Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Assessment of PLA2 Activities and Fusion Assays
2.1.2. Protein Fractionation, Gel Electrophoresis and Western Blotting
2.2. Methods
2.2.1. Molecular Analysis to Detect CSC Associated PLA2 Activities
2.2.2. Quantitative Lipid Analyses
2.2.3. Sequence Alignments
2.2.4. Sample Fractionation for 2DE Western Blotting
2.2.5. Molecular Analyses to Detect CV Associated PLA2 Activities
2.2.6. Effects of PLA2 Activities on Fusion Assays
3. Data Analyses
4. Results
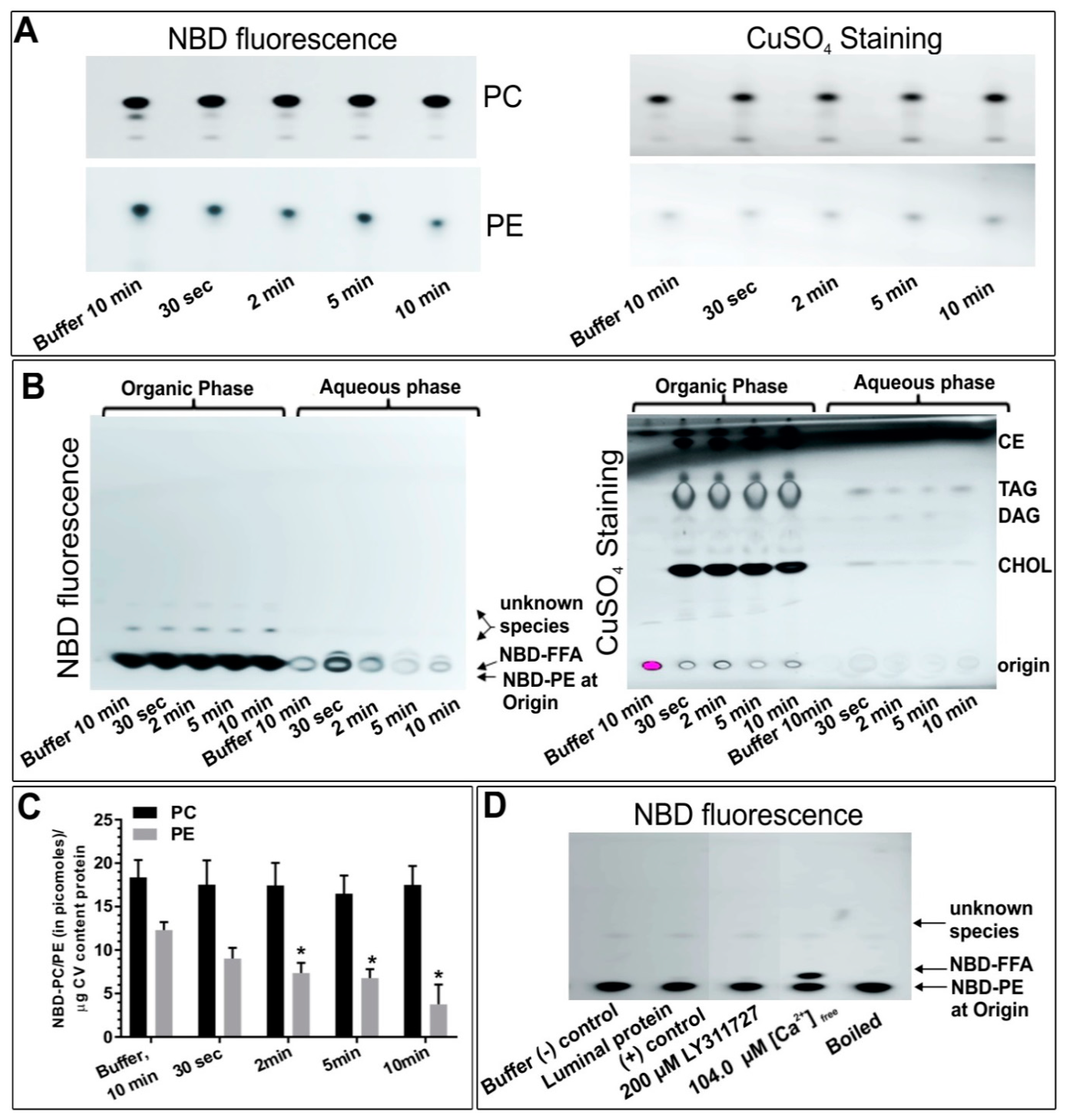
4.1. Identification of Endogenous PLA2 Activities on CSC and Their Effects on CSC Fusion
4.2. In-Silico Analysis
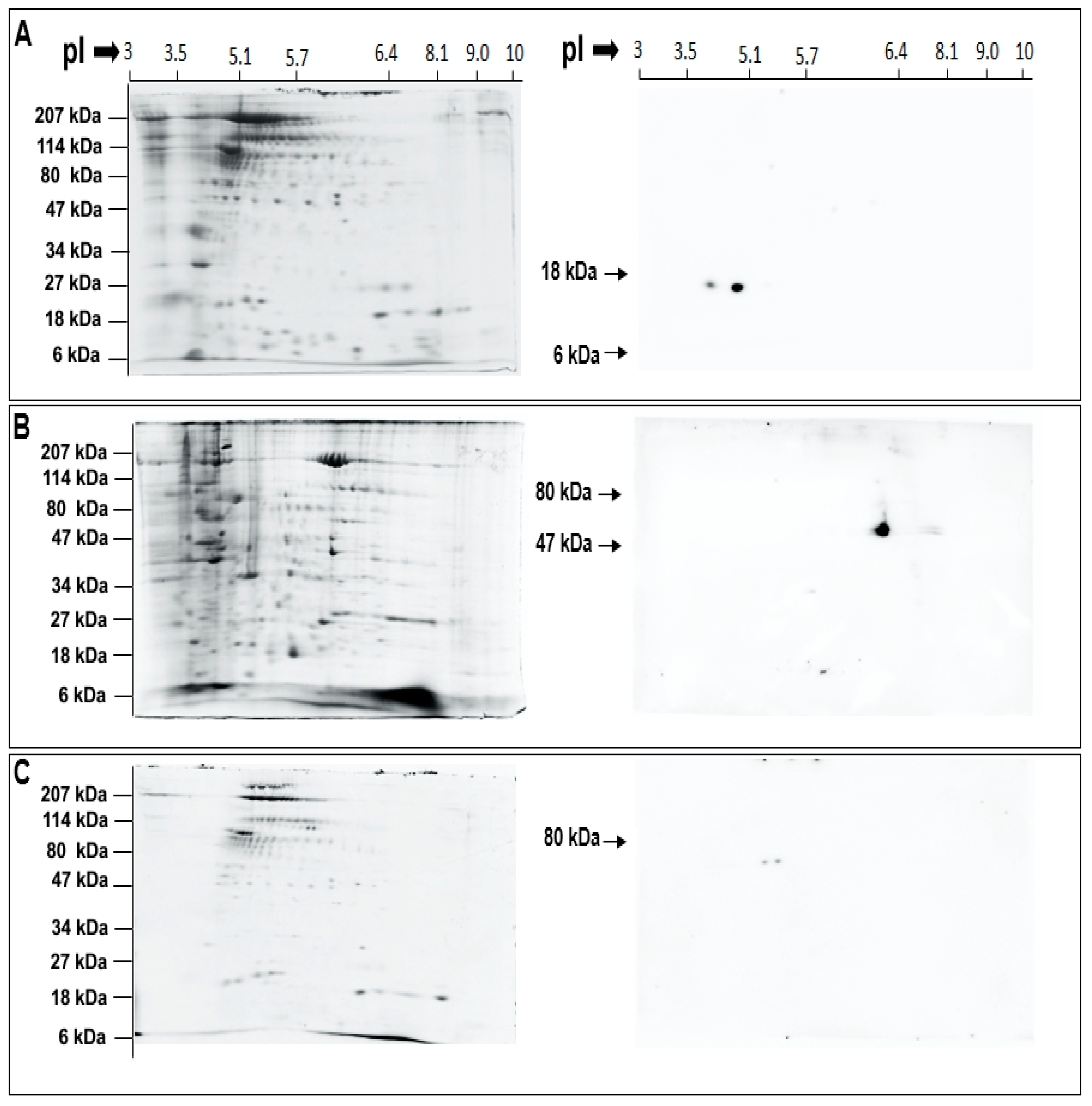
4.3. Western Blotting and Assays to Confirm Identity and Localization of PLA2 Isozymes
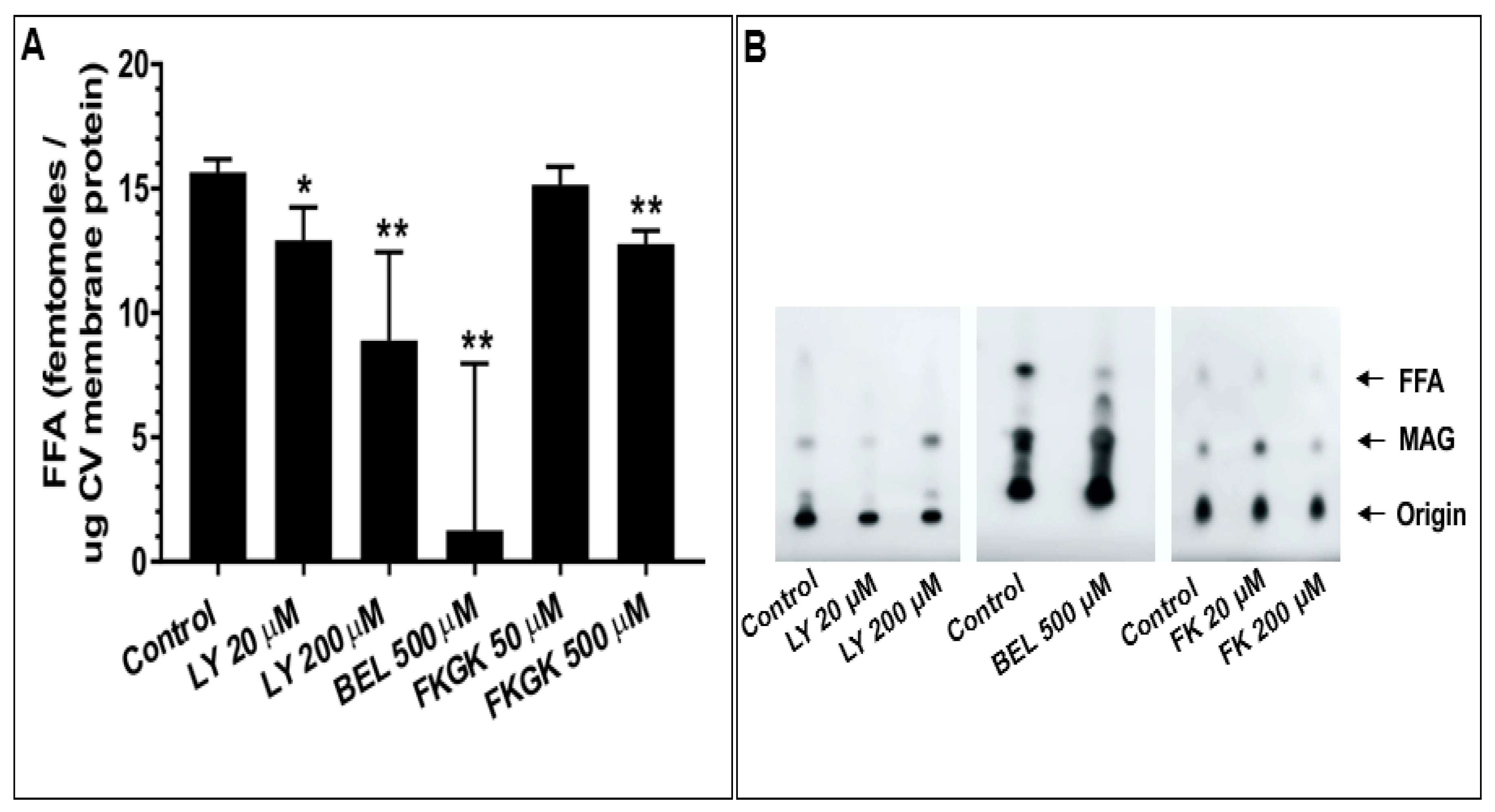
4.4. The Effect of PLA2 Activities on CV-CV Fusion
5. Discussion
5.1. PLA2 Activity and Their Effects on CSC Fusion
5.2. Bioinformatics
5.3. Top-Down Proteomics Identify CV Associated PLA2 Isozymes
5.4. CV Associated PLA2 Isozymes Regulate Docking and/or Priming
5.5. Concluding Remarks
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARA | arachidonic acid |
| [Ca2+]free | free calcium ion concentration |
| cCBB | colloidal Coomassie brilliant blue G-250 dye |
| CHAPS | 3-[(3-Cholamidopropyl) dimethylammonio]-1-propanesulfonate hydrate |
| CHOL | cholesterol |
| CE | cholesterol ester |
| CSC | cell surface complexes |
| CV | cortical vesicles |
| FFA | free fatty acid |
| DAG | diacylglycerol |
| LPC | lyso-phosphatidylcholine |
| PC, PE, PI, PG, PS | phosphatidyl-choline, -ethanolamine, -inositol, -glycerol, -serine |
| MAG | monoacylglycerol |
| pI | isoelectric point |
| PED6 | (N-((6-(2,4-dinitrophenyl) amino) hexanoyl)-2-(4,4-difluoro-5,7-dimethyl-4-Bora-3a,4a-diaza-s-Indacene-3-pentanoyl)-1-hexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt |
| SV | synaptic vesicles |
| TAG | triacylglycerol |
References
- Frye, R.A.; Holz, R.W. The relationship between arachidonic acid release and catecholamine secretion from cultured bovine adrenal chromaffin cells. J. Neurochem. 1984, 43, 146–150. [Google Scholar] [CrossRef]
- Frye, R.A.; Holz, R.W. Arachidonic acid release and catecholamine secretion from digitonin-treated chromaffin cells: Effects of micromolar calcium, phorbol ester and protein alkylating agents. J. Neurochem. 1985, 44, 265–273. [Google Scholar] [CrossRef]
- Juhl, K.; Hoy, M.; Olsen, H.L.; Bokvist, K.; Efanov, A.M.; Hoffmann, E.K.; Gromada, J. cPLA2 alpha-evoked formation of arachidonic acid and lysophospholipids is required for exocytosis in mouse pancreatic beta-cells. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E73–E81. [Google Scholar] [CrossRef]
- Latham, C.F.; Osborne, S.L.; Cryle, M.J.; Meunier, F.A. Arachidonic acid potentiates exocytosis and allows neuronal SNARE complex to interact with Munc18a. J. Neurochem. 2007, 100, 1543–1554. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Murakami, M.; Atsumi, G.; Imai, K.; Prados, P.; Inoue, K.; Kudo, I. Release of secretory phospholipase A2 from rat neuronal cells and its possible function in the regulation of catecholamine secretion. Biochem. J. 1996, 318, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Moskowitz, N.; Schook, W.; Puszkin, S. Interaction of brain synaptic vesicles induced by endogenous Ca2+ -dependent phospholipase A2. Science 1982, 216, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.; Berman, J.D.; Middleton, W.; Brendle, J. Botulinum toxin inhibits arachidonic acid release associated with acetylcholine release from PC12 cells. J. Biol. Chem. 1993, 268, 11057–11064. [Google Scholar] [PubMed]
- Ray, P.; Ishida, H.; Millard, C.B.; Petrali, J.P.; Ray, R. Phospholipaise A2 and arachidonic acid-mediated mechanism of neuroexocytosis: A possible target of botidinum neurotoxin A other then SNAP-25. J. Appl. Toxicol. 1999, 19, S27–S28. [Google Scholar] [CrossRef]
- Karli, U.O.; Schafer, T.; Burger, M.M. Fusion of neurotransmitter vesicles with target membrane is calcium independent in a cell-free system. Proc. Natl. Acad. Sci. USA 1990, 87, 5912–5915. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L.; Chanturiya, A.; Green, J.; Zimmerberg, J. The hemifusion intermediate and its conversion to complete fusion: Regulation by membrane composition. Biophys. J. 1995, 69, 922–929. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Vogel, S.S.; Sokoloff, A.; Onaran, H.O.; Leikina, E.A.; Zimmerberg, J. Lysolipids reversibly inhibit Ca2+-, GTP- and pH-dependent fusion of biological membranes. FEBS Lett. 1993, 318, 71–76. [Google Scholar] [CrossRef]
- Reese, C.; Mayer, A. Transition from hemifusion to pore opening is rate limiting for vacuole membrane fusion. J. Cell Biol. 2005, 171, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Uriarte, S.M.; Powell, D.W.; Luerman, G.C.; Merchant, M.L.; Cummins, T.D.; Jog, N.R.; Ward, R.A.; McLeish, K.R. Comparison of Proteins Expressed on Secretory Vesicle Membranes and Plasma Membranes of Human Neutrophils. J. Immunol. 2008, 180, 5575–5581. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, J.L.; Bark, S.J.; Funkelstein, L.; Mosier, C.; Yap, A.; Kazemi-Esfarjani, P.; La Spada, A.R.; Sigurdson, C.; O’Connor, D.T.; Hook, V. Proteomics of Dense Core Secretory Vesicles Reveal Distinct Protein Categories for Secretion of Neuroeffectors for Cell−Cell Communication. J. Proteome Res. 2010, 9, 5002–5024. [Google Scholar] [CrossRef]
- Brunner, Y.; Couté, Y.; Iezzi, M.; Foti, M.; Fukuda, M.; Hochstrasser, D.F.; Wollheim, C.B.; Sanchez, J.-C. Proteomics Analysis of Insulin Secretory Granules. Mol. Cell. Proteom. 2007, 6, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Schvartz, D.; Brunner, Y.; Coute, Y.; Foti, M.; Wollheim, C.B.; Sanchez, J.C. Improved characterization of the insulin secretory granule proteomes. J. Proteom. 2012, 75, 4620–4631. [Google Scholar] [CrossRef] [PubMed]
- Chock, S.P.; Schmauder-Chock, E.A.; Cordella-Miele, E.; Miele, L.; Mukherjee, A.B. The localization of phospholipase A2 in the secretory granule. Biochem. J. 1994, 300, 619–622. [Google Scholar] [CrossRef]
- Bingham, C.O.; Fijneman, R.J.A.; Friend, D.S.; Goddeau, R.P.; Rogers, R.A.; Austen, K.F.; Arm, J.P. Low Molecular Weight Group IIA and Group V Phospholipase A2 Enzymes Have Different Intracellular Locations in Mouse Bone Marrow-derived Mast Cells. J. Biol. Chem. 1999, 274, 31476–31484. [Google Scholar] [CrossRef]
- Moskowitz, N.; Puszkin, S.; Schook, W. Characterization of brain synaptic vesicle phospholipase A2 activity and its modulation by calmodulin, prostaglandin E2, prostaglandin F2 alpha, cyclic AMP and ATP. J. Neurochem. 1983, 41, 1576–1586. [Google Scholar] [CrossRef]
- Moskowitz, N.; Schook, W.; Puszkin, S. Regulation of endogenous calcium-dependent synaptic membrane phospholipase A2. Brain Res. 1984, 290, 273–280. [Google Scholar] [CrossRef]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular Anatomy of a Trafficking Organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Marco, M.; Jacqueline, B.; Carsten, C.; Michael, K.; Herbert, Z.; Walter, V. Immunoisolation of two synaptic vesicle pools from synaptosomes: A proteomics analysis. J. Neurochem. 2005, 95, 1732–1745. [Google Scholar] [CrossRef]
- Heo, S.; Diering, G.H.; Na, C.H.; Nirujogi, R.S.; Bachman, J.L.; Pandey, A.; Huganir, R.L. Identification of long-lived synaptic proteins by proteomic analysis of synaptosome protein turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E3827–E3836. [Google Scholar] [CrossRef]
- Boyken, J.; Grønborg, M.; Riedel, D.; Urlaub, H.; Jahn, R.; Chua, J.J.E. Molecular Profiling of Synaptic Vesicle Docking Sites Reveals Novel Proteins but Few Differences between Glutamatergic and GABAergic Synapses. Neuron 2013, 78, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 1997, 22, 1–2. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Yang, H.C.; Rosenberger, T.A.; Horrocks, L.A. Phospholipase A2 and its role in brain tissue. J. Neurochem. 1997, 69, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Kudo, I.; Murakami, M.; Hara, S.; Inoue, K. Mammalian non-pancreatic phospholipases A2. Biochim. Biophys. Acta (Bba) Lipids Lipid Metab. 1993, 1170, 217–231. [Google Scholar] [CrossRef]
- Mounier, C.M.; Ghomashchi, F.; Lindsay, M.R.; James, S.; Singer, A.G.; Parton, R.G.; Gelb, M.H. Arachidonic Acid Release from Mammalian Cells Transfected with Human Groups IIA and X Secreted Phospholipase A2 Occurs Predominantly during the Secretory Process and with the Involvement of Cytosolic Phospholipase A2-α. J. Biol. Chem. 2004, 279, 25024–25038. [Google Scholar] [CrossRef]
- Dabral, D.; Coorssen, J.R. Phospholipase A2: Potential roles in native membrane fusion. Int. J. Biochem. Cell Biol. 2017, 85, 1–5. [Google Scholar] [CrossRef]
- Raveh, A.; Valitsky, M.; Shani, L.; Coorssen, J.R.; Blank, P.S.; Zimmerberg, J.; Rahamimoff, R. Observations of Calcium Dynamics in Cortical Secretory Vesicles. Cell Calcium 2012, 52, 217–225. [Google Scholar] [CrossRef]
- Estévez-Herrera, J.; Domínguez, N.; Pardo, M.R.; González-Santana, A.; Westhead, E.W.; Borges, R.; Machado, J.D. ATP: The crucial component of secretory vesicles. Proc. Natl. Acad. Sci. USA 2016, 113, E4098–E4106. [Google Scholar] [CrossRef]
- Winkler, H.; Westhead, E. Th molecular organization of adrenal chromaffin granules. Neuroscience 1980, 5, 1803–1823. [Google Scholar] [CrossRef]
- Jaime, S.; Laura, V.; Marcial, C.; Esther, H.S.; Fonteriz, R.I.; Lobaton, C.D.; Mayte, M.; Alfredo, M.; Javier, A. Calcium dynamics in bovine adrenal medulla chromaffin cell secretory granules. Eur. J. Neurosci. 2008, 28, 1265–1274. [Google Scholar] [CrossRef]
- Balsinde, J.; Balboa, M.A.; Dennis, E.A. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J. Biol. Chem. 1997, 272, 29317–29321. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Ali, T.; Ashley, J.W.; Bone, R.N.; Hancock, W.D.; Lei, X. Calcium-independent phospholipases A2 and their roles in biological processes and diseases. J. Lipid Res. 2015, 56, 1643–1668. [Google Scholar] [CrossRef]
- Six, D.A.; Dennis, E.A. The expanding superfamily of phospholipase A(2) enzymes: Classification and characterization. Biochim. Biophys. Acta 2000, 1488, 1–19. [Google Scholar] [CrossRef]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Hirabayashi, T.; Yamamoto, K. Recent progress in phospholipase A2 research: From cells to animals to humans. Prog. Lipid Res. 2011, 50, 152–192. [Google Scholar] [CrossRef]
- Abbineni, P.S.; Wright, E.P.; Rogasevskaia, T.P.; Killingsworth, M.C.; Malladi, C.S.; Coorssen, J.R. The Sea Urchin Egg and Cortical Vesicles as Model Systems to Dissect the Fast, Ca2+-Triggered Steps of Regulated Exocytosis. In Exocytosis Methods; Thorn, P., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 221–241. [Google Scholar]
- Zimmerberg, J.; Coorssen, J.R.; Vogel, S.S.; Blank, P.S. Sea urchin egg preparations as systems for the study of calcium-triggered exocytosis. J. Physiol. 1999, 520, 15–21. [Google Scholar] [CrossRef]
- Sodergren, E.; Weinstock, G.M.; Davidson, E.H.; Cameron, R.A.; Gibbs, R.A.; Angerer, R.C.; Angerer, L.M.; Arnone, M.I.; Burgess, D.R.; Burke, R.D.; et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science 2006, 314, 941–952. [Google Scholar] [CrossRef]
- Tahara, M.; Coorssen, J.R.; Timmers, K.; Blank, P.S.; Whalley, T.; Scheller, R.; Zimmerberg, J. Calcium Can Disrupt the SNARE Protein Complex on Sea Urchin Egg Secretory Vesicles without Irreversibly Blocking Fusion. J. Biol. Chem. 1998, 273, 33667–33673. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Tahara, M.; Zimmerberg, J. Biochemical and Functional Studies of Cortical Vesicle Fusion: The SNARE Complex and Ca2+ Sensitivity. J. Cell Biol. 1998, 143, 1845–1857. [Google Scholar] [CrossRef]
- Szule, J.A.; Jarvis, S.E.; Hibbert, J.E.; Spafford, J.D.; Braun, J.E.A.; Zamponi, G.W.; Wessel, G.M.; Coorssen, J.R. Calcium-triggered Membrane Fusion Proceeds Independently of Specific Presynaptic Proteins. J. Biol. Chem. 2003, 278, 24251–24254. [Google Scholar] [CrossRef] [PubMed]
- Sean, C.; David, L.; Gary, W. Members of the SNARE hypothesis are associated with cortical granule exocytosis in the sea urchin egg. Mol. Reprod. Dev. 1997, 48, 106–118. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Chen, X.; Backlund, P.S.; Zimmerberg, J. Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell Sci. 2003, 116, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.; Hodel, A.; Whitaker, M. In vitro exocytosis in sea urchin eggs requires a synaptobrevin-related protein. J. Cell Sci. 1997, 110, 1555–1561. [Google Scholar] [PubMed]
- Rogasevskaia, T.; Coorssen, J.R. Sphingomyelin-enriched microdomains define the efficiency of native Ca2+-triggered membrane fusion. J. Cell Sci. 2006, 119, 2688–2694. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Churchward, M.A.; Coorssen, J.R. Anionic lipids in Ca(2+)-triggered fusion. Cell Calcium 2012, 52, 259–269. [Google Scholar] [CrossRef]
- Rogasevskaia, T.P.; Coorssen, J.R. The role of phospholipase D in regulated exocytosis. J. Biol. Chem. 2015, 290, 28683–28696. [Google Scholar] [CrossRef]
- Rogasevskaia, T.P.; Coorssen, J.R. A new approach to the molecular analysis of docking, priming and regulated membrane fusion. J. Chem. Biol. 2011, 4, 117–136. [Google Scholar] [CrossRef]
- Churchward, M.A.; Rogasevskaia, T.; Höfgen, J.; Bau, J.; Coorssen, J.R. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci. 2005, 118, 4833–4848. [Google Scholar] [CrossRef]
- Churchward, M.A.; Rogasevskaia, T.; Brandman, D.M.; Khosravani, H.; Nava, P.; Atkinson, J.K.; Coorssen, J.R. Specific Lipids Supply Critical Negative Spontaneous Curvature—An Essential Component of Native Ca2+-Triggered Membrane Fusion. Biophys. J. 2008, 94, 3976–3986. [Google Scholar] [CrossRef]
- Abbineni, P.; Coorssen, J. Application of High-Throughput Assays to Examine Phospho-Modulation of the Late Steps of Regulated Exocytosis. High.-Throughput 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Abbineni, P.S.; Coorssen, J.R. Sphingolipids modulate docking, Ca2+ sensitivity and membrane fusion of native cortical vesicles. Int. J. Biochem. Cell Biol. 2018, 104, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Zimmerberg, J.; Blank, P.S.; Kolosova, I.; Cho, M.-S.; Tahara, M.; Coorssen, J.R. A stage-specific preparation to study the Ca2+-triggered fusion steps of exocytosis: Rationale and perspectives. Biochimie 2000, 82, 303–314. [Google Scholar] [CrossRef]
- Churchward, M.A.; Brandman, D.M.; Rogasevskaia, T.; Coorssen, J.R. Copper (II) sulfate charring for high sensitivity on-plate fluorescent detection of lipids and sterols: Quantitative analyses of the composition of functional secretory vesicles. J. Chem. Biol. 2008, 1, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Butt, R.H.; Coorssen, J.R. Postfractionation for Enhanced Proteomic Analyses: Routine Electrophoretic Methods Increase the Resolution of Standard 2D-PAGE. J. Proteome Res. 2005, 4, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building Phylogenetic Trees from Molecular Data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Backlund, P.S., Jr.; Zimmerberg, J. Quantitative femto- to attomole immunodetection of regulated secretory vesicle proteins critical to exocytosis. Anal. Biochem. 2002, 307, 54–62. [Google Scholar] [CrossRef]
- Gauci, V.J.; Padula, M.P.; Coorssen, J.R. Coomassie blue staining for high sensitivity gel-based proteomics. J. Proteom. 2013, 90, 96–106. [Google Scholar] [CrossRef]
- Noaman, N.; Abbineni, P.S.; Withers, M.; Coorssen, J.R. Coomassie staining provides routine (sub)femtomole in-gel detection of intact proteoforms: Expanding opportunities for genuine Top-down Proteomics. Electrophoresis 2017, 38, 3086–3099. [Google Scholar] [CrossRef]
- Balsinde, J.; Bianco, I.D.; Ackermann, E.J.; Conde-Frieboes, K.; Dennis, E.A. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc. Natl. Acad. Sci. USA 1995, 92, 8527–8531. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, Cloning, Expression and Purification of Three Novel Human Calcium-independent Phospholipase A2 Family Members Possessing Triacylglycerol Lipase and Acylglycerol Transacylase Activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Litsky, M.L.; Farooqui, T.; Horrocks, L.A. Inhibitors of intracellular phospholipase A2 activity: Their neurochemical effects and therapeutical importance for neurological disorders. Brain Res. Bull. 1999, 49, 139–153. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Bucher, D.; Cao, J.; Li, S.; Yang, S.-W.; Kokotos, G.; Woods, V.L.; McCammon, J.A.; Dennis, E.A. Fluoroketone Inhibition of Ca2+-Independent Phospholipase A2 through Binding Pocket Association Defined by Hydrogen/Deuterium Exchange and Molecular Dynamics. J. Am. Chem. Soc. 2013, 135, 1330–1337. [Google Scholar] [CrossRef]
- Tibes, U. Phospholipase A2 inhibitors in development. Expert Opin. Investig. Drugs 1997, 6, 279–298. [Google Scholar] [CrossRef]
- Baker, P.F.; Whitaker, M.J. Influence of ATP and calcium on the cortical reaction in sea urchin eggs. Nature 1978, 276, 513. [Google Scholar] [CrossRef] [PubMed]
- Vacquier, V.D. The isolation of intact cortical granules from sea urchin eggs: Calcium lons trigger granule discharge. Dev. Biol. 1975, 43, 62–74. [Google Scholar] [CrossRef]
- Balsinde, J.; Dennis, E.A. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J. Biol. Chem. 1996, 271, 31937–31941. [Google Scholar] [CrossRef]
- Ramanadham, S.; Wolf, M.J.; Jett, P.A.; Gross, R.W.; Turk, J. Characterization of an ATP-stimulatable Ca(2+)-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: A possible molecular component of the beta-cell fuel sensor. Biochemistry 1994, 33, 7442–7452. [Google Scholar] [CrossRef] [PubMed]
- Cameron, R.A. Comparing the Human and Sea Urchin Genomes. In eLS; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Bradham, C.A.; Foltz, K.R.; Beane, W.S.; Arnone, M.I.; Rizzo, F.; Coffman, J.A.; Mushegian, A.; Goel, M.; Morales, J.; Geneviere, A.-M.; et al. The sea urchin kinome: A first look. Dev. Biol. 2006, 300, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Ramanadham, S.; Hsu, F.F.; Zhang, S.; Jin, C.; Bohrer, A.; Song, H.; Bao, S.; Ma, Z.; Turk, J. Apoptosis of insulin-secreting cells induced by endoplasmic reticulum stress is amplified by overexpression of group VIA calcium-independent phospholipase A2 (iPLA2 beta) and suppressed by inhibition of iPLA2 beta. Biochemistry 2004, 43, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA2β. Biochim. Biophys. Acta (Bba) Mol. Cell Biol. Lipids 2010, 1801, 547–558. [Google Scholar] [CrossRef]
- Larsson, P.K.A.; Claesson, H.-E.; Kennedy, B.P. Multiple Splice Variants of the Human Calcium-independent Phospholipase A2 and Their Effect on Enzyme Activity. J. Biol. Chem. 1998, 273, 207–214. [Google Scholar] [CrossRef]
- Song, H.; Ramanadham, S.; Bao, S.; Hsu, F.-F.; Turk, J. A Bromoenol Lactone Suicide Substrate Inactivates Group VIA Phospholipase A(2) by Generating a Diffiusible Bromomethyl Keto Acid That Alkylates Cysteine Thiols. Biochemistry 2006, 45, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Darrow, A.L.; Olson, M.W.; Xin, H.; Burke, S.L.; Smith, C.; Schalk-Hihi, C.; Williams, R.; Bayoumy, S.S.; Deckman, I.C.; Todd, M.J.; et al. A novel fluorogenic substrate for the measurement of endothelial lipase activity. J. Lipid Res. 2011, 52, 374–382. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, K.P.; Rhee, H.J.; Das, S.; Rafter, J.D.; Oh, Y.S.; Cho, W. Internalized group V secretory phospholipase A2 acts on the perinuclear membranes. J. Biol. Chem. 2002, 277, 9358–9365. [Google Scholar] [CrossRef] [PubMed]
- Coorssen, J.R. Phospholipase activation and secretion: Evidence that PLA2, PLC and PLD are not essential to exocytosis. Am. J. Physiol. Cell Physiol. 1996, 270, C1153–C1163. [Google Scholar] [CrossRef] [PubMed]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A2 regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar] [CrossRef]
- Peterson, M.E.; Chen, F.; Saven, J.G.; Roos, D.S.; Babbitt, P.C.; Sali, A. Evolutionary constraints on structural similarity in orthologs and paralogs. Protein Sci. A Publ. Protein Soc. 2009, 18, 1306–1315. [Google Scholar] [CrossRef]
- Hazen, S.L.; Stuppy, R.J.; Gross, R.W. Purification and characterization of canine myocardial cytosolic phospholipase A2. A calcium-independent phospholipase with absolute f1-2 regiospecificity for diradyl glycerophospholipids. J. Biol. Chem. 1990, 265, 10622–10630. [Google Scholar] [PubMed]
- Rosenthal, M.D.; Gordon, M.N.; Buescher, E.S.; Slusser, J.H.; Harris, L.K.; Franson, R.C. Human Neutrophils Store Type II 14-kDa Phospholipase A2 in Granules and Secrete Active Enzyme in Response to Soluble Stimuli. Biochem. Biophys. Res. Commun. 1995, 208, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Ishizaki, J.; Yokota, Y.; Higashino, K.; Ono, T.; Ikeda, M.; Fujii, N.; Kawamoto, K.; Hanasaki, K. Structures, enzymatic properties and expression of novel human and mouse secretory phospholipase A(2)s. J. Biol. Chem. 2000, 275, 5785–5793. [Google Scholar] [CrossRef]
- Zupan, L.A.; Steffens, D.L.; Berry, C.A.; Landt, M.; Gross, R.W. Cloning and expression of a human 14-3-3 protein mediating phospholipolysis. Identification of an arachidonoyl-enzyme intermediate during catalysis. J. Biol. Chem. 1992, 267, 8707–8710. [Google Scholar] [PubMed]
- Wilkins, M.R.; Lindskog, I.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Hochstrasser, D.F.; Appel, R.D. Detailed peptide characterization using PEPTIDEMASS—A World-Wide-Web-accessible tool. Electrophoresis 1997, 18, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Szule, J.A.; Fuller, N.L.; Rand, R.P. The effects of acyl chain length and saturation of diacylglycerols and phosphatidylcholines on membrane monolayer curvature. Biophys. J. 2002, 83, 977–984. [Google Scholar] [CrossRef]
- Furber, K.L.; Churchward, M.A.; Rogasevskaia, T.P.; Coorssen, J.R. Identifying Critical Components of Native Ca2+-triggered Membrane Fusion. Ann. N. Y. Acad. Sci. 2009, 1152, 121–134. [Google Scholar] [CrossRef]
- Furber, K.L.; Dean, K.T.; Coorssen, J.R. Dissecting the mechanism of Ca2+ triggered membrane fusion: Probing protein function using thiol reactivity. Clin. Exp. Pharmacol. Physiol. 2010, 37, 208–217. [Google Scholar] [CrossRef]
- Furber, K.L.; Brandman, D.M.; Coorssen, J.R. Enhancement of the Ca(2+)-triggering steps of native membrane fusion via thiol-reactivity. J. Chem. Biol. 2009, 2, 27–37. [Google Scholar] [CrossRef]
- Spessard, G.O. ACD Labs/LogP dB 3.5 and ChemSketch 3.5. J. Chem. Inf. Comput. Sci. 1998, 38, 1250–1253. [Google Scholar] [CrossRef]
- Chernomordik, L.; Kozlov, M.M.; Zimmerberg, J. Lipids in biological membrane fusion. J. Membarin Biol. 1995, 146, 1–14. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Kozlov, M.M. Mechanics of membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 675–683. [Google Scholar] [CrossRef]
- Kozlov, M.M.; Leikin, S.L.; Chernomordik, L.V.; Markin, V.S.; Chizmadzhev, Y.A. Stalk mechanism of vesicle fusion. Eur. Biophys. J. 1989, 17, 121–129. [Google Scholar] [CrossRef]
- Kozlovsky, Y.; Kozlov, M.M. Stalk Model of Membrane Fusion: Solution of Energy Crisis. Biophys. J. 2002, 82, 882–895. [Google Scholar] [CrossRef]
- Fuller, N.; Rand, R.P. The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys. J. 2001, 81, 243–254. [Google Scholar] [CrossRef]
- Gillot, I.; Ciapa, B.; Payan, P.; Sardet, C. The calcium content of cortical granules and the loss of calcium from sea urchin eggs at fertilization. Dev. Biol. 1991, 146, 396–405. [Google Scholar] [CrossRef]
- Mahapatra, N.R.; Mahata, M.; Hazra, P.P.; McDonough, P.M.; O’Connor, D.T.; Mahata, S.K. A Dynamic Pool of Calcium in Catecholamine Storage Vesicles: Exploration in living cells by a novel vesicle-targeted chromogranin a-aequorin chimeric photoprotein. J. Biol. Chem. 2004, 279, 51107–51121. [Google Scholar] [CrossRef] [PubMed]
- Leitner, J.W.; Sussman, K.E.; Vatter, A.E.; Schneider, F.H. Adenine nucleotides in the secretory granule fraction of rat islets. Endocrinology 1975, 96, 662–677. [Google Scholar] [CrossRef] [PubMed]
- Brash, A.R. Arachidonic acid as a bioactive molecule. J. Clin. Investig. 2001, 107, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.A. Fatty acid transport: Difficult or easy? J. Lipid Res. 1998, 39, 467–481. [Google Scholar]
- Alder-Baerens, N.; Lisman, Q.; Luong, L.; Pomorski, T.; Holthuis, J.C.M. Loss of P4 ATPases Drs2p and Dnf3p Disrupts Aminophospholipid Transport and Asymmetry in Yeast Post-Golgi Secretory Vesicles. Mol. Biol. Cell 2006, 17, 1632–1642. [Google Scholar] [CrossRef] [PubMed]
- Galli, C.; Risé, P.; Marangoni, F. Fate of exogenous arachidonic acid in THP-1 cells: Incorporation in cell lipids and conversion to other N-6 fatty acids. ProstaglandinsLeukot. Essent. Fat. Acids 1995, 52, 103–106. [Google Scholar] [CrossRef]
- Ibarguren, M.; López, D.J.; Escribá, P.V. The effect of natural and synthetic fatty acids on membrane structure, microdomain organization, cellular functions and human health. Biochim. Biophys. Acta (Bba) Biomembr. 2014, 1838, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F.; Ahmed, N.; Chen, R. Promotion of hexagonal phase formation and lipid mixing by fatty acids with varying degrees of unsaturation. Chem. Phys. Lipids 1991, 57, 75–80. [Google Scholar] [CrossRef]
- Darios, F.; Davletov, B. Omega-3 and omega-6 fatty acids stimulate cell membrane expansion by acting on syntaxin 3. Nature 2006, 440, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Dulubova, I.; Sugita, S.; Hill, S.; Hosaka, M.; Fernandez, I.; Sudhof, T.C.; Rizo, J. A conformational switch in syntaxin during exocytosis: Role of munc18. EMBO J. 1999, 18, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Chernomordik, L. Non-bilayer lipids and biological fusion intermediates. Chem. Phys. Lipids 1996, 81, 203–213. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Species | Percent Identities | Percent Similarities | Percent Identities | Percent Similarities | |
|---|---|---|---|---|---|
| Urchin Isozymes | (Full Length) | (at Histidine Catalytic Site) | |||
| sPLA2 | Human | 39 | 50 | 68 | 67 |
| Murine | 36 | 46 | 74 | 74 | |
| (Full Length) | (at Serine Catalytic Site) | ||||
| iPLA2 | Human | 36 | 52 | 100 | 100 |
| Murine | 36 | 53 | 100 | 100 | |
| A | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PE (Picomoles/µg CSC Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | PC (Picomoles/µg CSC Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | ||
| Control | 29.5 | 7.5 | 5 | Control | 47.7 | 11.0 | 6 | ||||
| LY311727 | 20 µM | ND | NA | NA | NA | LY311727 | 20 µM | 66.6 | 3.0 | 0.38 | 2 |
| LY311727 | 200 µM | ND | NA | NA | NA | LY311727 | 200 µM | 38.9 | 2.2 | 0.68 | 2 |
| BEL | 100 µM | ND | NA | NA | NA | BEL | 100 µM | ND | ND | NA | NA |
| BEL | 500 µM | 28.0 | 7.5 | 0.90 | 3 | BEL | 500 µM | 77.2 | 5.0 | 0.31 | 3 |
| FKGK-11 | 50 µM | 51.0 | 7.6 | 0.11 | 3 | FKGK-11 | 50 µM | 63.2 | 19.1 | 0.47 | 3 |
| FKGK-11 | 200 µM | 31.8 | 9.3 | 0.85 | 3 | FKGK-11 | 200 µM | 50.7 | 8.9 | 0.86 | 3 |
| B | |||||||||||
| PE (Picomoles/µg CV Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | PC (Picomoles/µg CV Membrane Protein) | Mean | SEM | Student t-Test | Sample Size (n) | ||
| Control | 46.0 | 5.5 | 11 | Control | 75.1 | 11.8 | 13 | ||||
| LY311727 | 20 µM | ND | NA | NA | NA | LY311727 | 20 µM | 96.4 | 26.3 | 0.5 | 3 |
| LY311727 | 200 µM | 70.5 | 5.1 | 0.05 | 3 | LY311727 | 200 µM | 114.2 | 19.9 | 0.1 | 6 |
| BEL | 100 µM | ND | NA | NA | 1 | BEL | 100 µM | ND | NA | NA | NA |
| BEL | 500 µM | 46.6 | 12.7 | 0.96 | 5 | BEL | 500 µM | 57.3 | 7.2 | 0.4 | 5 |
| FKGK-11 | 50 µM | 40.4 | 12.9 | 0.66 | 3 | FKGK-11 | 50 µM | 53.5 | 26.0 | 0.4 | 3 |
| FKGK-11 | 200 µM | 22.5 | 6.7 | 0.06 | 3 | FKGK-11 | 200 µM | 42.4 | 16.5 | 0.2 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dabral, D.; Coorssen, J.R. Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis. Cells 2019, 8, 303. https://doi.org/10.3390/cells8040303
Dabral D, Coorssen JR. Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis. Cells. 2019; 8(4):303. https://doi.org/10.3390/cells8040303
Chicago/Turabian StyleDabral, Deepti, and Jens R Coorssen. 2019. "Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis" Cells 8, no. 4: 303. https://doi.org/10.3390/cells8040303
APA StyleDabral, D., & Coorssen, J. R. (2019). Combined Targeted Omic and Functional Assays Identify Phospholipases A2 that Regulate Docking/Priming in Calcium-Triggered Exocytosis. Cells, 8(4), 303. https://doi.org/10.3390/cells8040303