iPla2β Deficiency Suppresses Hepatic ER UPR, Fxr, and Phospholipids in Mice Fed with MCD Diet, Resulting in Exacerbated Hepatic Bile Acids and Biliary Cell Proliferation


Abstract
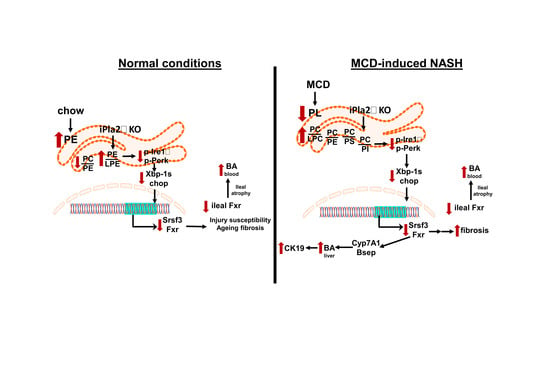
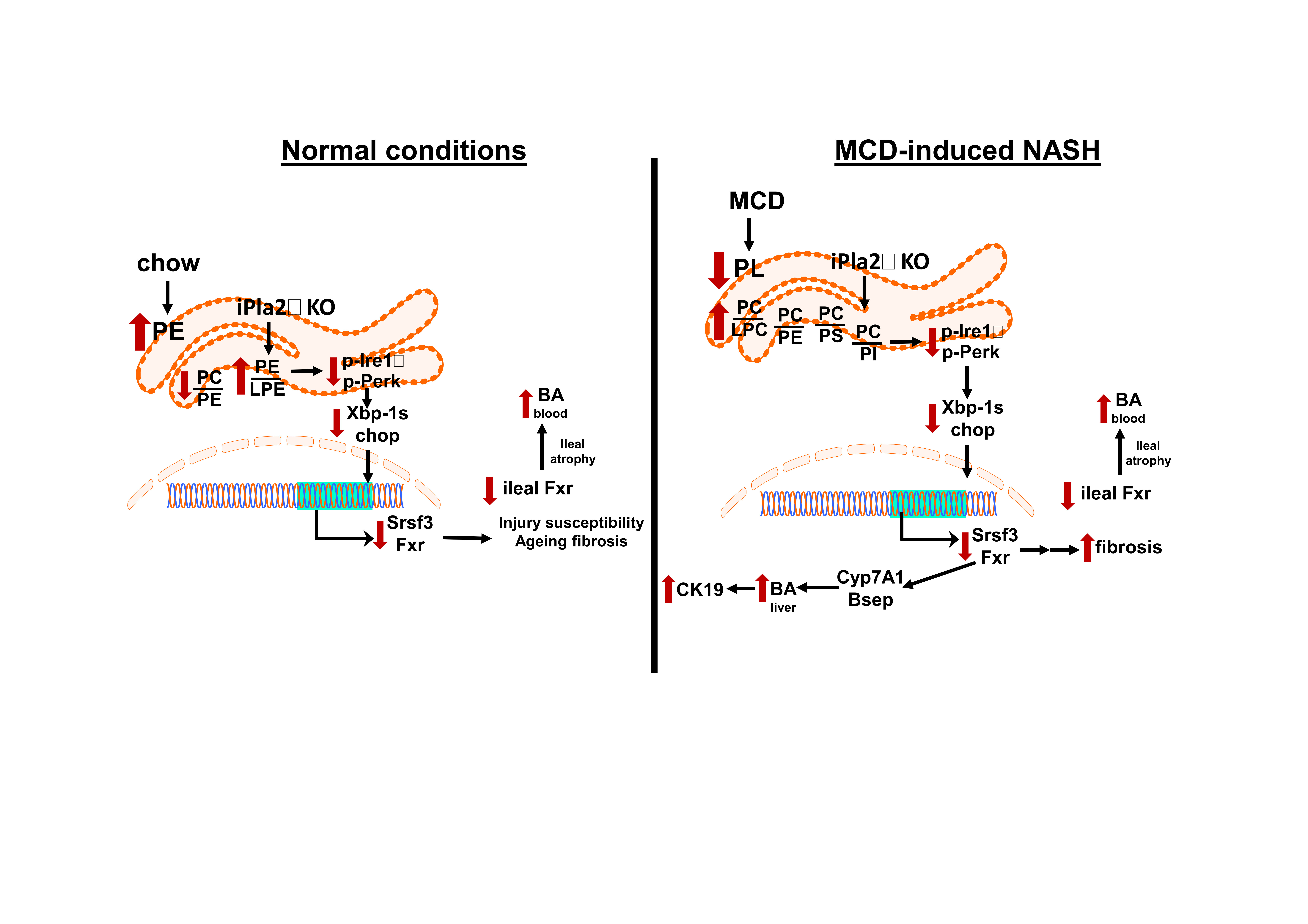{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods and Methods
2.1. Animals and Feeding
2.2. Isolation of Hepatic ER
2.3. Western Blot Analyses
2.4. Histology and Immunohistochemistry (IHC)
2.5. Hepatic Profiling of Fatty Acids and PL
2.6. BA Profiling by LC-MS/MS and Liver Cholesterol
2.7. Gene Expression by RT-PCR
2.8. Statistics
3. Results
3.1. iPla2β Deficiency during MCD Induced Liver Fibrosis without Affecting Steatosis
3.2. Effects of iPla2β Deficiency on Hepatic ER UPR in Mice Fed with Chow or MCD
3.3. Altered ER Membrane Phospholipids by iPla2β Deficiency in Mice Fed with Chow or MCD
3.4. iPla2β Deficiency under MCD Altered Hepatic and Intestinal Fxr and BA Transport Genes
3.5. iPla2β Deficiency during MCD Exacerbated Muricholic Acid and Its Tauro-Conjugate in Liver and Peripheral Blood
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Atf6 | activating transcription factor 6 |
| BiP | binding immunoglobulin protein |
| BA | bile acids |
| Bsep | bile salt export pump |
| CA | cholic acid |
| Chop | CCAAT-enhancer-binding protein homologous protein |
| CK19 | cytokeratin 19 |
| Cyp7a1 | cholesterol 7α-hydroxylase |
| ER | endoplasmic reticulum |
| eIF2α | eukaryotic translation initiation factor 2α |
| Fxr | farnesoid X-activated receptor |
| Fgf-15 | fibroblast growth factor 15 |
| Gadph | glyceraldehyde-3-phosphate dehydrogenase |
| H&E | hematoxylin-eosin |
| HFD | high-fat-diet |
| IHC | immunohistochemistry |
| Ire-1α | inositol-requiring enzyme-1α |
| iPla2β | group VIA calcium-independent phospholipaseA2 |
| LC-MS/MS | liquid-chromatography mass spectrometry |
| LPC | lysophosphatidylcholine |
| LPE | lysophosphatidylethanolamine |
| MCA | muricholic acid |
| Mrp3 | multidrug resistance protein 3 |
| MUFA | monounsaturated fatty acids |
| MCD | methionine-and choline-deficiency |
| MCP-1 | monocyte chemoattractant protein-1 |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PI | phosphatidylinositol |
| PS | phosphatidylserine |
| PUFA | polyunsaturated fatty acids |
| Scd1 | stearoyl-CoA desaturase1 |
| Srsf3 | serine/arginine-rich splicing factor3 |
| SM | sphingomyelin |
| TCA | tauro-cholic acid |
| TLCA | tauro-lithocholic acid |
| TMCA | tauro-muricholic acid |
| UDCA | ursodeoxycholic acid |
| UPR | unfolded protein response |
| Xbp-1s | spliced X-box binding protein-1 |
| WT | wild-type. |
References
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mohan, S. Non-alcoholic fatty liver disease in lean subjects: Characteristics and Implications. J. Clin. Transl. Hepatol. 2017, 5, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Wattacheril, J.; Sanyal, A.J. Lean NAFLD: An underrecognized outlier. Curr. Hepatol. Rep. 2016, 15, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.J. Prevalence and risk factors for non-alcoholic fatty liver disease in Asian people who are not obese. J. Gastroenterol. Hepatol. 2012, 27, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Negro, F.; Hallaji, S.; Younossi, Y.; Lam, B.; Srishord, M. Nonalcoholic fatty liver disease in lean individuals in the United States. Medicine (Baltim.) 2012, 91, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Golabi, P.; Paik, J.; Fukui, N.; Locklear, C.T.; de Avilla, L.; Younossi, Z.M. Patients with Lean nonalcoholic fatty liver disease are metabolically abnormal and have a higher risk for mortality. Clin. Diabetes 2019, 37, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Vos, B.; Moreno, C.; Nagy, N.; Féry, F.; Cnop, M.; Vereerstraeten, P.; Devière, J.; Adler, M. Lean non-alcoholic fatty liver disease (lean-NAFLD): A major cause of cryptogenic liver disease. Acta Gastroenterol. Belg. 2011, 74, 389–394. [Google Scholar] [PubMed]
- Kim, D.; Kim, W.; Joo, S.K.; Kim, J.H.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Predictors of nonalcoholic steatohepatitis and significant fibrosis in non-obese nonalcoholic fatty liver disease. Liver Int. 2019, 39, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.; Eder, S.K.; Felder, T.K.; Kedenko, L.; Paulweber, B.; Stadlmayr, A.; Huber-Schonauer, U.; Niederseer, D.; Stickel, F.; Auer, S.; et al. Clinical and metabolic characterization of lean caucasian subjects with non-alcoholic fatty liver. Am. J. Gastroenterol. 2017, 112, 102–110. [Google Scholar] [CrossRef]
- Nakatsuka, A.; Matsuyama, M.; Yamaguchi, S.; Katayama, A.; Eguchi, J.; Murakami, K.; Teshigawara, S.; Ogawa, D.; Wada, N.; Yasunaka, T.; et al. Insufficiency of phosphatidylethanolamine N-methyltransferase is risk for lean non-alcoholic steatohepatitis. Sci. Rep. 2016, 6, 21721. [Google Scholar] [CrossRef]
- Tanaka, N.; Matsubara, T.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J. Disruption of phospholipid and bile acid homeostasis in mice with nonalcoholic steatohepatitis. Hepatology 2012, 56, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Turk, J.; White, T.D.; Nelson, A.J.; Lei, X.; Ramanadham, S. iPLA2β and its role in male fertility, neurological disorders, metabolic disorders, and inflammation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Hu, C.; Jiang, F.; Zhang, R.; Wang, J.; Tang, S.; Peng, D.; Chen, M.; Bao, Y.; Jia, W. Genetic variants of PLA2G6 are associated with Type 2 diabetes mellitus and triglyceride levels in a Chinese population. Diabet. Med. 2015, 32, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wang, J.; Jiao, L.; Utaipan, T.; Tuma-Kellner, S.; Schmitz, G.; Liebisch, G.; Stremmel, W.; Chamulitrat, W. iPla2beta deficiency attenuates obesity and hepatic steatosis in ob/ob mice through hepatic fatty-acyl phospholipid remodeling. Biochim. Biophys. Acta 2016, 1861, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Otto, A.C.; Gan-Schreier, H.; Zhu, X.; Tuma-Kellner, S.; Staffer, S.; Ganzha, A.; Liebisch, G.; Chamulitrat, W. Group VIA phospholipase A2 deficiency in mice chronically fed with high-fat-diet attenuates hepatic steatosis by correcting a defect of phospholipid remodeling. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 662–676. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Gan-Schreier, H.; Otto, A.C.; Cheng, Y.; Staffer, S.; Tuma-Kellner, S.; Ganzha, A.; Liebisch, G.; Chamulitrat, W. iPla2β deficiency in mice fed with MCD diet does not correct the defect of phospholipid remodeling but attenuates hepatocellular injury via an inhibition of lipid uptake genes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; Almeida Pereira, T.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef]
- Henkel, A.S.; Dewey, A.M.; Anderson, K.A.; Olivares, S.; Green, R.M. Reducing endoplasmic reticulum stress does not improve steatohepatitis in mice fed a methionine- and choline-deficient diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G54–G59. [Google Scholar] [CrossRef]
- Jiao, L.; Gan-Schreier, H.; Zhu, X.; Wei, W.; Tuma-Kellner, S.; Liebisch, G.; Stremmel, W.; Chamulitrat, W. Ageing sensitized by iPla2beta deficiency induces liver fibrosis and intestinal atrophy involving suppression of homeostatic genes and alteration of intestinal lipids and bile acids. Biochim. Biophys. Acta 2017, 1862, 1520–1533. [Google Scholar] [CrossRef]
- Song, H.; Bao, S.; Lei, X.; Jin, C.; Zhang, S.; Turk, J.; Ramanadham, S. Evidence for proteolytic processing and stimulated organelle redistribution of iPLA (2) beta. Biochim. Biophys. Acta 2010, 1801, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Cariello, M.; Piccinin, E.; Garcia-Irigoyen, O.; Sabbà, C.; Moschetta, A. Nuclear receptor FXR, bile acids and liver damage: Introducing the progressive familial intrahepatic cholestasis with FXR mutations. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864 Pt 4, 1308–1318. [Google Scholar] [CrossRef]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Appleby, R.N.; Moghul, I.; Khan, S.; Yee, M.; Manousou, P.; Neal, T.D.; Walters, J.R.F. Non-alcoholic fatty liver disease is associated with dysregulated bile acid synthesis and diarrhea: A prospective observational study. PLoS ONE 2019, 14, e0211348. [Google Scholar] [CrossRef] [PubMed]
- Croze, E.M.; Morré, D.J. Isolation of plasma membrane, Golgi apparatus, and endoplasmic reticulum fractions from single homogenates of mouse liver. J. Cell. Physiol. 1984, 119, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Gan-Schreier, H.; Tuma-Kellner, S.; Stremmel, W.; Chamulitrat, W. Sensitization to autoimmune hepatitis in group VIA calcium-independent phospholipase A2-null mice led to duodenal villous atrophy with apoptosis, goblet cell hyperplasia and leaked bile acids. Biochim. Biophys. Acta 2015, 1852, 1646–1657. [Google Scholar] [CrossRef]
- Lombardi, B.; Rao, N.K. Acute hemorrhagic pancreatic necrosis in mice. Influence of the age and sex of the animals and of dietary ethionine, choline, methionine, and adenine sulfate. Am. J. Pathol. 1975, 81, 87–100. [Google Scholar]
- Fischer, L.M.; daCosta, K.A.; Kwock, L.; Stewart, P.W.; Lu, T.S.; Stabler, S.P.; Allen, R.H.; Zeisel, S.H. Sex and menopausal status influence human dietary requirements for the nutrient choline. Am. J. Clin. Nutr. 2007, 85, 1275–1285. [Google Scholar] [CrossRef]
- Hampton, R.Y. ER stress response: Getting the UPR hand on misfolded proteins. Curr. Biol. 2000, 10, R518–R521. [Google Scholar] [CrossRef]
- Liu, X.; Guo, G.L.; Kong, B.; Hilburn, D.B.; Hubchak, S.C.; Park, S.; LeCuyer, B.; Hsieh, A.; Wang, L.; Fang, D.; et al. Farnesoid X receptor signaling activates the hepatic X-box binding protein 1 pathway in vitro and in mice. Hepatology 2018, 68, 304–331. [Google Scholar] [CrossRef]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef]
- Gao, X.; van der Veen, J.N.; Vance, J.E.; Thiesen, A.; Vance, D.E.; Jacobs, R.L. Lack of phosphatidylethanolamine N-methyltransferase alters hepatic phospholipid composition and induces endoplasmic reticulum stress. Biochim. Biophys. Acta 2015, 1852, 2689–2699. [Google Scholar] [CrossRef]
- Cicione, C.; Degirolamo, C.; Moschetta, A. Emerging role of fibroblast growth factors 15/19 and 21 as metabolic integrators in the liver. Hepatology 2012, 56, 2404–2411. [Google Scholar] [CrossRef]
- Aguilar-Olivos, N.E.; Carrillo-Córdova, D.; Oria-Hernández, J.; Sánchez-Valle, V.; Ponciano-Rodríguez, G.; Ramírez-Jaramillo, M.; Chablé-Montero, F.; Chávez-Tapia, N.C.; Uribe, M.; Méndez-Sánchez, N. The nuclear receptor FXR, but not LXR, up-regulates bile acid transporter expression in non-alcoholic fatty liver disease. Ann. Hepatol. 2015, 14, 487–493. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Alpini, G.; Ueno, Y.; Glaser, S.S.; Marzioni, M.; Phinizy, J.L.; Francis, H.; Lesage, G. Bile acid feeding increased proliferative activity and apical bile acid transporter expression in both small and large rat cholangiocytes. Hepatology 2001, 34, 868–876. [Google Scholar] [CrossRef]
- Fabris, L.; Spirli, C.; Cadamuro, M.; Fiorotto, R.; Strazzabosco, M. Emerging concepts in biliary repair and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G102–G116. [Google Scholar] [CrossRef]
- Lionarons, D.A.; Heger, M.; van Golen, R.F.; Alles, L.K.; van der Mark, V.A.; Kloek, J.J.; de Waart, D.R.; Marsman, H.A.; Rusch, H.; Verheij, J.; et al. Simple steatosis sensitizes cholestatic rats to liver injury and dysregulates bile salt synthesis and transport. Sci. Rep. 2016, 6, 31829. [Google Scholar] [CrossRef]
- Volmer, R.; Ron, D. Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hänelt, I.; Hummer, G.; Ernst, R. Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 2017, 67, 673–684. [Google Scholar] [CrossRef]
- Thibault, G.; Shui, G.; Kim, W.; McAlister, G.C.; Ismail, N.; Gygi, S.P.; Wenk, M.R.; Ng, D.T. The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol. Cell 2012, 48, 16–27. [Google Scholar] [CrossRef]
- Promlek, T.; Ishiwata-Kimata, Y.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 2011, 22, 3520–3532. [Google Scholar] [CrossRef]
- Hou, N.S.; Gutschmidt, A.; Choi, D.Y.; Pather, K.; Shi, X.; Watts, J.L.; Hoppe, T.; Taubert, S. Activation of the endoplasmic reticulum unfolded protein response by lipid disequilibrium without disturbed proteostasis in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E2271–E2280. [Google Scholar] [CrossRef]
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of european descent. Gastroenterology 2016, 150, 1219–1230. [Google Scholar] [CrossRef]
- Guna, A.; Volkmar, N.; Christianson, J.C.; Hegde, R.S. The ER membrane protein complex is a transmembrane domain insertase. Science 2018, 359, 470–473. [Google Scholar] [CrossRef]
- Ward, K.E.; Ropa, J.P.; Adu-Gyamfi, E.; Stahelin, R.V. C2 domain membrane penetration by group IVA cytosolic phospholipase A2 induces membrane curvature changes. J. Lipid Res. 2012, 53, 2656–2666. [Google Scholar] [CrossRef]
- Drin, G.; Antonny, B. Amphipathic helices and membrane curvature. FEBS Lett. 2010, 584, 1840–1847. [Google Scholar] [CrossRef]
- Vanni, S.; Hirose, H.; Barelli, H.; Antonny, B.; Gautier, R. A sub-nanometre view of how membrane curvature and composition modulate lipid packing and protein recruitment. Nat. Commun. 2014, 5, 4916. [Google Scholar] [CrossRef]
- Sen, S.; Jumaa, H.; Webster, N.J. Splicing factor SRSF3 is crucial for hepatocyte differentiation and metabolic function. Nat. Commun. 2013, 4, 1336. [Google Scholar] [CrossRef]
- Wu, W.; Liu, X.; Peng, X.; Xue, R.; Ji, L.; Shen, X.; Chen, S.; Gu, J.; Zhang, S. Bile acids override steatosis in farnesoid X receptor deficient mice in a model of non-alcoholic steatohepatitis. Biochem. Biophys. Res. Commun. 2014, 448, 50–55. [Google Scholar] [CrossRef]
- Fiorucci, S.; Antonelli, E.; Rizzo, G.; Renga, B.; Mencarelli, A.; Riccardi, L.; Orlandi, S.; Pellicciari, R.; Morelli, A. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004, 127, 1497–1512. [Google Scholar] [CrossRef]
- Okushin, K.; Tsutsumi, T.; Enooku, K.; Fujinaga, H.; Kado, A.; Shibahara, J.; Fukayama, M.; Moriya, K.; Yotsuyanagi, H.; Koike, K. The intrahepatic expression levels of bile acid transporters are inversely correlated with the histological progression of nonalcoholic fatty liver disease. J. Gastroenterol. 2016, 51, 808–818. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Ridolfi, F.; Hannivoort, R.; Saccomanno, S.; Homan, M.; De Minicis, S.; Jansen, P.L.; Candelaresi, C.; Benedetti, A.; Moshage, H. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 2005, 128, 1042–1055. [Google Scholar] [CrossRef]
- Kordes, C.; Sawitza, I.; Götze, S.; Herebian, D.; Häussinger, D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J. Clin. Investig. 2014, 124, 5503–5515. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef]
- Devisscher, L.; Scott, C.L.; Lefere, S.; Raevens, S.; Bogaerts, E.; Paridaens, A.; Verhelst, X.; Geerts, A.; Guilliams, M.; Van Vlierberghe, H. Non-alcoholic steatohepatitis induces transient changes within the liver macrophage pool. Cell. Immunol. 2017, 322, 74–83. [Google Scholar] [CrossRef]
- Rinella, M.E.; Elias, M.S.; Smolak, R.R.; Fu, T.; Borensztajn, J.; Green, R.M. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J. Lipid Res. 2008, 49, 1068–1076. [Google Scholar] [CrossRef]
- Sha, W.; da Costa, K.A.; Fischer, L.M.; Milburn, M.V.; Lawton, K.A.; Berger, A.; Jia, W.; Zeisel, S.H. Metabolomic profiling can predict which humans will develop liver dysfunction when deprived of dietary choline. FASEB J. 2010, 24, 2962–2975. [Google Scholar] [CrossRef]
- Blanchard, H.; Taha, A.Y.; Cheon, Y.; Kim, H.W.; Turk, J.; Rapoport, S.I. iPLA2β knockout mouse, a genetic model for progressive human motor disorders, develops age-related neuropathology. Neurochem. Res. 2014, 39, 1522–1532. [Google Scholar] [CrossRef]
- Malaguti, M.C.; Melzi, V.; Di Giacopo, R.; Monfrini, E.; Di Biase, E.; Franco, G.; Borellini, L.; Trezzi, I.; Monzio Compagnoni, G.; Fortis, P.; et al. A novel homozygous PLA2G6 mutation causes dystonia-parkinsonism. Parkinsonism Relat. Disord. 2015, 21, 337–339. [Google Scholar] [CrossRef]
- Iodice, A.; Spagnoli, C.; Salerno, G.G.; Frattini, D.; Bertani, G.; Bergonzini, P.; Pisani, F.; Fusco, C. Infantile neuroaxonal dystrophy and PLA2G6-associated neurodegeneration: An update for the diagnosis. Brain Dev. 2017, 39, 93–100. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ming, Y.; Zhu, X.; Tuma-Kellner, S.; Ganzha, A.; Liebisch, G.; Gan-Schreier, H.; Chamulitrat, W. iPla2β Deficiency Suppresses Hepatic ER UPR, Fxr, and Phospholipids in Mice Fed with MCD Diet, Resulting in Exacerbated Hepatic Bile Acids and Biliary Cell Proliferation. Cells 2019, 8, 879. https://doi.org/10.3390/cells8080879
Ming Y, Zhu X, Tuma-Kellner S, Ganzha A, Liebisch G, Gan-Schreier H, Chamulitrat W. iPla2β Deficiency Suppresses Hepatic ER UPR, Fxr, and Phospholipids in Mice Fed with MCD Diet, Resulting in Exacerbated Hepatic Bile Acids and Biliary Cell Proliferation. Cells. 2019; 8(8):879. https://doi.org/10.3390/cells8080879
Chicago/Turabian StyleMing, Yanan, Xingya Zhu, Sabine Tuma-Kellner, Alexandra Ganzha, Gerhard Liebisch, Hongying Gan-Schreier, and Walee Chamulitrat. 2019. "iPla2β Deficiency Suppresses Hepatic ER UPR, Fxr, and Phospholipids in Mice Fed with MCD Diet, Resulting in Exacerbated Hepatic Bile Acids and Biliary Cell Proliferation" Cells 8, no. 8: 879. https://doi.org/10.3390/cells8080879
APA StyleMing, Y., Zhu, X., Tuma-Kellner, S., Ganzha, A., Liebisch, G., Gan-Schreier, H., & Chamulitrat, W. (2019). iPla2β Deficiency Suppresses Hepatic ER UPR, Fxr, and Phospholipids in Mice Fed with MCD Diet, Resulting in Exacerbated Hepatic Bile Acids and Biliary Cell Proliferation. Cells, 8(8), 879. https://doi.org/10.3390/cells8080879