Effects of Cigarette Smoke on TSPO-related Mitochondrial Processes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Exposure of H1299 Cells to Cigarette Smoke
2.3. The Effect of CS Exposure on TSPO and cAMP Levels
2.4. ADP/ATP Ratio
2.5. Cardiolipin Peroxidation Levels
2.6. Collapse of the Mitochondrial Membrane Potential (ΔΨm)
2.7. Cellular Cytotoxicity Measurement by LDH Enzyme Activity
2.8. Apoptosis and Necrosis Levels as Measured by FACS
2.9. Statistical Analysis
3. Results
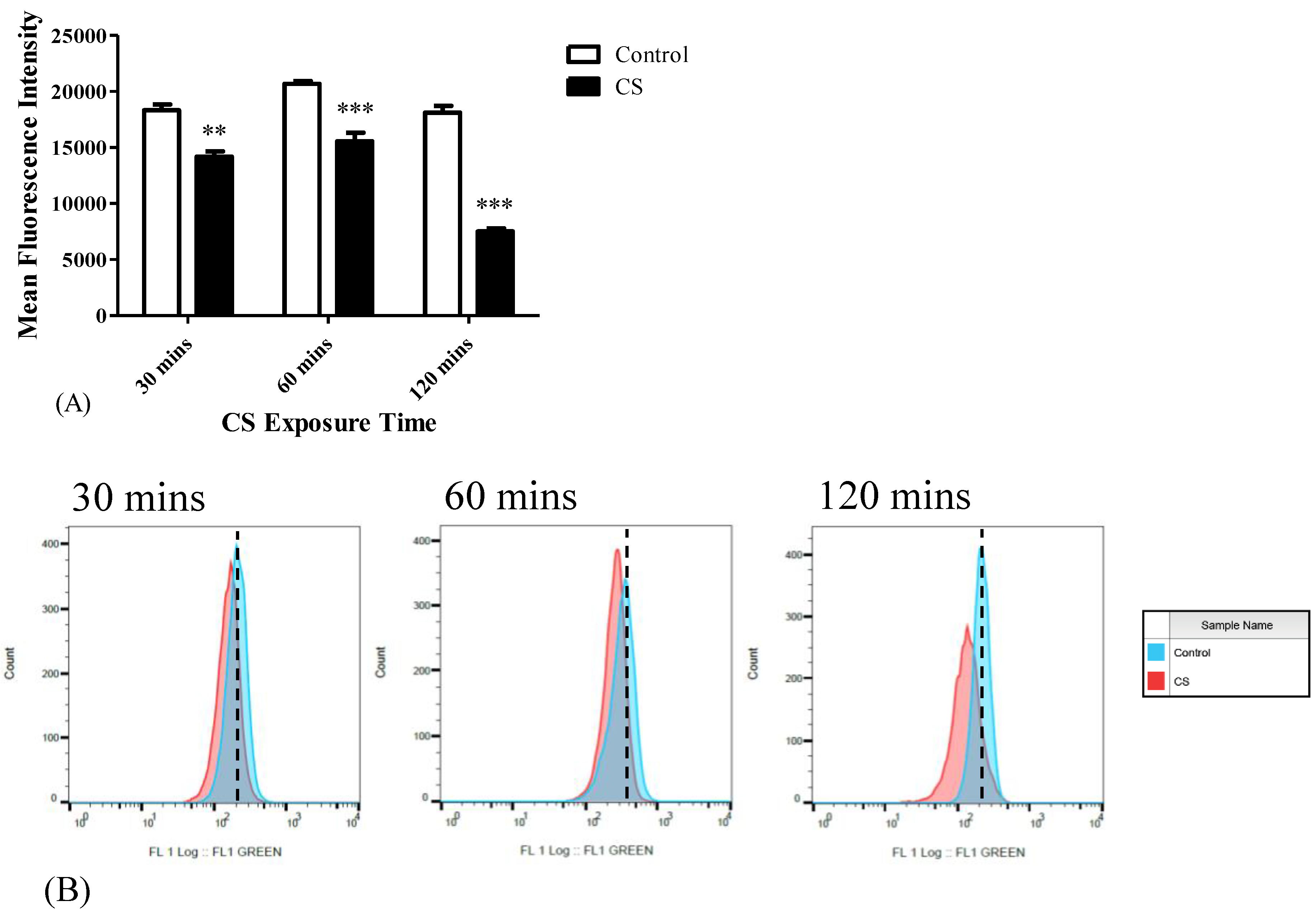
3.1. TSPO Levels
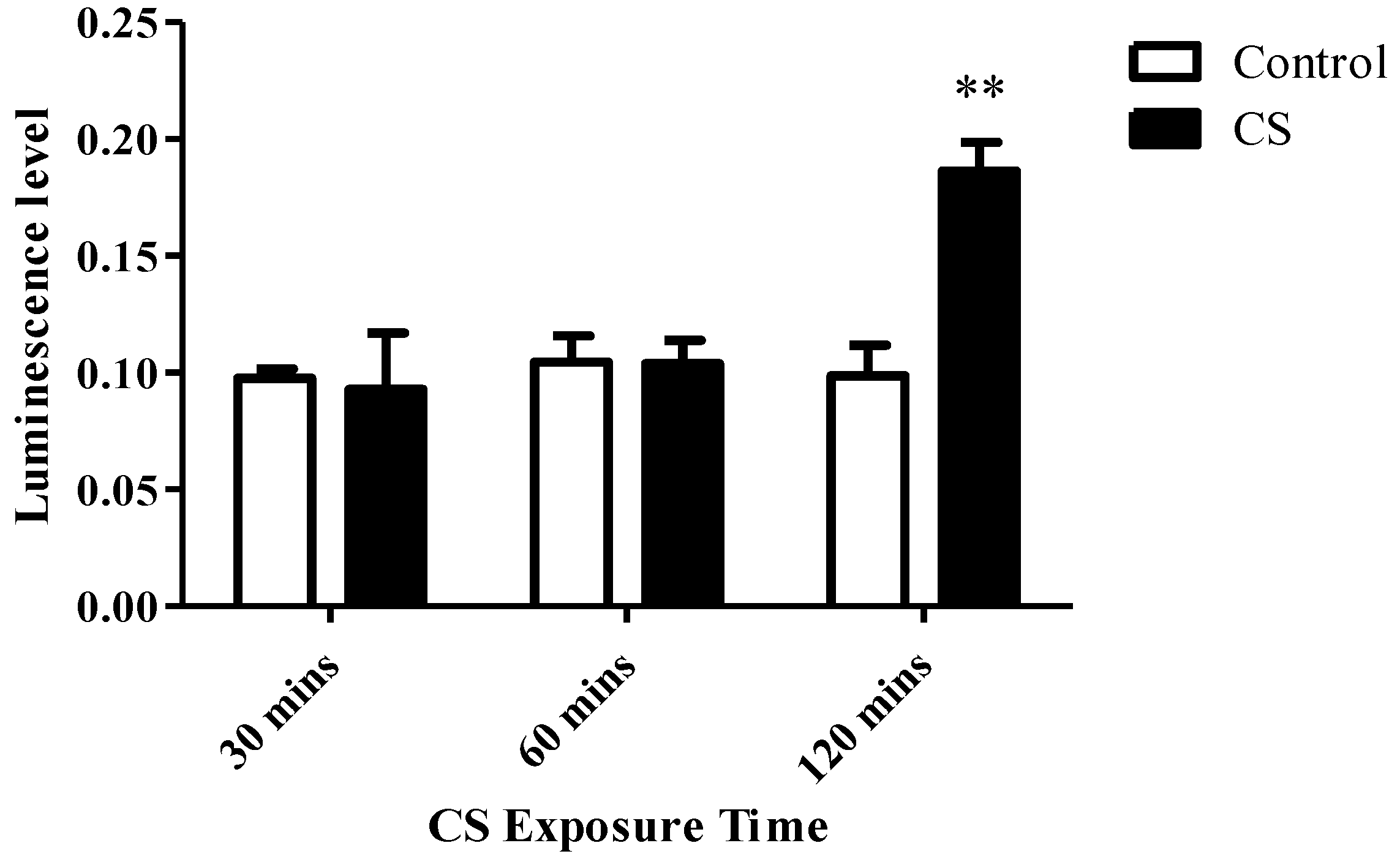
3.2. ADP/ATP Ratio
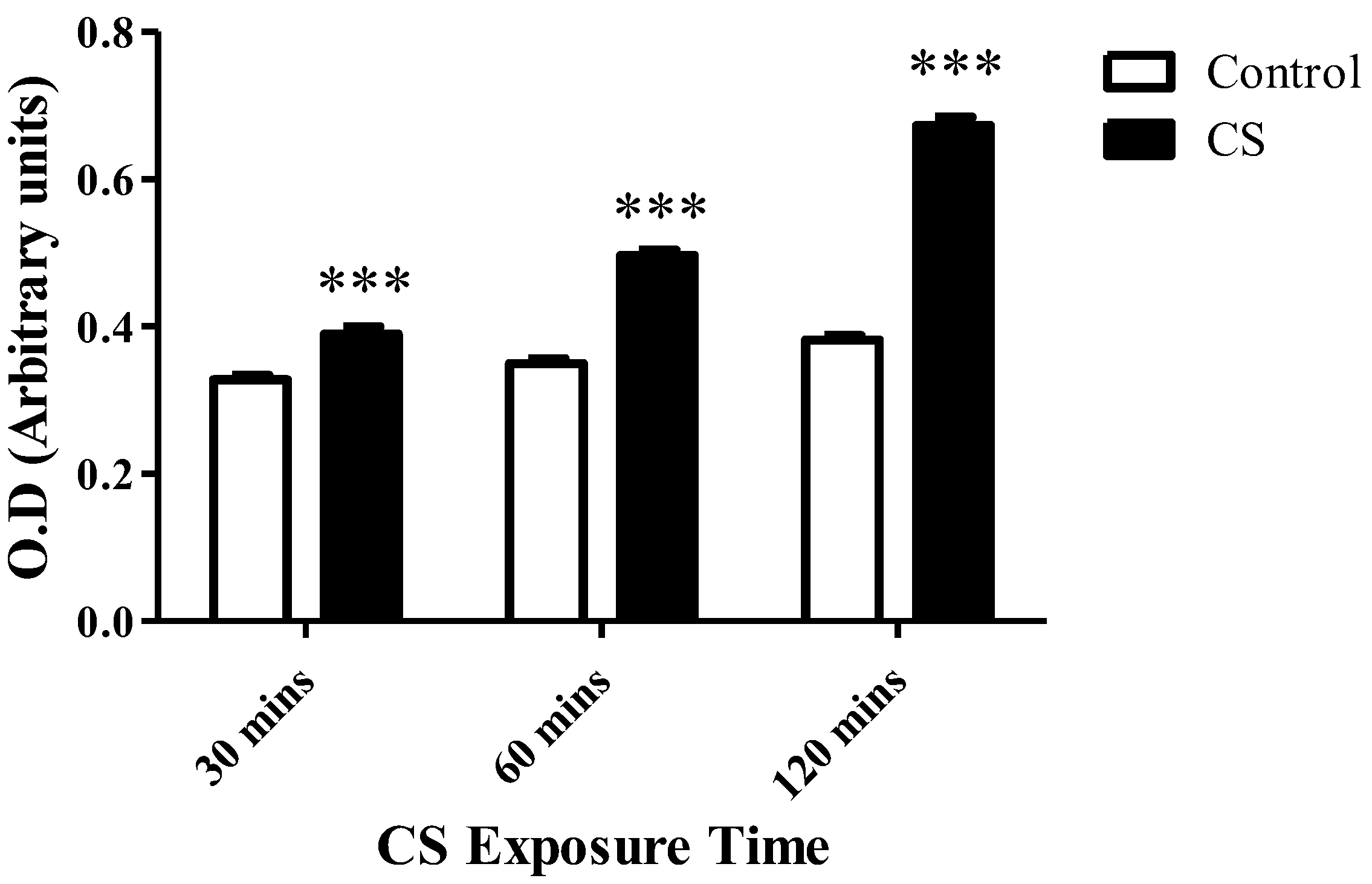
3.3. Cardiolipin Levels
3.4. Collapse of the Mitochondrial Membrane Potential (ΔΨm)
3.5. Cellular Cytotoxicity (LDH)
3.6. Apoptosis and Necrosis Levels
3.7. cAMP Levels
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Report on the Global Tobacco Epidemic 2011. 2011. Available online: https://www.who.int/tobacco/global_report/2011/en/ (accessed on 7 April 2015).
- Edwards, R. The problem of tobacco smoking. BMJ 2004, 328, 217. [Google Scholar] [CrossRef] [PubMed]
- Caram, L.M.; Ferrari, R.; Bertani, A.L.; Garcia, T.; Mesquita, C.B.; Knaut, C.; Tanni, S.E.; Godoy, I. Smoking and Early COPD as Independent Predictors of Body Composition, Exercise Capacity, and Health Status. PLoS ONE 2016, 11, e0164290. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO report on the global tobacco epidemic 2017. 2017. Available online: https://www.who.int/tobacco/global_report/2017/en/ (accessed on 7 April 2015).
- Assadollahi, V.; Mohammadi, E.; Fathi, F.; Hassanzadeh, K.; Erfan, M.B.K.; Soleimani, F.; Banafshi, O.; Yosefi, F.; Allahvaisi, O. Effects of cigarette smoke condensate on proliferation and pluripotency gene expression in mouse embryonic stem cells. J. Cell Biochem. 2019, 120, 4071–4080. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.; Ramasundarahettige, C.; Landsman, V.; Rostron, B.; Thun, M.; Anderson, R.N.; McAfee, T.; Peto, R. 21st-century hazards of smoking and benefits of cessation in the United States. N. Engl. J. Med. 2013, 368, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Crofton, J.; Bjartveit, K. Smoking as a risk factor for chronic airways disease. Chest 1989, 96, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P. Cancer, cigarette smoking and premature death in Europe: A review including the Recommendations of European Cancer Experts Consensus Meeting, Helsinki, October 1996. Lung Cancer 1997, 17, 1–60. [Google Scholar] [CrossRef]
- Yanbaeva, D.G.; Dentener, M.A.; Creutzberg, E.C.; Wesseling, G.; Wouters, E.F. Systemic effects of smoking. Chest 2007, 131, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Katz, Y.; Ben-Baruch, G.; Kloog, Y.; Menczer, J.; Gavish, M. Increased density of peripheral benzodiazepine-binding sites in ovarian carcinomas as compared with benign ovarian tumours and normal ovaries. Clin. Sci. 1990, 78, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Katz, Y.; Eitan, A.; Gavish, M. Increase in Peripheral Benzodiazepine Binding Sites in Colonic Adenocarcinoma. Oncology 1990, 47, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Lazzarini, R.; Malucelli, B.E.; Palermo-Neto, J. Reduction of acute inflammation in rats by diazepam: role of peripheral benzodiazepine receptors and corticosterone. Immunopharmacol. Immunotoxicol. 2001, 23, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Boutin, H.; Chauveau, F.; Thominiaux, C.; Gregoire, M.-C.; James, M.L.; Trebossen, R.; Hantraye, P.; Dollé, F.; Tavitian, B.; Kassiou, M. 11C-DPA-713: A Novel Peripheral Benzodiazepine Receptor PET Ligand for In Vivo Imaging of Neuroinflammation. J. Nucl. Med. 2007, 48, 573–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azrad, M.; Zeineh, N.; Weizman, A.; Veenman, L.; Gavish, M. The TSPO Ligands 2-Cl-MGV-1, MGV-1, and PK11195 Differentially Suppress the Inflammatory Response of BV-2 Microglial Cell to LPS. Int. J. Mol. Sci. 2019, 20, 594. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapere, J.-J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.-R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- De Souza, E.B.; Anholt, R.R.H.; Murphy, K.M.M.; Snyder, S.H.; Kuhar, M.J. Peripheral-Type Benzodiazepine Receptors in Endocrine Organs: Autoradiographic Localization in Rat Pituitary, Adrenal, and Testis. Endocrinology 1985, 116, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Gavish, M.; Bachman, I.; Shoukrun, R.; Katz, Y.; Veenman, L.; Weisinger, G.; Weizman, A. Enigma of the peripheral benzodiazepine receptor. Pharmacol. Rev. 1999, 51, 629–650. [Google Scholar]
- Zeno, S.; Zaaroor, M.; Leschiner, S.; Veenman, L.; Gavish, M. CoCl(2) induces apoptosis via the 18 kDa translocator protein in U118MG human glioblastoma cells. Biochemistry 2009, 48, 4652–4661. [Google Scholar] [CrossRef] [PubMed]
- Gavish, M.; Bar-Ami, S.; Weizman, R. The endocrine system and mitochondrial benzodiazepine receptors. Mol. Cell. Endocrinol. 1992, 88, 1–13. [Google Scholar] [CrossRef]
- Rupprecht, R.; Papadopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef]
- Veenman, L.; Gavish, M. The peripheral-type benzodiazepine receptor and the cardiovascular system. Implications for drug development. Pharmacol. Ther. 2006, 110, 503–524. [Google Scholar] [CrossRef]
- Veenman, L.; Papadopoulos, V.; Gavish, M. Channel-like functions of the 18-kDa translocator protein (TSPO): Regulation of apoptosis and steroidogenesis as part of the host-defense response. Curr. Pharm. Des. 2007, 13, 2385–2405. [Google Scholar] [CrossRef]
- Veenman, L.; Gavish, M. The Role of 18 kDa Mitochondrial Translocator Protein (TSPO) in Programmed Cell Death, and Effects of Steroids on TSPO Expression. Curr. Mol. Med. 2012, 12, 398–412. [Google Scholar] [PubMed]
- Zeno, S.; Veenman, L.; Katz, Y.; Bode, J.; Gavish, M.; Zaroor, M. The 18 kDa Mitochondrial Translocator Protein (TSPO) Prevents Accumulation of Protoporphyrin IX. Involvement of Reactive Oxygen Species (ROS). Curr. Mol. Med. 2012, 12, 494–501. [Google Scholar] [PubMed]
- Caballero, B.; Veenman, L.; Gavish, M. Role of mitochondrial translocator protein (18 kDa) on mitochondrial-related cell death processes. Recent Patents Endocrine Metab. Immune Drug Discov. 2013, 7, 86–101. [Google Scholar] [CrossRef]
- Veenman, L.; Shandalov, Y.; Gavish, M. VDAC activation by the 18 kDa translocator protein (TSPO), implications for apoptosis. J. Bioenerg. Biomembr. 2008, 40, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Veenman, L.; Alten, J.; Linnemannstöns, K.; Shandalov, Y.; Zeno, S.; Lakomek, M.; Gavish, M.; Kugler, W. Potential involvement of F(0)F(1)- ATP(synth)ase and reactive oxygen species in apoptosis induction by the antineoplastic agent erucylphosphohomocholine in glioblastoma cell lines: A mechanism for induction of apoptosis via the 18 kDa mitochondrial translocator protein. Apoptosis 2010, 15, 753–768. [Google Scholar] [PubMed]
- Veenman, L.; Gavish, M.; Kugler, W. Apoptosis induction by erucylphosphohomocholine via the 18 kDa mitochondrial translocator protein: Implications for cancer treatment. Anticancer Agents Med. Chem. 2014, 14, 559–577. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.; Wong, L.-J. Diagnosis of mitochondrial myopathies. Mol. Genet. Metab. 2013, 110, 35–41. [Google Scholar] [CrossRef]
- Shuvaev, S.; Fox, M.A.; Parker, D. Monitoring of the ADP/ATP Ratio by Induced Circularly Polarised Europium Luminescence. Angew. Chem. Int. Ed. Engl. 2018, 57, 7488–7492. [Google Scholar] [CrossRef]
- Hunter, T. Why nature chose phosphate to modify proteins. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2513–2516. [Google Scholar] [CrossRef]
- Insel, P.A.; Zhang, L.; Murray, F.; Yokouchi, H.; Zambon, A.C. Cyclic AMP is both a pro-apoptotic and anti-apoptotic second messenger. Acta Physiol. (Oxf. Engl.) 2012, 204, 277–287. [Google Scholar] [CrossRef]
- Cross, T.G.; Scheel-Toellner, D.; Henriquez, N.V.; Deacon, E.; Salmon, M.; Lord, J.M. Serine/Threonine Protein Kinases and Apoptosis. Exp. Cell Res. 2000, 256, 34–41. [Google Scholar] [CrossRef]
- Lerner, A.; Kim, D.H.; Lee, R. The cAMP Signaling Pathway as a Therapeutic Target in Lymphoid Malignancies. Leuk. Lymphoma 2000, 37, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Reznick, A.Z.; E Cross, C.; Hu, M.L.; Suzuki, Y.J.; Khwaja, S.; Safadi, A.; A Motchnik, P.; Packer, L.; Halliwell, B. Modification of plasma proteins by cigarette smoke as measured by protein carbonyl formation. Biochem. J. 1992, 286, 607–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, C.E.; O’Neil, C.A.; Reznick, A.Z.; Hu, M.-L.; Marcocci, L.; Packer, L.; Frei, B. Cigarette Smoke Oxidation of Human Plasma Constituents. Ann. New York Acad. Sci. 1993, 686, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Nagler, R.; Ben-Izhak, O.; Savulescu, D.; Krayzler, E.; Akrish, S.; Leschiner, S.; Otradnov, I.; Zeno, S.; Veenman, L.; Gavish, M. Oral cancer, cigarette smoke and mitochondrial 18kDa translocator protein (TSPO)—In vitro, in vivo, salivary analysis. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2010, 1802, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Legrand, C.; Bour, J.M.; Jacob, C.; Capiaumont, J.; Martial, A.; Marc, A.; Wudtke, M.; Kretzmer, G.; Demangel, C.; Duval, D. Lactate dehydrogenase (LDH) activity of the cultured eukaryotic cells as marker of the number of dead cells in the medium [corrected]. J. Biotechnol. 1992, 25, 231–243. [Google Scholar] [CrossRef]
- Avezov, K.; Reznick, A.Z.; Aizenbud, D. Oxidative damage in keratinocytes exposed to cigarette smoke and aldehydes. Toxicol. Vitr. 2014, 28, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cheng, K.S.; Wang, Y.W.; Chen, Y.F.; Wong, K.L.; Su, T.H.; Chan, P.; Leung, Y.M. Perturbation of Akt Signaling, Mitochondrial Potential, and ADP/ATP Ratio in Acidosis-Challenged Rat Cortical Astrocytes. J. Cell. Biochem. 2017, 118, 1108–1117. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Lansiaux, A.; Wattez, N.; Hildebrand, M.-P.; Léonce, S.; Pierre, A.; Hickman, J.A.; Bailly, C. Induction of apoptosis in HL-60 leukemia and B16 melanoma cells by the acronycine derivative S23906-1. Biochem. Pharmacol. 2002, 63, 1443–1452. [Google Scholar] [CrossRef]
- Ferlini, C.; Scambia, G. Assay for apoptosis using the mitochondrial probes, Rhodamine123 and 10-N-nonyl acridine orange. Nat. Protoc. 2007, 2, 3111–3114. [Google Scholar] [CrossRef]
- Weiner, D.; Levy, Y.; Khankin, E.V.; Reznick, A.Z. Inhibition of salivary amylase activity by cigarette smoke aldehydes. J. Physiol. Pharmacol. 2008, 59, 727–737. [Google Scholar] [PubMed]
- Ha, J.-H.; Lee, J.-T.; Cho, I.-H.; Chun, K.-A.; Park, G.-E.; Choi, H.-C.; Lee, K.-Y.; Kim, S.-H.; Suk, K.; Kim, I.-K.; et al. Upregulation of PBR mRNA expression in human neuroblastoma cells by flavonoids. Phytomedicine 2007, 14, 232–235. [Google Scholar] [CrossRef] [PubMed]
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General. 2014. Available online: http://www.cdc.gov/tobacco/data_statistics/sgr/50th anniversary/index.htm (accessed on 7 April 2015).
- Chen, Z.; Wang, D.; Liu, X.; Pei, W.; Li, J.; Cao, Y.; Zhang, J.; An, Y.; Nie, J.; Tong, J. Oxidative DNA damage is involved in cigarette smoke-induced lung injury in rats. Environ. Health Prev. Med. 2015, 20, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Tapia, C.; Pérez, M.J. Possible role of mitochondrial permeability transition pore in the pathogenesis of Huntington disease. Biochem. Biophys. Res. Commun. 2017, 483, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Avezov, K.; Reznick, A.Z.; Aizenbud, D. Time and dose effects of cigarette smoke and acrolein on protein carbonyl formation in HaCaT keratinocytes. Adv. Exp. Med. Biol. 2015, 849, 57–64. [Google Scholar] [PubMed]
- Hasnis, E.; Reznick, A.Z.; Pollack, S.; Klein, Y.; Nagler, R.M. Synergistic effect of cigarette smoke and saliva on lymphocytes—The mediatory role of volatile aldehydes and redox active iron and the possible implications for oral cancer. Int. J. Biochem. Cell Biol. 2004, 36, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Krayzler, E.; Nagler, R.M. Carbonyl levels and survival rates in oral cancer cells exposed to cigarette smoke. Anticancer. Res. 2015, 35, 1961–1965. [Google Scholar] [PubMed]
- Levin, E.; Premkumar, A.; Veenman, L.; Kugler, W.; Leschiner, S.; Spanier, I.; Weisinger, G.; Lakomek, M.; Weizman, A.; Snyder, S.H.; et al. The peripheral-type benzodiazepine receptor and tumorigenicity: Isoquinoline binding protein (IBP) antisense knockdown in the C6 glioma cell line. Biochemistry 2005, 44, 9924–9935. [Google Scholar] [CrossRef] [PubMed]
- Andrè, E.; Campi, B.; Materazzi, S.; Trevisani, M.; Amadesi, S.; Massi, D.; Creminon, C.; Vaksman, N.; Nassini, R.; Civelli, M.; et al. Cigarette smoke-induced neurogenic inflammation is mediated by alpha,beta-unsaturated aldehydes and the TRPA1 receptor in rodents. J. Clin. Invest. 2008, 118, 2574–2582. [Google Scholar]
- Lin, A.H.; Liu, M.H.; Ko, H.K.; Perng, D.W.; Lee, T.S.; Kou, Y.R. Lung Epithelial TRPA1 Transduces the Extracellular ROS into Transcriptional Regulation of Lung Inflammation Induced by Cigarette Smoke: The Role of Influxed Ca2+. Mediators Inflamm. 2015, 2015, 148367. [Google Scholar] [CrossRef] [PubMed]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeineh, N.; Nagler, R.; Gabay, M.; Weizman, A.; Gavish, M. Effects of Cigarette Smoke on TSPO-related Mitochondrial Processes. Cells 2019, 8, 694. https://doi.org/10.3390/cells8070694
Zeineh N, Nagler R, Gabay M, Weizman A, Gavish M. Effects of Cigarette Smoke on TSPO-related Mitochondrial Processes. Cells. 2019; 8(7):694. https://doi.org/10.3390/cells8070694
Chicago/Turabian StyleZeineh, Nidal, Rafael Nagler, Martin Gabay, Abraham Weizman, and Moshe Gavish. 2019. "Effects of Cigarette Smoke on TSPO-related Mitochondrial Processes" Cells 8, no. 7: 694. https://doi.org/10.3390/cells8070694
APA StyleZeineh, N., Nagler, R., Gabay, M., Weizman, A., & Gavish, M. (2019). Effects of Cigarette Smoke on TSPO-related Mitochondrial Processes. Cells, 8(7), 694. https://doi.org/10.3390/cells8070694