; Subramaniam, S.; Piguet, V. HIV-1 activates Cdc42 and induces membrane extensions in immature dendritic cells to facilitate cell-to-cell virus propagation. Blood 2011, 118, 4841–4852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengfeld, J.; Wang, Q.; Zohlman, A.; Salvarezza, S.; Morgan, S.; Ren, J.; Kato, K.; Rodriguez-Boulan, E.; Liu, B. Protein kinase C δ regulates the release of collagen type I from vascular smooth muscle cells via regulation of Cdc42. Mol. Biol. Cell 2012, 23, 1955–1963. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Kenney, S.R.; Phillips, G.K.; Simpson, D.; Schroeder, C.E.; Nöth, J.; Romero, E.; Swanson, S.; Waller, A.; Strouse, J.J.; et al. Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Biol. Chem. 2013, 288, 8531–8543. [Google Scholar] [CrossRef] [PubMed]
- Friesland, A.; Zhao, Y.; Chen, Y.H.; Wang, L.; Zhou, H.; Lu, Q. Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. USA 2013, 110, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Muhoza, D.; Adams, P.D. Two small molecules, ZCL278 and AZA197 show promise in influencing protein interactions involving the Ras-related protein cell division cycle 42 [Cdc42] to modulate its oncogenic potential. Open J. Biophys. 2017, 7, 71–81. [Google Scholar] [CrossRef]
- An, Y.; Liu, T.; Liu, X.; Zhao, L.; Wang, J. Rac1 and Cdc42 play important roles in arsenic neurotoxicity in primary cultured rat cerebellar astrocytes. Biol. Trace Elem. Res. 2016, 170, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Gao, Y.; An, Y.; Fu, X.; Li, Y.; Sun, D.; Wang, J. Neuroglobin plays a protective role in arsenite-induced cytotoxicity by inhibition of Cdc42 and Rac1GTPases in rat cerebellar granule neurons. Cell. Physiol. Biochem. 2015, 36, 1613–1627. [Google Scholar] [CrossRef]
- Biro, M.; Munoz, M.A.; Weninger, W. Targeting Rho-GTPases in immune cell migration and inflammation. Br. J. Pharmacol. 2014, 171, 5491–5506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphries-Bickley, T.; Castillo-Pichardo, L.; Hernandez-O’Farrill, E.; Borrero-Garcia, L.D.; Forestier-Roman, I.; Gerena, Y.; Blanco, M.; Rivera-Robles, M.J.; Rodriguez-Medina, J.R.; Cubano, L.A.; et al. Characterization of a dual Rac/Cdc42 inhibitor MBQ-167 in metastatic cancer. Mol. Cancer Ther. 2017, 16, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Zins, K.; Lucas, T.; Reichl, P.; Abraham, D.; Aharinejad, S. A Rac1/Cdc42 GTPase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS ONE 2013, 8, e74924. [Google Scholar] [CrossRef]
- Guo, Y.; Kenney, S.R.; Muller, C.Y.; Adams, S.; Rutledge, T.; Romero, E.; Murray-Krezan, C.; Prekeris, R.; Sklar, L.A.; Hudson, L.G.; et al. R-Ketorolac targets Cdc42 and Rac1 and alters ovarian cancer cell behaviors critical for invasion and metastasis. Mol. Cancer Ther. 2015, 14, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Mao, K.; Liu, Z.; Dinh-Xuan, A.T. The role of the RhoA/Rho kinase pathway in angiogenesis and its potential value in prostate cancer (Review). Oncol. Lett. 2014, 8, 1907–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, X.; Marchioni, F.; Sipes, N.; Evelyn, C.R.; Jerabek-Willemsen, M.; Duhr, S.; Seibel, W.; Wortman, M.; Zheng, Y. Rational design of small molecule inhibitors targeting RhoA subfamily Rho GTPases. Chem. Biol. 2012, 19, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Cho, S.J.; Aksoy, B.A.; Park, D.J.; Schultz, N.; Ryeom, S.W.; Yoon, S.S. Chemotherapy resistance in diffuse type gastric adenocarcinoma is mediated by RhoA activation in cancer stem-like cells. Clin. Cancer Res. 2016, 22, 971–983. [Google Scholar] [CrossRef]
- Shang, X.; Marchioni, F.; Evelyn, C.R.; Sipes, N.; Zhou, X.; Seibel, W.; Wortman, M.; Zheng, Y. Small-molecule inhibitors targeting G-protein-coupled Rho guanine nucleotide exchange factors. Proc. Natl. Acad. Sci. USA 2013, 110, 3155–3160. [Google Scholar] [CrossRef]
- Chang, L.C.; Huang, T.H.; Chang, C.S.; Tsai, Y.R.; Lin, R.H.; Lee, P.W.; Hsu, M.F.; Huang, L.J.; Wang, J.P. Signaling mechanisms of inhibition of phospholipase D activation by CHS-111 in formyl peptide-stimulated neutrophils. Biochem. Pharmacol. 2011, 81, 269–278. [Google Scholar] [CrossRef]
- He, H.; Dai, F.; Yu, L.; She, X.; Zhao, Y.; Jiang, J.; Chen, X.; Zhao, S. Identification and characterization of nine novel human small GTPases showing variable expressions in liver cancer tissues. Gene Expr. 2002, 10, 231–242. [Google Scholar] [CrossRef]
- Shimada, K.; Uzawa, K.; Kato, M.; Endo, Y.; Shiiba, M.; Bukawa, H.; Yokoe, H.; Seki, N.; Tanzawa, H. Aberrant expression of RAB1A in human tongue cancer. Br. J. Cancer 2005, 92, 1915–1921. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Liu, X.F.; Wu, H.C.; Zou, S.B.; Wang, J.Y.; Ni, P.H.; Chen, X.H.; Fan, Q.S. Rab5a overexpression promoting ovarian cancer cell proliferation may be associated with APPL1-related epidermal growth factor signaling pathway. Cancer Sci. 2010, 101, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jia, Q.; Wang, Y.; Li, F.; Jia, Z.; Wan, Y. Rab40b upregulation correlates with the prognosis of gastric cancer by promoting migration, invasion, and metastasis. Med. Oncol. 2015, 32, 126. [Google Scholar] [CrossRef]
- Hou, R.; Jiang, L.; Yang, Z.; Wang, S.; Liu, Q. Rab14 is overexpressed in ovarian cancers and promotes ovarian cancer proliferation through Wnt pathway. Tumor Biol. 2016, 37, 16005–16013. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, S.D.; Marchi, F.A.; Xu, B.; Bijian, K.; Alobaid, F.; Mlynarek, A.; Rogatto, S.R.; Hier, M.; Kowalski, L.P.; Alaoui-Jamali, M.A. Predominant Rab-GTPase amplicons contributing to oral squamous cell carcinoma progression to metastasis. Oncotarget 2015, 6, 21950–21963. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, A.; Maynard, D.; Pauwels, P.; Braems, G.; Denys, H.; van den Broecke, R.; Lambert, J.; van Belle, S.; Cocquyt, V.; Gespach, C.; et al. Effect of the secretory small GTPase Rab27B on breast cancer growth, invasion, and metastasis. J. Natl. Cancer Inst. 2010, 102, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Huo, Y.; Zheng, Z.; Jiang, X.; Deng, H.; Chen, Y.; Lian, Q.; Ge, R.; Deng, H. Down-regulation of Ras-related protein Rab 5C-dependent endocytosis and glycolysis in cisplatin-resistant ovarian cancer cell lines. Mol. Cell. Proteom. 2014, 13, 3138–3151. [Google Scholar] [CrossRef] [PubMed]
- Recchi, C.; Seabra, M.C. Novel functions for Rab GTPases in multiple aspects of tumour progression. Biochem. Soc. Trans. 2012, 40, 1398–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelofs, A.J.; Hulley, P.A.; Meijer, A.; Ebetino, F.H.; Russell, R.G.; Shipman, C.M. Selective inhibition of Rab prenylation by a phosphonocarboxylate analogue of risedronate induces apoptosis, but not S-phase arrest, in human myeloma cells. Int. J. Cancer 2006, 119, 1254–1261. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, S.; Jiang, Y.; Kawamura, K.; Shingyoji, M.; Tada, Y.; Sekine, I.; Takiguchi, Y.; Tatsumi, K.; Kobayashi, H.; Shimada, H.; et al. Zoledronic acid induces apoptosis and S-phase arrest in mesothelioma through inhibiting Rab family proteins and topoisomerase II actions. Cell Death Dis. 2014, 5, e1517. [Google Scholar] [CrossRef]
- Boissier, S.; Magnetto, S.; Frappart, L.; Cuzin, B.; Ebetino, F.H.; Delmas, P.D.; Clezardin, P. Bisphosphonates inhibit prostate and breast carcinoma cell adhesion to unmineralized and mineralized bone extracellular matrices. Cancer Res. 1997, 57, 3890–3894. [Google Scholar]
- Boissier, S.; Ferreras, M.; Peyruchaud, O.; Magnetto, S.; Ebetino, F.H.; Colombel, M.; Delmas, P.; Delaissé, J.M.; Clézardin, P. Bisphosphonates inhibit breast and prostate carcinoma cell invasion, an early event in the formation of bone metastases. Cancer Res. 2000, 60, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.G.J.; Daubiné, F.; Lundy, M.W.; Rogers, M.J.; Ebetino, F.H.; Clézardin, P. Lowering bone mineral affinity of bisphosphonates as a therapeutic strategy to optimize skeletal tumor growth inhibition In vivo. Cancer Res. 2008, 68, 8945–8953. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Name of the Inhibitor | Mechanism of Action | Model | Global Outcomes | Reference |
|---|---|---|---|---|
| LM11 | Inhibition of ArfGEF binding to Arf1 | Breast cancer cell lines cultured in vitro and breast cancer xenografts in zebrafish | Inhibition of cell proliferation, invasion and metastasis | [38] |
| Breast adenocarcinoma cells cultured in vitro | Reduction of cell migration in a dose-dependent manner, cell adhesion to matrix and cell proliferation | [13] | ||
| Exo2 | Inhibition of ArfGEF activity | Prostate cancer cells cultured in vitro | Suppression of cell proliferation, invasion and migration and induction of programmed cell death through apoptosis | [44] |
| BFA | Hindering of Arf1 and GEF joining | Anaplastic large cell carcinoma in vitro | Reduction of cell proliferation | [49] |
| Lung, colon, melanoma, ovarian, renal, prostate, breast and central nervous system tumors in vitro | Increment of cell death and reduction of their proliferation | [50] | ||
| Acetylated BFA derivatives | Hindering of Arf1 and GEF joining | Esophagus squamous cell carcinoma in vitro | Increment of cell death in a sharper way than BFA | [51] |
| Ester derivatives of BFA | Hindering of Arf1 and GEF joining | Lung, colon, melanoma, ovarian, renal, prostate, breast and central nervous system tumors in vitro | Increment of cell death and reduction of their proliferation in a sharper way than BFA | [50] |
| C15 BFA derivatives | Hindering of Arf1 and GEF joining | Lung, colon, ovarian, renal, prostate, breast, leukemia, melanoma and central nervous system tumors in vitro | Increase of cell death, which is stronger than BFA | [52] |
| AMF-26/M-COPA | Impairment of ArfGEF activity | Breast cancer xenografts in vivo | Induction of complete reversion in the growth of these xenografts | [54] |
| Melanoma cells both in vitro and in vivo models | Inhibition of angiogenesis, proliferation and tumor growth through the suppression of VEGFR1/2. | [56] | ||
| Neoplastic mast cells cultured in vitro | Suppression of cell proliferation and resistance to imatinib through the abolishment of Kit signaling | [57] | ||
| SecinH3 | Inhibition of ArfGEF binding to Arf1 | Breast xenografts in vivo | Reduction of tumor growth, aggressiveness and metastasis | [63] |
| Non-small cell lung cancer cell lines in vitro | Inhibition of cell proliferation and reduction of cell resistance to gefitinib | [64] | ||
| Colorectal cancer models both in vivo and in vitro | Decrease cell proliferation, migration and proliferation through the abolishment of ARNO-dependent signaling | [65] | ||
| M69 | Block of ArfGEF activity | Acute T cell leukemia cells cultured in vitro | Disturbance of intracellular adhesion through restructuration of actin skeleton | [67] |
| Name of the Inhibitor | Mechanism of Action | Model | Global Outcomes | Reference |
|---|---|---|---|---|
| Bisphenol A | Disruption of the binding between Ras and SOS. | Cervical cancer cells cultured in vitro | Decrease in cell proliferation | [73] |
| SCH-53870 derivates | Disruption of the binding between Ras and SOS. | NIH3T3 mouse fibroblast in vitro | Decrease in cell proliferation both in normal and KRas-overexpressing cells | [74] |
| SAH-SOS1 | Disruption of the binding between Ras and SOS. | Pancreatic, lung and colon cancer cells cultured in vitro bearing different KRAS mutants | Decrease in cell proliferation in a dose-dependent manner, independently of the KRAS mutant which bears the cells. | [75] |
| SCH-54292 | Hindering of the binding between Ras and SOS | NIH3T3 mouse fibroblast in vitro | Inhibition of cell proliferation | [78] |
| MCP110 | Inhibition of Raf and Ras-binding | Colon cancer models both in vivo and in vitro | Impediment of cell proliferation both in vitro and in vivo and synergy with other chemotherapeutic drugs, such as paclitaxel or vincristine | [88] |
| Colon cancer cells cultured in vitro | Arrest of cell cycle in G1 phase through the abolishment of cyclin D1 levels | [87] | ||
| MCP1 | Inhibition of Raf and Ras binding | Multiple myeloma cells cultured in vitro | Reduction of cancer cell growth through the induction of intrinsic apoptosis | [87] |
| MCP1 and MCP110 | Inhibition of Raf and Ras binding | Multiple cancer cell lines defined by the National Cancer Institute (NCI) (Weinstein et al., 1997) | Reduction of cell proliferation | [87] |
| Enantiomeric iridium(III) metal-based compound | Inhibition of Ras and Raf interaction | Human kidney xenografts in vivo and kidney, breast, lung, prostatic, ovarian, melanoma and erythroleukemic cancer cell lines in vitro | Inhibition of cell cancer proliferation and reduction of tumor volume without affecting mice global weight | [83] |
| Sulindac sulfide | Hindering of Raf activation by Ras | NIH3T3 mouse fibroblast in vitro and Saos epithelial cells | Abolishment of Ras-dependent malignant transformation | [84] |
| Brest cancer cells in vitro | Inhibition of E2-derivated pro-proliferative outcomes | [85] | ||
| Sema4D | Stimulation of Ras-GAP activity | Adrenal gland phaeochromocytoma cells cultured in vitro | Reduction of cell migration through inhibition of β1 integrin activation | [91] |
| Salirasib | Inhibition of Ras anchorage to cytoplasmic membranes | Pancreatic cell xenografts in vivo | Inhibition of tumor growth dose-dependently and stimulation of gemcitabine antiproliferative effects | [94] |
| Lung cancer models both in vivo and in vitro | Inhibition of cell proliferation and tumor growth | [95] | ||
| Hepatocellular carcinoma models both in vivo and in vitro | Inhibition of cell proliferation through the arrest of cell cycle and the induction of apoptosis | [96] | ||
| Pancreatic cancer cells cultured in vitro | Reduction of cell proliferation through the arrest of cell cycle | [97] | ||
| Prostate cancer cells cultured in vitro | Enhancement of Exo2 effects on cell proliferation, migration and invasion. | [44] | ||
| Lung cancer patients | Common used doses and schedule failed in the inhibition of cell proliferation | [98] |
| Name of the Inhibitor | Mechanism of Action | Model | Global Outcomes | Reference |
|---|---|---|---|---|
| NSC23766 | Inhibition of RacGEF binding to Rac | Prostate cancer cells cultured in vitro | Reduction of cell proliferation and their invasive characteristics | [109] |
| Pancreatic cancer cells in vitro | Increase of sensibility to radiotherapy | [111] | ||
| NSCLC models both in vitro and in vivo | Inhibition of cell proliferation and migration. Increment of cell sensibility to gefitinib. | [110] | ||
| NSC23766 analogs | Inhibition of RacGEF binding to Rac | High-metastatic breast cancer cells cultured in vitro | Inhibition of cell proliferation in a sharper way than NSC23766 does | [112] |
| EHT 1864 | Inhibition of RacGEF activity | Breast cancer cells cultured in vitro | Inhibition of cell proliferation stimulated by estrogen signaling | [120] |
| Breast cancer cells cultured in vitro | Sensitization of cancer cells to tamoxifen | [120] | ||
| NIH3T3 mouse fibroblast in vitro | Inhibition of Rac1-derived malignant cell transformation | [119] | ||
| Breast cancer tumors biopsied from patients’ samples | Restraining of cell invasion and proliferation through programmed cell death induction | [121] | ||
| EHop-016 | Inhibition of Vav1 and -2 activity and its binding with Rac | Metastatic breast cancer cells cultured in vitro | Reduction of cell viability and migration through the inhibition of Rac-derived actin structures | [113] |
| Human and murine leukemic cell models both in vitro and in vivo and patient-derived cells | Increment of overall survival due to the inhibition of cell growth and survival | [115] | ||
| Myxofibrosarcoma cell lines cultured in vitro and xenografts tumors cultured in vivo | Inhibition of cell growth through the induction of apoptosis and suppression of the generation of lung metastasis | [116] | ||
| Breast cancer xenografts models with EHop-016 intraperitoneal administration | Repression of tumor growth, metastasis and angiogenesis | [114] | ||
| YM1B | Repression of RacGEF binding to Rac | Breast cancer cells cultured in vitro | Reduction of cell migration and invasion | [122] |
| BART | Repression of RacGEF activity | Pancreatic cancer cell lines cultured in vitro | Inhibition of cell motility and invasion through the regulation of actin cytoskeleton | [124] |
| Migrastatin analogs | Repression of Rac activity | High metastatic breast cancer cells in vivo xenograft models | Blockage of cell migration and metastasis through the inhibition of lamellipodia formation | [125] |
| Secramine A | Repression of Cdc42 shuttling between cytoplasm and cell membrane | ALCL cells cultured in vitro | Repression of cell proliferation through the induction of programmed cell death in ALK-positive cells | [133] |
| ZCL278 | Inhibition of ITSN and Cdc42 binding | Prostate cancer cell lines cultured in vitro | Inhibition of cell motility and migration mediated by actin filaments | [139] |
| ML141 or CID2950007 | Inhibition of GTP binding to Cdc42 | Ovarian cancer cells cultured in vitro | Inhibition of cell motility and invasion without affecting to its viability | [138] |
| MBQ-167 | Inhibition of GEF binding to Rac/Cdc42 | Breast cancer cells cultured in vitro and xenografts models in vivo | Repression of cell migration, metastasis and proliferation | [144] |
| AZA1 | Prevention of RacGEF binding to Cdc42/Rac | Prostatic cancer models both in vivo and in vitro | Decrease in cell proliferation through the induction of apoptosis in vitro. Reduction of tumor growth and improvement of mice survival in vivo | [145] |
| R-ketorolac | Inhibition of nucleotide docking | Ovarian cancer cell lines and primary patient-derived cells in vitro | Reduction in cell proliferation and growth | [146] |
| Rhosin | Inhibition of RhoAGEF binding to RhoA | Breast cancer cells cultured in vitro | Inhibition of cell proliferation, migration and invasion | [148] |
| Diffuse gastric cancer spheroids cultured in vitro | Inhibition of cell proliferation, migration and invasion. Sensitization of cells to cisplatin | [149] | ||
| Y16 | Hindering of RhoA and LARG joining | Breast cancer cells cultured in vitro | Reduction of cell proliferation and spheroid formation both alone and in combination with Rhosin | [150] |
| Biphosphonate derivatives | Inhibits Rab prenylation. | Melanoma cells cultured in vitro | Inhibition of cell proliferation through cell cycle arrest in S phase | [161] |
| Mesothelioma cells cultured in vitro | Induction of cell apoptosis due to the inhibition of topoisomerase II and Rab6 | [162] | ||
| Prostate and breast cancer cell lines cultured in vitro | Inhibition of cell adhesion to extracellular matrix | [163] | ||
| Prostate and breast cancer cell lines cultured in vitro | Inhibition of cell invasion and metastasis through the repression of MMPs activity | [164] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto-Dominguez, N.; Parnell, C.; Teng, Y. Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges. Cells 2019, 8, 255. https://doi.org/10.3390/cells8030255
Prieto-Dominguez N, Parnell C, Teng Y. Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges. Cells. 2019; 8(3):255. https://doi.org/10.3390/cells8030255
Chicago/Turabian StylePrieto-Dominguez, Néstor, Christopher Parnell, and Yong Teng. 2019. "Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges" Cells 8, no. 3: 255. https://doi.org/10.3390/cells8030255
APA StylePrieto-Dominguez, N., Parnell, C., & Teng, Y. (2019). Drugging the Small GTPase Pathways in Cancer Treatment: Promises and Challenges. Cells, 8(3), 255. https://doi.org/10.3390/cells8030255