Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Animals and Treatment
2.2. Repetitive Mild Traumatic Brain Injury
2.3. Antibodies and Reagents
2.4. Immunoblotting
2.5. Immunofluorescence Staining
2.6. Reactive Oxygen Species (ROS) Assay
2.7. Fluoro-Jade B (FJB) Staining
2.8. Measurement of Inflammatory Markers Using ELISA
2.9. Aβ1–42 ELISA Analysis
2.10. Beam Walking Test
2.11. Lipid Per Oxidation (LPO) Assay
2.12. Nissl Staining
2.13. Assessment of Brain Lesion Volume
2.14. Morris Water Maze (MWM) Test
2.15. Y-Maze Test
2.16. Statistical Analysis
3. Results
3.1. Melatonin Treatment Ameliorates Oxidative Stress and Regulates the Expression of p-AMPK and Downstream Signaling in TBI Mouse Brains
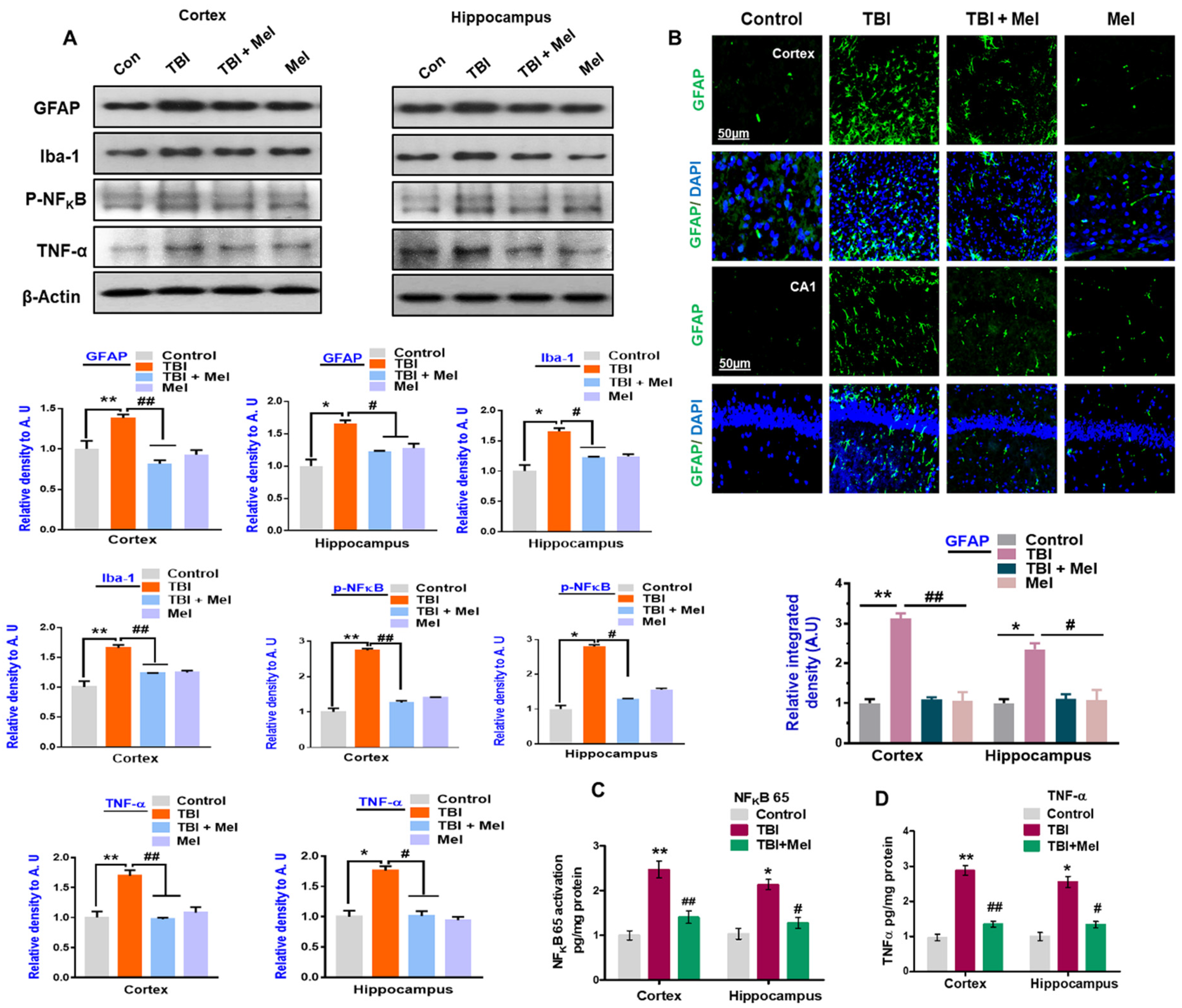
3.2. Melatonin Reduced TBI-Induced Neuroinflammation in rmTBI Mouse Brains
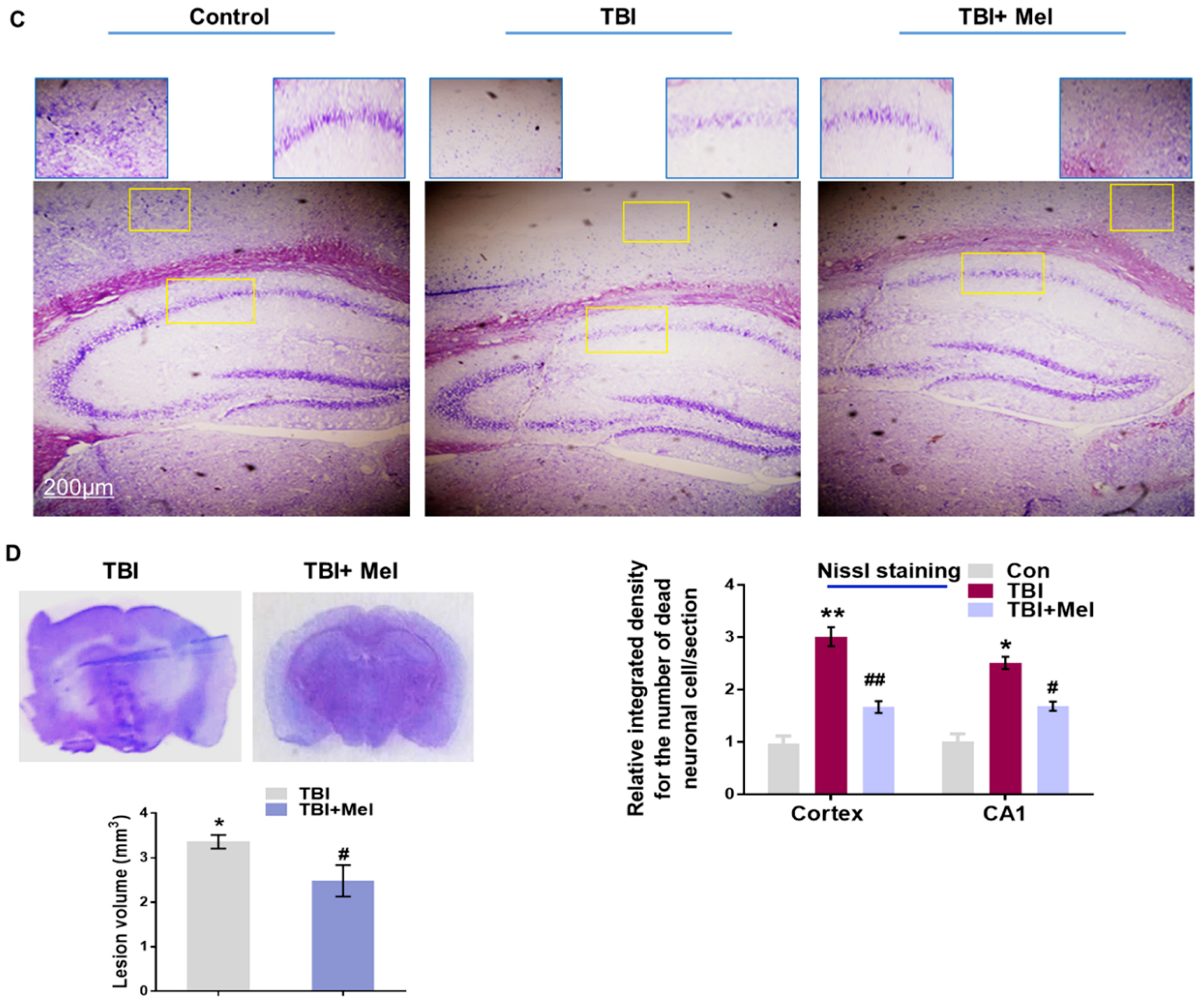
3.3. Melatonin Regulated p-JNK, and Reduced Apoptotic Cell Death and Lesion Volume in an rmTBI Mouse Model
3.4. Melatonin Ameliorated TBI-Induced AD-Like Pathological Changes in the Mouse Brain
3.5. Melatonin Rescued Synaptic Protein Loss in the rmTBI Mouse Model
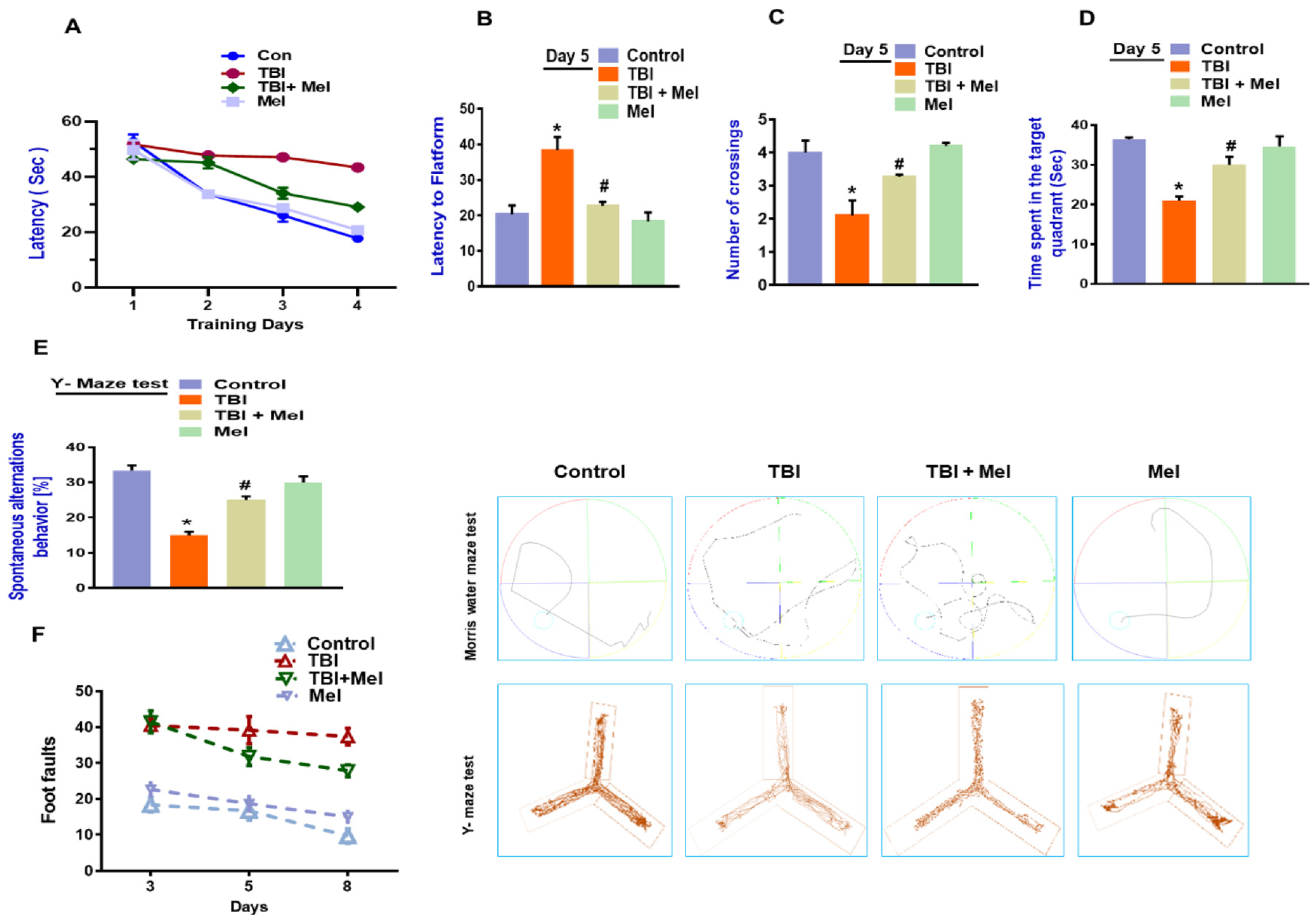
3.6. Melatonin Rescued Memory Dysfunction in a rmTBI Mouse Model
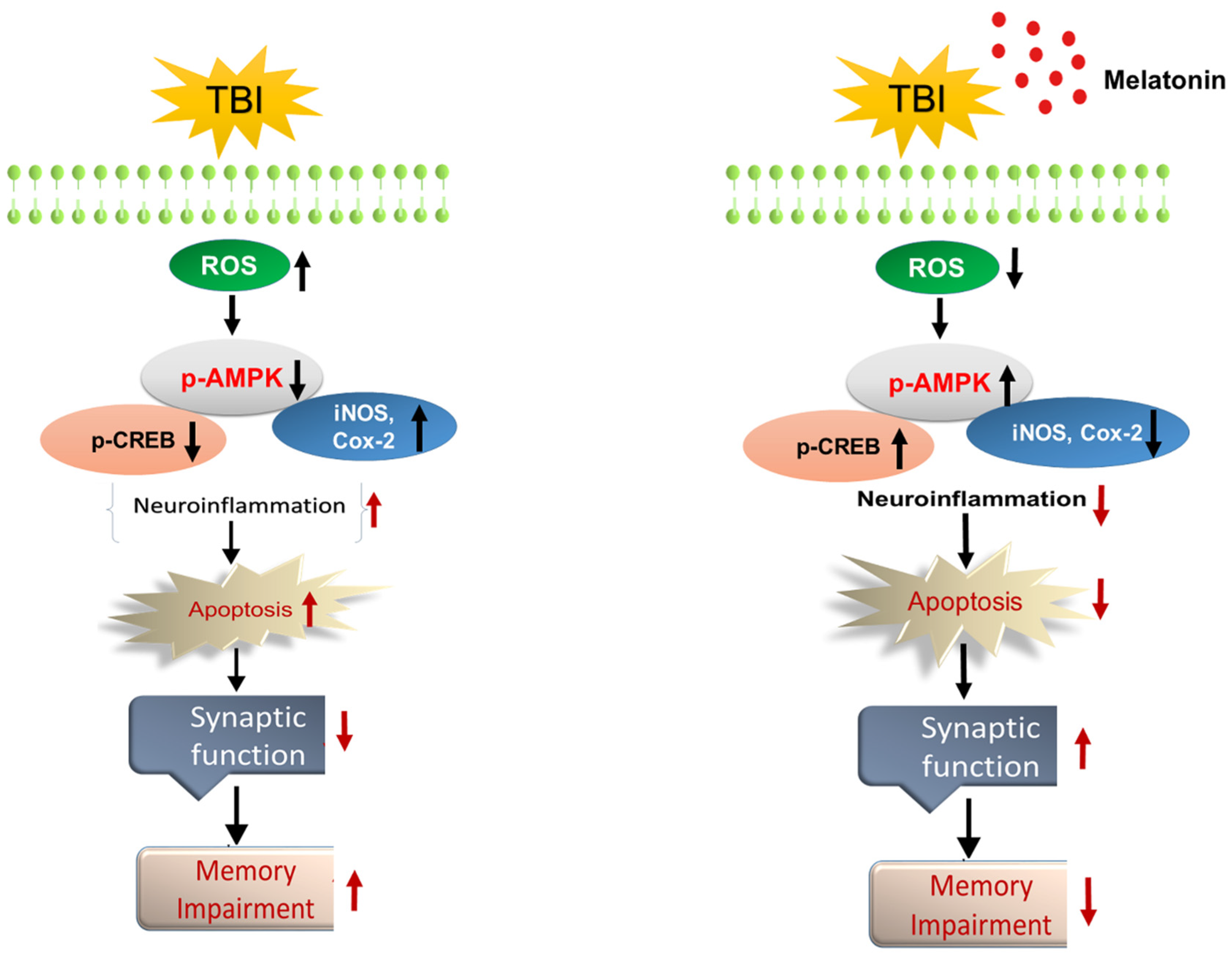
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamada, K.H.; Kozlowski, D.A.; Seidl, S.E.; Lance, S.; Wieschhaus, A.J.; Sundivakkam, P.; Tiruppathi, C.; Chishti, I.; Herman, I.M.; Kuchay, S.M.; et al. Targeted gene inactivation of calpain-1 suppresses cortical degeneration due to traumatic brain injury and neuronal apoptosis induced by oxidative stress. J. Biol. Chem. 2012, 287, 13182–13193. [Google Scholar] [CrossRef] [PubMed]
- Domise, M.; Vingtdeux, V. AMPK in Neurodegenerative Diseases. Exp. Suppl. 2016, 107, 153–177. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.L.; Kobori, N.; Zhao, J.; Rozas, N.S.; Hylin, M.J.; Moore, A.N.; Dash, P.K. Traumatic brain injury decreases AMP-activated protein kinase activity and pharmacological enhancement of its activity improves cognitive outcome. J. Neurochem. 2016, 139, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, A.; Ali, T.; Rehman, S.U.; Kim, M.O. Phytomedicine-Based Potent Antioxidant, Fisetin Protects CNS-Insult LPS-Induced Oxidative Stress-Mediated Neurodegeneration and Memory Impairment. J. Clin. Med. 2019, 8, 850. [Google Scholar] [CrossRef]
- Chang, J.-C.; Lien, C.-F.; Lee, W.-S.; Chang, H.-R.; Hsu, Y.-C.; Luo, Y.-P.; Jeng, J.-R.; Hsieh, J.-C.; Yang, K.-T. Intermittent Hypoxia Prevents Myocardial Mitochondrial Ca2+ Overload and Cell Death during Ischemia/Reperfusion: The Role of Reactive Oxygen Species. Cells 2019, 8, 564. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Hayashi, T.; Miyamoto, L.; Yonemitsu, S.; Nakano, M.; Tanaka, S.; Ebihara, K.; Masuzaki, H.; Hosoda, K.; Inoue, G. Possible involvement of the α1 isoform of 5′ AMP-activated protein kinase in oxidative stress-stimulated glucose transport in skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2004, 287, E166–E173. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.-C.; Chen, H.-M.; Chen, Y.-C.; Chang, C.-P.; Chang, C.; Chern, Y. AMPK-α1 functions downstream of oxidative stress to mediate neuronal atrophy in Huntington’s disease. Biochim. Et Biophys. Acta (Bba)-Mol. Basis Dis. 2014, 1842, 1668–1680. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, Y.W.; Lee, I.K.; Kim, J.Y.; Kang, Y.J.; Park, S.Y. AMP-activated protein kinase activation by 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) inhibits palmitate-induced endothelial cell apoptosis through reactive oxygen species suppression. J. Pharmacol. Sci. 2008, 106, 394–403. [Google Scholar] [CrossRef]
- Ceolotto, G.; Gallo, A.; Papparella, I.; Franco, L.; Murphy, E.; Iori, E.; Pagnin, E.; Fadini, G.P.; Albiero, M.; Semplicini, A.; et al. Rosiglitazone reduces glucose-induced oxidative stress mediated by NAD(P)H oxidase via AMPK-dependent mechanism. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2627–2633. [Google Scholar] [CrossRef]
- Liou, C.-J.; Lee, Y.-K.; Ting, N.-C.; Chen, Y.-L.; Shen, S.-C.; Wu, S.-J.; Huang, W.-C. Protective Effects of Licochalcone A Ameliorates Obesity and Non-Alcoholic Fatty Liver Disease Via Promotion of the Sirt-1/AMPK Pathway in Mice Fed a High-Fat Diet. Cells 2019, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Leem, Y.-H.; Park, J.-E.; Kim, D.-Y.; Kim, H.-S. Neuroprotective Effect of β-Lapachone in MPTP-Induced Parkinson’s Disease Mouse Model: Involvement of Astroglial p-AMPK/Nrf2/HO-1 Signaling Pathways. Biomol. Ther. 2019, 27, 178. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K. CREB and cAMP response element-mediated gene expression in the ischemic brain. FEBS J. 2007, 274, 3210–3217. [Google Scholar] [CrossRef] [PubMed]
- Lonze, B.E.; Riccio, A.; Cohen, S.; Ginty, D.D. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron 2002, 34, 371–385. [Google Scholar] [CrossRef]
- Nguyên, T.L.-A.; De Walque, S.; Veithen, E.; Dekoninck, A.; Martinelli, V.; De Launoit, Y.; Burny, A.; Harrod, R.; Van Lint, C. Transcriptional regulation of the bovine leukemia virus promoter by the cyclic AMP-response element modulator τ isoform. J. Biol. Chem. 2007, 282, 20854–20867. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chen, T.; Zhou, H.; Feng, Y.; Hoi, M.P.M.; Ma, D.; Zhao, C.; Zheng, Y.; Lee, S.M.Y. BHDPC Is a Novel Neuroprotectant That Provides Anti-neuroinflammatory and Neuroprotective Effects by Inactivating NF-kappaB and Activating PKA/CREB. Front. Pharmacol 2018, 9, 614. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Xu, Y.; Pearse, D.D. Cyclic AMP is a key regulator of M1 to M2a phenotypic conversion of microglia in the presence of Th2 cytokines. J. Neuroinflammation 2016, 13, 9. [Google Scholar] [CrossRef]
- Kerr, N.A.; de Rivero Vaccari, J.P.; Umland, O.; Bullock, M.R.; Conner, G.E.; Dietrich, W.D.; Keane, R.W. Human lung cell pyroptosis following traumatic brain injury. Cells 2019, 8, 69. [Google Scholar] [CrossRef]
- Singleton, Q.; Vaibhav, K.; Braun, M.; Patel, C.; Khayrullin, A.; Mendhe, B.; Lee, B.R.; Kolhe, R.; Kaiser, H.; Awad, M.E. Bone marrow derived extracellular vesicles activate osteoclast differentiation in traumatic brain injury induced bone loss. Cells 2019, 8, 63. [Google Scholar] [CrossRef]
- Cassidy, J.D.; Carroll, L.J.; Peloso, P.M.; Borg, J.; von Holst, H.; Holm, L.; Kraus, J.; Coronado, V.G. Incidence, risk factors and prevention of mild traumatic brain injury: Results of the WHO Collaborating Centre Task Force on Mild Traumatic Brain Injury. J. Rehabil. Med. 2004, 28–60. [Google Scholar] [CrossRef]
- Briones, T.L.; Woods, J.; Rogozinska, M. Decreased neuroinflammation and increased brain energy homeostasis following environmental enrichment after mild traumatic brain injury is associated with improvement in cognitive function. Acta Neuropathol. Commun. 2013, 1, 57. [Google Scholar] [CrossRef] [PubMed]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Byers, A.L.; Barnes, D.E.; Li, Y.; Boscardin, J.; Yaffe, K. Mild TBI and risk of Parkinson disease: A Chronic Effects of Neurotrauma Consortium Study. Neurology 2018, 90, e1771–e1779. [Google Scholar] [CrossRef] [PubMed]
- Bonnefont-Rousselot, D.; Collin, F. Melatonin: Action as antioxidant and potential applications in human disease and aging. Toxicology 2010, 278, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Jin, Y.; Park, K.; Choi, J.; Kang, H.; Lee, S.-R.; Hong, Y. Elevated Serum Melatonin under Constant Darkness Enhances Neural Repair in Spinal Cord Injury through Regulation of Circadian Clock Proteins Expression. J. Clin. Med. 2019, 8, 135. [Google Scholar] [CrossRef]
- Liu, X.; Gong, Y.; Xiong, K.; Ye, Y.; Xiong, Y.; Zhuang, Z.; Luo, Y.; Jiang, Q.; He, F. Melatonin mediates protective effects on inflammatory response induced by interleukin-1 beta in human mesenchymal stem cells. J. Pineal Res. 2013, 55, 14–25. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Li, T.; Zhang, L.; Ding, Y.; Zhu, L.; He, J.; Zhou, M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: The Nrf2-ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 2014, 73, 1–11. [Google Scholar] [CrossRef]
- Wu, H.J.; Wu, C.; Niu, H.J.; Wang, K.; Mo, L.J.; Shao, A.W.; Dixon, B.J.; Zhang, J.M.; Yang, S.X.; Wang, Y.R. Neuroprotective Mechanisms of Melatonin in Hemorrhagic Stroke. Cell Mol. Neurobiol 2017, 37, 1173–1185. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.O. Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3beta pathway in the mouse hippocampus. J. Pineal Res. 2015, 59, 47–59. [Google Scholar] [CrossRef]
- Lin, Y.W.; Chen, T.Y.; Hung, C.Y.; Tai, S.H.; Huang, S.Y.; Chang, C.C.; Hung, H.Y.; Lee, E.J. Melatonin protects brain against ischemia/reperfusion injury by attenuating endoplasmic reticulum stress. Int. J. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Ali, T.; Kim, M.W.; Khan, A.; Jo, M.H.; Rehman, S.U.; Khan, M.S.; Abid, N.B.; Khan, M.; Ullah, R.; et al. Adiponectin homolog novel osmotin protects obesity/diabetes-induced NAFLD by upregulating AdipoRs/PPARalpha signaling in ob/ob and db/db transgenic mouse models. Metab. Clin. Exp. 2019, 90, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Rehman, S.U.; Amin, F.U.; Kim, M.O. Enhanced neuroprotection of anthocyanin-loaded PEG-gold nanoparticles against Abeta1-42-induced neuroinflammation and neurodegeneration via the NF-KB /JNK/GSK3β signaling pathway. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Badshah, H.; Ali, T.; Shafiq-ur, R.; Faiz-ul, A.; Ullah, F.; Kim, T.H.; Kim, M.O. Protective Effect of Lupeol Against Lipopolysaccharide-Induced Neuroinflammation via the p38/c-Jun N-Terminal Kinase Pathway in the Adult Mouse Brain. J. Neuroimmune Pharmacol. 2016, 11, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, M.J.; Rehman, S.U.; Ahmad, A.; Kim, M.O. Anthocyanin-Loaded PEG-Gold Nanoparticles Enhanced the Neuroprotection of Anthocyanins in an Abeta1-42 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6490–6506. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Xie, W.; Meng, X.; Zhai, Y.; Dong, X.; Zhang, X.; Sun, G.; Sun, X. Notoginsenoside R1 Ameliorates Diabetic Retinopathy through PINK1-Dependent Activation of Mitophagy. Cells 2019, 8, 213. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-kappaB Signaling in an Abeta Mouse Model. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Ikram, M.; Saeed, K.; Khan, A.; Muhammad, T.; Khan, M.S.; Jo, M.G.; Rehman, S.U.; Kim, M.O. Natural Dietary Supplementation of Curcumin Protects Mice Brains against Ethanol-Induced Oxidative Stress-Mediated Neurodegeneration and Memory Impairment via Nrf2/TLR4/RAGE Signaling. Nutrients 2019, 11, 1082. [Google Scholar] [CrossRef]
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.-Y.; Hwang, H.; Rhim, H.; Kim, M.O.J.M.n. Gintonin Mitigates MPTP-Induced Loss of Nigrostriatal Dopaminergic Neurons and Accumulation of α-Synuclein via the Nrf2/HO-1 Pathway. Mol. Neurobiol. 2018, 1–17. [Google Scholar] [CrossRef]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflammation 2018, 15, 119. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wei, X.T.; Guan, J.H.; Yuan, J.W.; Peng, Y.T.; Song, L.; Liu, Y.H. Inhibition of c-Jun N-terminal kinase prevents blood-brain barrier disruption and normalizes the expression of tight junction proteins clautin-5 and ZO-1 in a rat model of subarachnoid hemorrhage. Acta Neurochir. 2012, 154, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of c-Jun N-Terminal Kinase Protects Against Brain Damage and Improves Learning and Memory After Traumatic Brain Injury in Adult Mice. Cerebral Cortex. 2018, 28, 2854–2872. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a Citrus Flavonoid, Attenuates LPS-Induced Neuroinflammation, Apoptosis and Memory Impairments by Modulating TLR4/NF-κB Signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, T.; Rehman, S.U.; Khan, M.S.; Alam, S.I.; Ikram, M.; Muhammad, T.; Saeed, K.; Badshah, H.; Kim, M.O. Neuroprotective effect of quercetin against the detrimental effects of LPS in the adult mouse brain. Front. Pharmacol. 2018, 9, 1383. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Xu, D.; Ji, Y.; Yin, F.; Cui, J.; Su, R.; Wang, Y.; Zhu, Y.; Wei, S.; Lai, J. LiCl Pretreatment Ameliorates Adolescent Methamphetamine Exposure-Induced Long-Term Alterations in Behavior and Hippocampal Ultrastructure in Adulthood in Mice. J. Neuropsychopharmacol. 2019, 22, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress in Neurodegenerative Diseases: From a Mitochondrial Point of View. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Fiorelli, S.; Porro, B.; Cosentino, N.; Di Minno, A.; Manega, C.M.; Fabbiocchi, F.; Niccoli, G.; Fracassi, F.; Barbieri, S.; Marenzi, G. Activation of Nrf2/HO-1 Pathway and Human Atherosclerotic Plaque Vulnerability: An In Vitro and In Vivo Study. Cells 2019, 8, 356. [Google Scholar] [CrossRef]
- Qin, L.; Bouchard, R.; Pugazhenthi, S. Regulation of cyclic AMP response element-binding protein during neuroglial interactions. J. Neurochemistry 2016, 136, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.I.; Ur Rehman, S.; Ok Kim, M. Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-kappaB Signaling Pathway after Brain Injury. J. Clin. Med. 2019, 8, 271. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ali, T.; Alam, S.I.; Ullah, R.; Zeb, A.; Lee, K.W.; Rutten, B.P.F.; Kim, M.O. Ferulic Acid Rescues LPS-Induced Neurotoxicity via Modulation of the TLR4 Receptor in the Mouse Hippocampus. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef]
- Merlo, L.; Cimino, F.; Angileri, F.F.; La Torre, D.; Conti, A.; Cardali, S.M.; Saija, A.; Germano, A. Alteration in synaptic junction proteins following traumatic brain injury. J. Neurotrauma 2014, 31, 1375–1385. [Google Scholar] [CrossRef]
- Wu, A.; Ying, Z.; Gomez-Pinilla, F. Omega-3 fatty acids supplementation restores mechanisms that maintain brain homeostasis in traumatic brain injury. J. Neurotrauma 2007, 24, 1587–1595. [Google Scholar] [CrossRef]
- Borriello, M.; Iannuzzi, C.; Sirangelo, I. Pinocembrin Protects from AGE-Induced Cytotoxicity and Inhibits Non-Enzymatic Glycation in Human Insulin. Cells 2019, 8, 385. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, K.-H.; Park, J.B.; Suh, S.W. The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury. Cells 2019, 8, 405. [Google Scholar] [CrossRef]
- Saito, M.; Saito, M.; Das, B.C. Involvement of AMP-activated protein kinase in neuroinflammation and neurodegeneration in the adult and developing brain. Int. J. Dev. Neurosci. 2019. [Google Scholar] [CrossRef]
- Thomson, D.M.; Herway, S.T.; Fillmore, N.; Kim, H.; Brown, J.D.; Barrow, J.R.; Winder, W.W. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J. Appl. Physiol 2008, 104, 429–438. [Google Scholar] [CrossRef] [Green Version]
- Atkins, C.M.; Oliva, A.A., Jr.; Alonso, O.F.; Pearse, D.D.; Bramlett, H.M.; Dietrich, W.D. Modulation of the cAMP signaling pathway after traumatic brain injury. Exp. Neurol. 2007, 208, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Peixoto, C.A.; Oliveira, W.H.; Araujo, S.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef]
- Calvo, J.R.; Gonzalez-Yanes, C.; Maldonado, M. The role of melatonin in the cells of the innate immunity: A review. J. Pineal Res. 2013, 55, 103–120. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-κB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef]
- Xu, J.; Wu, L.; Zhang, Y.; Gu, H.; Huang, Z.; Zhou, K.; Yin, X. Activation of AMPK by OSU53 protects spinal cord neurons from oxidative stress. Oncotarget 2017, 8, 112477–112486. [Google Scholar] [CrossRef] [Green Version]
- Ogeturk, M.; Kus, I.; Pekmez, H.; Yekeler, H.; Sahin, S.; Sarsilmaz, M. Inhibition of carbon tetrachloride-mediated apoptosis and oxidative stress by melatonin in experimental liver fibrosis. Toxicol. Ind. Health 2008, 24, 201–208. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, Y.; Chuang, D.M. Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J. Neurotrauma 2012, 29, 2342–2351. [Google Scholar] [CrossRef]
- Olsson, A.; Csajbok, L.; Ost, M.; Hoglund, K.; Nylen, K.; Rosengren, L.; Nellgard, B.; Blennow, K. Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J. Neurol. 2004, 251, 870–876. [Google Scholar] [CrossRef]
- Ou, Z.; Kong, X.; Sun, X.; He, X.; Zhang, L.; Gong, Z.; Huang, J.; Xu, B.; Long, D.; Li, J.; et al. Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav. Immun. 2018, 69, 351–363. [Google Scholar] [CrossRef]
- Cai, Z.; Li, B.; Li, K.; Zhao, B. Down-regulation of amyloid-beta through AMPK activation by inhibitors of GSK-3beta in SH-SY5Y and SH-SY5Y-AbetaPP695 cells. J. Alzheimer’s Dis. 2012, 29, 89–98. [Google Scholar] [CrossRef]
- Lahiri, D.K. Melatonin affects the metabolism of the beta-amyloid precursor protein in different cell types. J. Pineal Res. 1999, 26, 137–146. [Google Scholar] [CrossRef]
- McGuire, J.L.; Ngwenya, L.B.; McCullumsmith, R.E. Neurotransmitter changes after traumatic brain injury: An update for new treatment strategies. Mol. Psychiatry 2018. [Google Scholar] [CrossRef]
- Zihl, J.; Almeida, O. Neuropsychology of neuroendocrine dysregulation after traumatic brain injury. J. Clin. Med. 2015, 4, 1051–1062. [Google Scholar] [CrossRef]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Ok Kim, M. Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells 2019, 8, 760. https://doi.org/10.3390/cells8070760
Rehman SU, Ikram M, Ullah N, Alam SI, Park HY, Badshah H, Choe K, Ok Kim M. Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells. 2019; 8(7):760. https://doi.org/10.3390/cells8070760
Chicago/Turabian StyleRehman, Shafiq Ur, Muhammad Ikram, Najeeb Ullah, Sayed Ibrar Alam, Hyun Young Park, Haroon Badshah, Kyonghwan Choe, and Myeong Ok Kim. 2019. "Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling" Cells 8, no. 7: 760. https://doi.org/10.3390/cells8070760
APA StyleRehman, S. U., Ikram, M., Ullah, N., Alam, S. I., Park, H. Y., Badshah, H., Choe, K., & Ok Kim, M. (2019). Neurological Enhancement Effects of Melatonin against Brain Injury-Induced Oxidative Stress, Neuroinflammation, and Neurodegeneration via AMPK/CREB Signaling. Cells, 8(7), 760. https://doi.org/10.3390/cells8070760