JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases

{kind=link}
{kind=link}
{kind=link}
{kind=link}
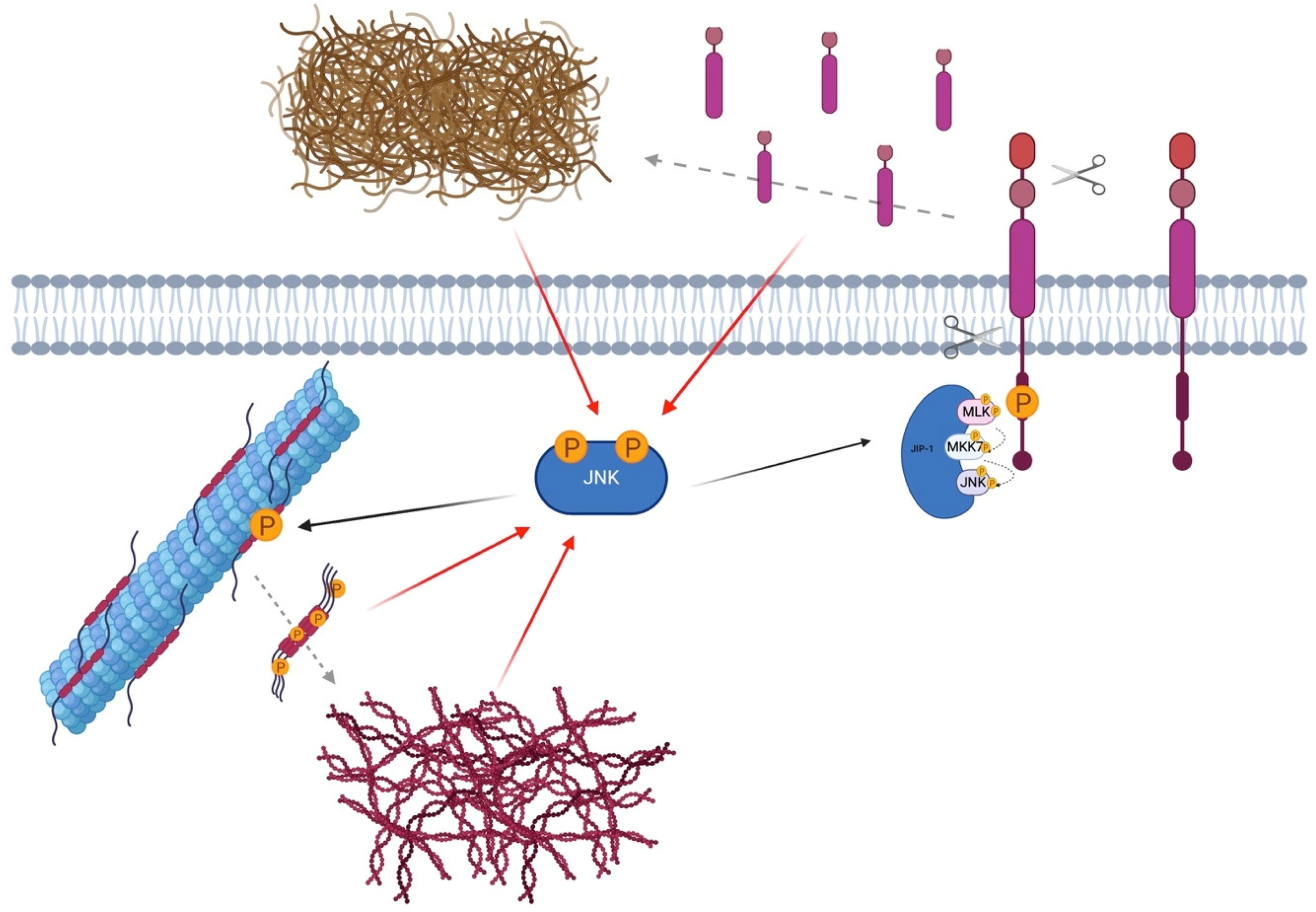{kind=link}
Abstract
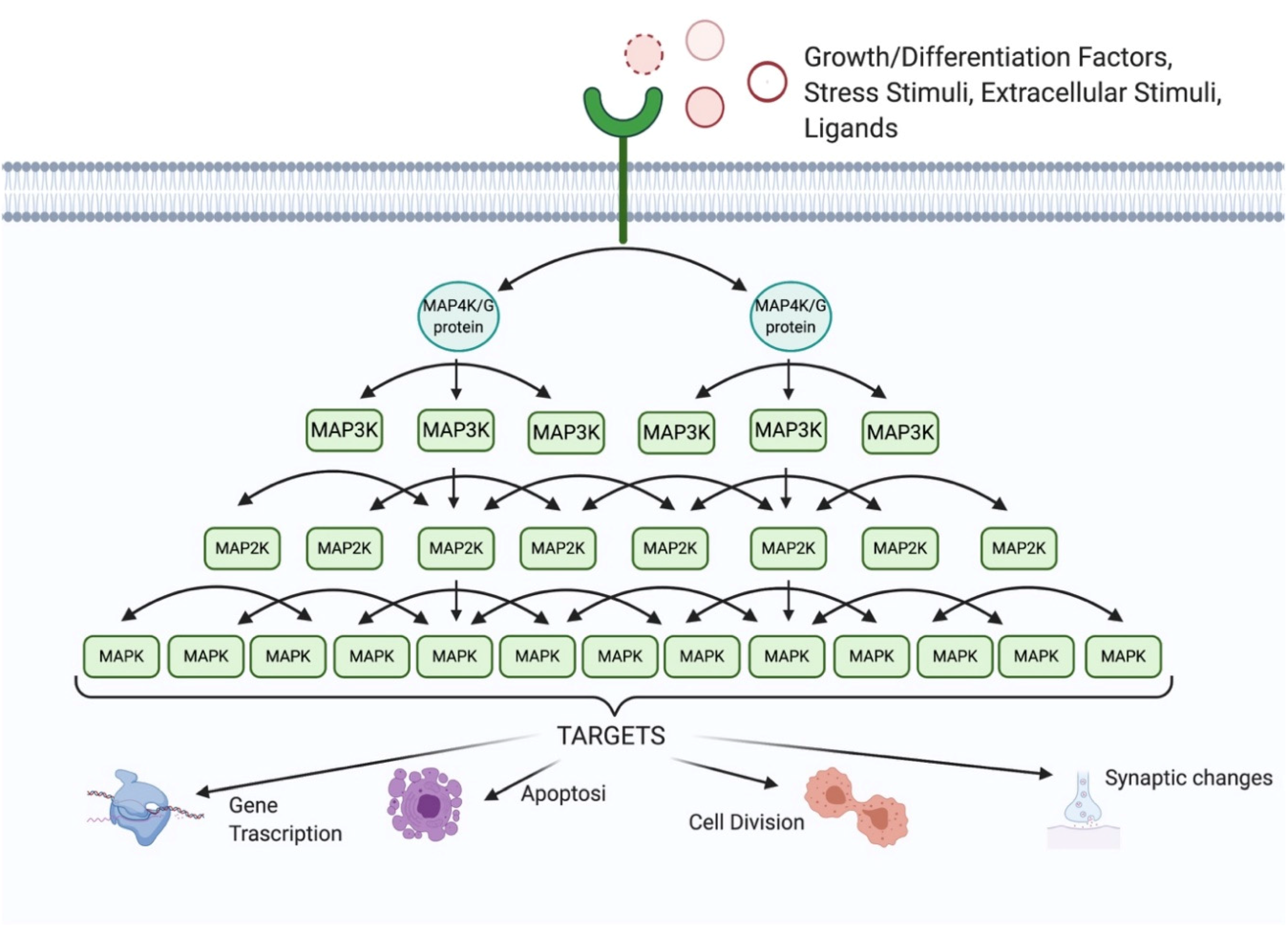
:1. The MAPK Family
2. The JNK Family
3. The JNK3 Isoform
4. The JNK Scaffold Proteins
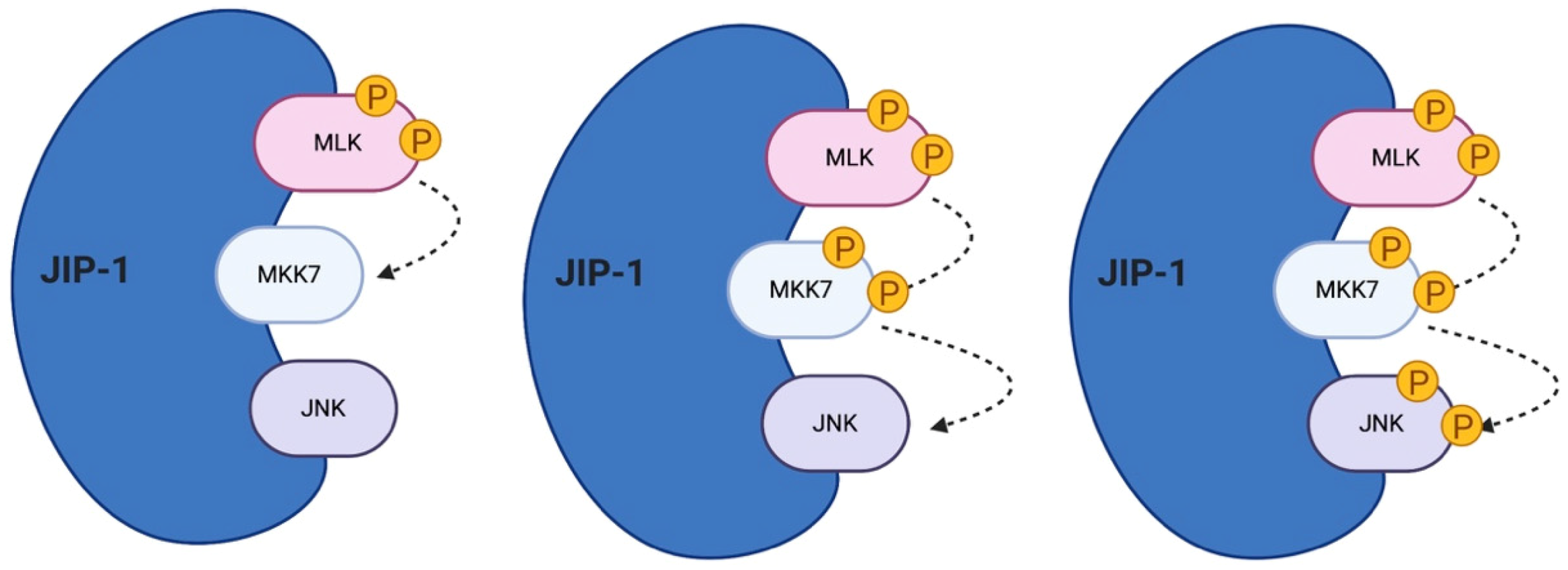
5. The Role of JIP Scaffold Proteins
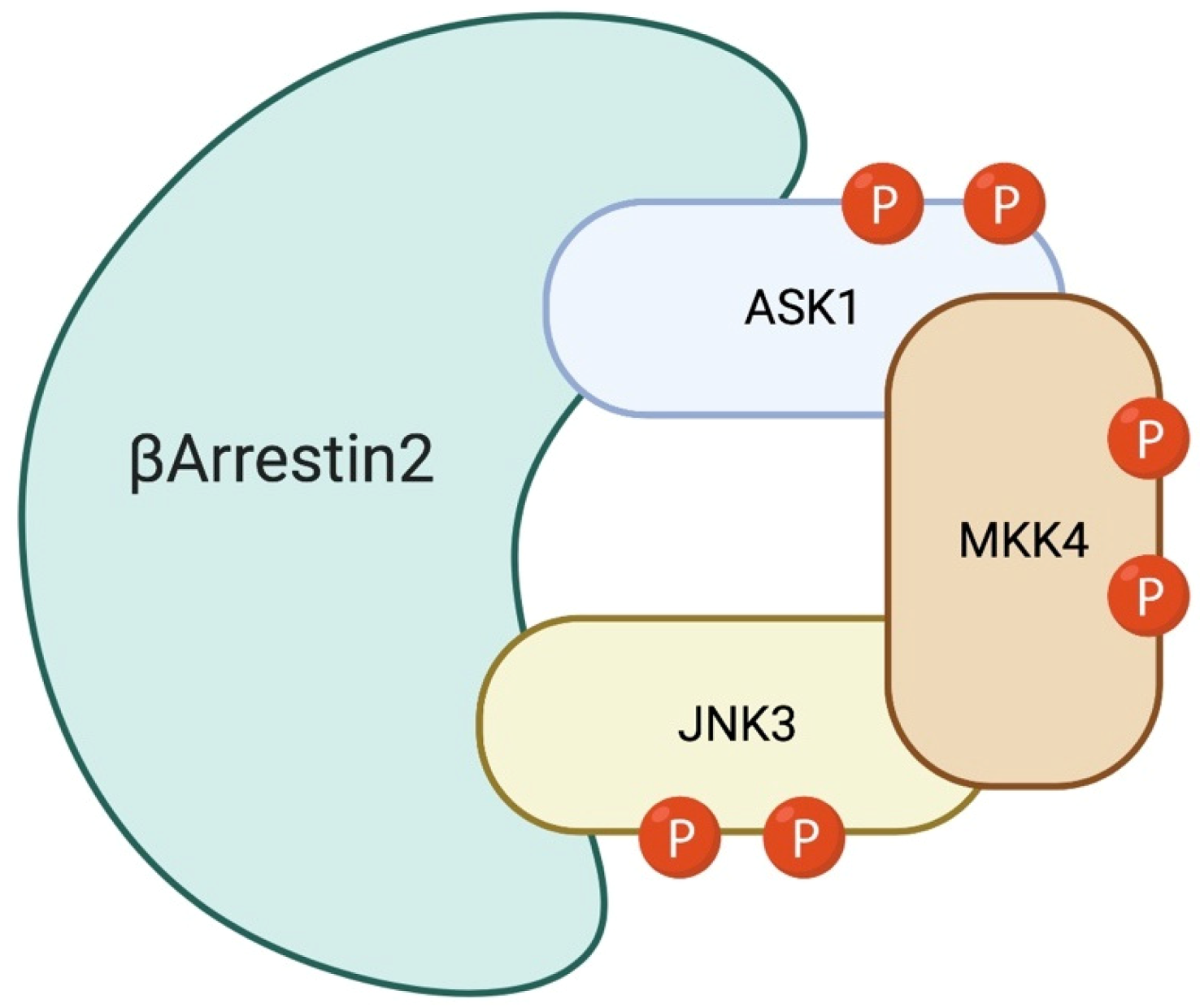
6. The Role of β-Arrestin-2 Scaffold Protein
7. JNK3 in the Central Nervous System
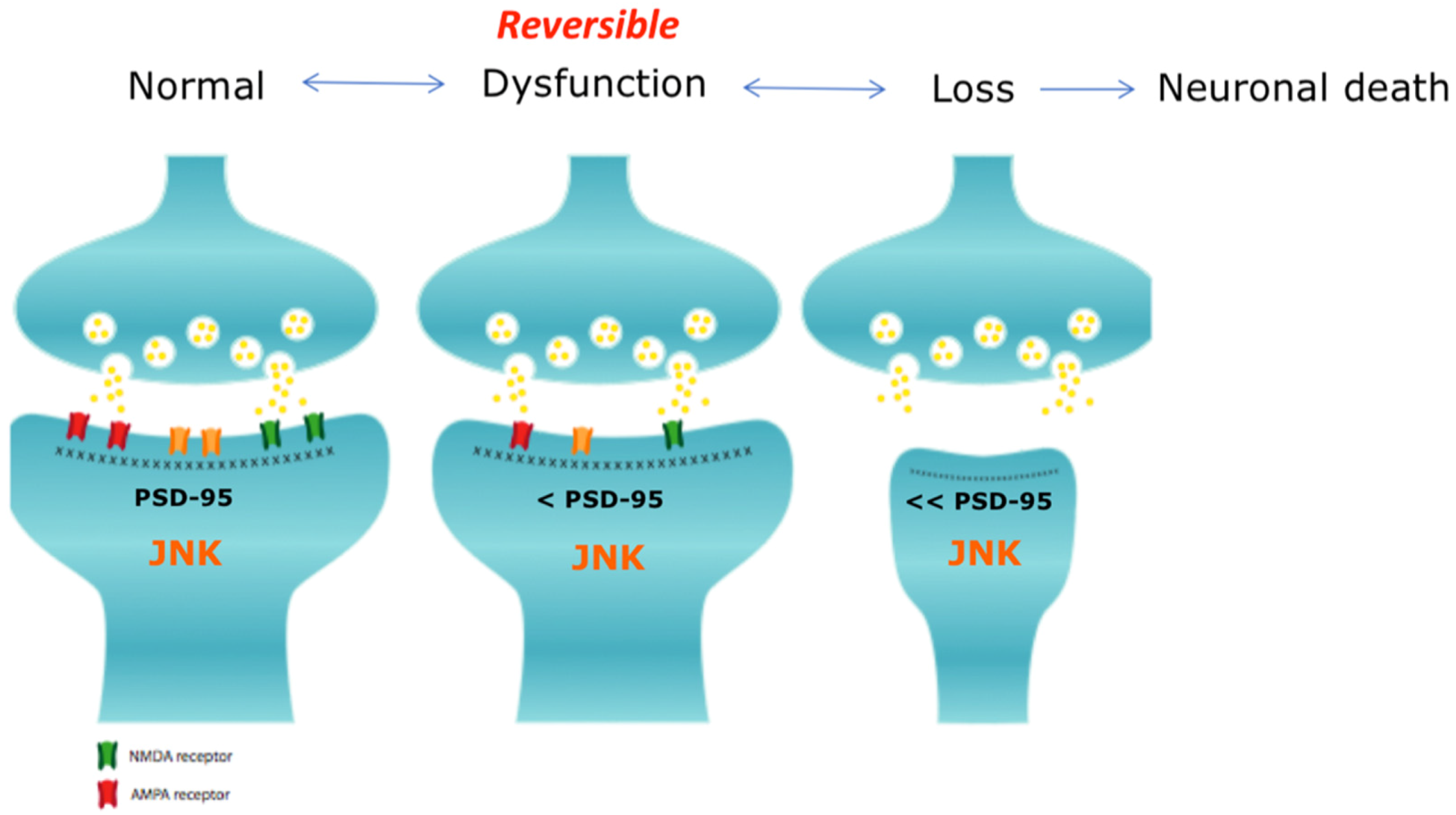
8. JNK3 is a Key Player in Synaptic Dysfunction
9. JNK3 in Brain Diseases
9.1. Ischemia
9.2. Epilepsy
9.3. Optic Neuropathies (ON)
9.4. Parkinson’s Disease (PD)
9.5. Huntington Disease (HD)
9.6. Alzheimer’s Disease (AD)
9.7. Schizophrenia
9.8. Amyotrophic Lateral Sclerosis (ALS)
9.9. Spinal Muscular Atrophy (SMA)
9.10. Neurodevelopmental Disorders
10. JNK3 as Therapeutic Target Against Neurodegenerative and Neurodevelopment Brain Diseases
11. Tuning JNK3 Pathway by Using JIP-1 and β-Arrestin-2
12. JNK3 as Biomarker
13. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaeffer, H.J.; Weber, M.J. Mitogen-activated protein kinases: Specific messages from ubiquitous messengers. Mol. Cell. Biol. 1999, 19, 2435–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locasale, J.W.; Shaw, A.S.; Chakraborty, A.K. Scaffold proteins confer diverse regulatory properties to protein kinase cascades. Proc. Natl. Acad. Sci. USA 2007, 104, 13307–13312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, H.; Tsutsui, J.; Takekawa, M.; Witten, E.A.; Saito, H. Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol. Cell. Biol. 2002, 22, 4544–4555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadee, D.N.; Yuasa, T.; Kyriakis, J.M. Direct activation of mitogen-activated protein kinase kinase kinase MEKK1 by the Ste20p homologue GCK and the adapter protein TRAF2. Mol. Cell. Biol. 2002, 22, 737–749. [Google Scholar] [CrossRef] [Green Version]
- Tegethoff, S.; Behlke, J.; Scheidereit, C. Tetrameric oligomerization of IkappaB kinase gamma (IKKgamma) is obligatory for IKK complex activity and NF-kappaB activation. Mol. Cell. Biol. 2003, 23, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Polley, S.; Huang, D.-B.; Hauenstein, A.V.; Fusco, A.J.; Zhong, X.; Vu, D.; Schröfelbauer, B.; Kim, Y.; Hoffmann, A.; Verma, I.M.; et al. A structural basis for IκB kinase 2 activation via oligomerization-dependent trans auto-phosphorylation. PLoS Biol. 2013, 11, e1001581. [Google Scholar] [CrossRef] [Green Version]
- Widmann, C.; Gibson, S.; Jarpe, M.B.; Johnson, G.L. Mitogen-activated protein kinase: Conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999, 79, 143–180. [Google Scholar] [CrossRef]
- Lisnock, J.; Griffin, P.; Calaycay, J.; Frantz, B.; Parsons, J.; O’Keefe, S.J.; LoGrasso, P. Activation of JNK3 alpha 1 requires both MKK4 and MKK7: Kinetic characterization of in vitro phosphorylated JNK3 alpha 1. Biochemistry 2000, 39, 3141–3148. [Google Scholar] [CrossRef]
- Fleming, Y.; Armstrong, C.G.; Morrice, N.; Paterson, A.; Goedert, M.; Cohen, P. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogen-activated protein kinase kinase 4 (MKK4) and MKK7. Biochem. J. 2000, 352 Pt 1, 145–154. [Google Scholar] [CrossRef]
- Ferrell, J.E. Tripping the switch fantastic: How a protein kinase cascade can convert graded inputs into switch-like outputs. Trends Biochem. Sci. 1996, 21, 460–466. [Google Scholar] [CrossRef]
- Thattai, M.; van Oudenaarden, A. Attenuation of Noise in Ultrasensitive Signaling Cascades. Biophys. J. 2002, 82, 2943–2950. [Google Scholar] [CrossRef] [Green Version]
- Swain, P.S.; Siggia, E.D. The Role of Proofreading in Signal Transduction Specificity. Biophys. J. 2002, 82, 2928–2933. [Google Scholar] [CrossRef] [Green Version]
- Saito, J.; Toriumi, S.; Awano, K.; Ichijo, H.; Sasaki, K.; Kobayashi, T.; Tamura, S. Regulation of apoptosis signal-regulating kinase 1 by protein phosphatase 2Cepsilon. Biochem. J. 2007, 405, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Seternes, O.-M.; Kidger, A.M.; Keyse, S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 124–143. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, K.; Nishida, E. Regulation of MAP kinases by MAP kinase phosphatases. Biochim. Biophys. Acta 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martell, K.J.; Seasholtz, A.F.; Kwak, S.P.; Clemens, K.K.; Dixon, J.E. hVH-5: A protein tyrosine phosphatase abundant in brain that inactivates mitogen-activated protein kinase. J. Neurochem. 1995, 65, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [Green Version]
- Sabio, G.; Davis, R.J. cJun NH2-terminal kinase 1 (JNK1): Roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 2010, 35, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Chang, L.; Yamanishi, Y.; Karin, M.; Firestein, G.S. Joint damage and inflammation in c-Jun N-terminal kinase 2 knockout mice with passive murine collagen-induced arthritis. Arthritis Rheum. 2002, 46, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Guma, M.; Kashiwakura, J.; Crain, B.; Kawakami, Y.; Beutler, B.; Firestein, G.S.; Kawakami, T.; Karin, M.; Corr, M. JNK1 controls mast cell degranulation and IL-1{beta} production in inflammatory arthritis. Proc. Natl. Acad. Sci. USA 2010, 107, 22122–22127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alrashdan, Y.A.; Alkhouri, H.; Chen, E.; Lalor, D.J.; Poniris, M.; Henness, S.; Brightling, C.E.; Burgess, J.K.; Armour, C.L.; Ammit, A.J.; et al. Asthmatic airway smooth muscle CXCL10 production: Mitogen-activated protein kinase JNK involvement. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1118–L1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cellurale, C.; Girnius, N.; Jiang, F.; Cavanagh-Kyros, J.; Lu, S.; Garlick, D.S.; Mercurio, A.M.; Davis, R.J. Role of JNK in mammary gland development and breast cancer. Cancer Res. 2012, 72, 472–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Wu, W.; Fu, B.; Shi, L.; Wang, X.; Kuca, K. JNK signaling in cancer cell survival. Med. Res. Rev. 2019, 39, 2082–2104. [Google Scholar] [CrossRef]
- Yoon, S.O.; Park, D.J.; Ryu, J.C.; Ozer, H.G.; Tep, C.; Shin, Y.J.; Lim, T.H.; Pastorino, L.; Kunwar, A.J.; Walton, J.C.; et al. JNK3 perpetuates metabolic stress induced by Aβ peptides. Neuron 2012, 75, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharm. 2015, 6, 321. [Google Scholar] [CrossRef] [Green Version]
- Pan, J.; Li, H.; Zhang, B.; Xiong, R.; Zhang, Y.; Kang, W.-Y.; Chen, W.; Zhao, Z.-B.; Chen, S.-D. Small peptide inhibitor of JNK3 protects dopaminergic neurons from MPTP induced injury via inhibiting the ASK1-JNK3 signaling pathway. PLoS ONE 2015, 10, e0119204. [Google Scholar] [CrossRef] [Green Version]
- Kuan, C.-Y.; Whitmarsh, A.J.; Yang, D.D.; Liao, G.; Schloemer, A.J.; Dong, C.; Bao, J.; Banasiak, K.J.; Haddad, G.G.; Flavell, R.A.; et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 15184–15189. [Google Scholar] [CrossRef] [Green Version]
- Chintala, S.K.; Putris, N.; Geno, M. Activation of TLR3 promotes the degeneration of retinal ganglion cells by upregulating the protein levels of JNK3. Investig. Ophthalmol. Vis. Sci. 2015, 56, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Lemos, L.; Junyent, F.; Camins, A.; Castro-Torres, R.D.; Folch, J.; Olloquequi, J.; Beas-Zarate, C.; Verdaguer, E.; Auladell, C. Neuroprotective Effects of the Absence of JNK1 or JNK3 Isoforms on Kainic Acid-Induced Temporal Lobe Epilepsy-Like Symptoms. Mol. Neurobiol. 2018, 55, 4437–4452. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.W.; Choi, H.Y.; Joo, K.M.; Nam, D.-H. Tumor progression locus 2 (Tpl2) kinase as a novel therapeutic target for cancer: Double-sided effects of Tpl2 on cancer. Int. J. Mol. Sci. 2015, 16, 4471–4491. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, T.; Nishida, E. Docking interactions in the mitogen-activated protein kinase cascades. Pharmacol. Ther. 2002, 93, 193–202. [Google Scholar] [CrossRef]
- Schreiber, M.; Kolbus, A.; Piu, F.; Szabowski, A.; Möhle-Steinlein, U.; Tian, J.; Karin, M.; Angel, P.; Wagner, E.F. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [Google Scholar] [CrossRef]
- Sabapathy, K.; Hochedlinger, K.; Nam, S.Y.; Bauer, A.; Karin, M.; Wagner, E.F. Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation. Mol. Cell. 2004, 15, 713–725. [Google Scholar] [CrossRef]
- Huang, L.; Quan, X.; Liu, Z.; Ma, T.; Wu, Y.; Ge, J.; Zhu, S.; Yang, Y.; Liu, L.; Sun, Z.; et al. c-Jun gene-modified Schwann cells: Upregulating multiple neurotrophic factors and promoting neurite outgrowth. Tisue Eng. Part A. 2015, 21, 1409–1421. [Google Scholar] [CrossRef] [Green Version]
- Sanna, M.D.; Manassero, G.; Vercelli, A.; Herdegen, T.; Galeotti, N. The isoform-specific functions of the c-Jun N-terminal kinase (JNK) in a mouse model of antiretroviral-induced painful peripheral neuropathy. Eur. J. Pharmacol. 2020, 880, 173161. [Google Scholar] [CrossRef] [PubMed]
- Coffey, E.T.; Smiciene, G.; Hongisto, V.; Cao, J.; Brecht, S.; Herdegen, T.; Courtney, M.J. c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J. Neurosci. 2002, 22, 4335–4345. [Google Scholar] [CrossRef] [PubMed]
- Bruckner, S.R.; Tammariello, S.P.; Kuan, C.Y.; Flavell, R.A.; Rakic, P.; Estus, S. JNK3 contributes to c-Jun activation and apoptosis but not oxidative stress in nerve growth factor-deprived sympathetic neurons. J. Neurochem. 2001, 78, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Herdegen, T. A single c-Jun N-terminal kinase isoform (JNK3-p54) is an effector in both neuronal differentiation and cell death. J. Boil. Chem. 2002, 278, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Gu, Y.; Fox, T.; Coll, J.T.; Fleming, M.A.; Markland, W.; Caron, P.R.; Wilson, K.P.; Su, M.S. Crystal structure of JNK3: A kinase implicated in neuronal apoptosis. Structure 1998, 6, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Günther, S. New insights into the structural dynamics of the kinase JNK3. Sci. Rep. 2018, 8, 9435. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Whitmarsh, A.J. The beta-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 2008, 283, 15903–15911. [Google Scholar] [CrossRef] [Green Version]
- Peterson, Y.K.; Luttrell, L.M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. [Google Scholar] [CrossRef] [Green Version]
- Bonny, C.; Nicod, P.; Waeber, G. IB1, a JIP-1-related nuclear protein present in insulin-secreting cells. J. Biol. Chem. 1998, 273, 1843–1846. [Google Scholar] [CrossRef] [Green Version]
- Perry, N.A.; Kaoud, T.S.; Ortega, O.O.; Kaya, A.I.; Marcus, D.J.; Pleinis, J.M.; Berndt, S.; Chen, Q.; Zhan, X.; Dalby, K.N.; et al. Arrestin-3 scaffolding of the JNK3 cascade suggests a mechanism for signal amplification. Proc. Natl. Acad. Sci. USA 2019, 116, 810–815. [Google Scholar] [CrossRef] [Green Version]
- Willoughby, E.A.; Perkins, G.R.; Collins, M.K.; Whitmarsh, A.J. The JNK-interacting protein-1 scaffold protein targets MAPK phosphatase-7 to dephosphorylate JNK. J. Biol. Chem. 2003, 278, 10731–10736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.K.; Davis, R.J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J. The JIP family of MAPK scaffold proteins. Biochem. Soc. Trans. 2006, 34, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Kashef, K.; Lee, C.M.; Xu, H.; Reddy, E.P. Scaffold proteins of MAP-kinase modules. Oncogene 2007, 26, 3185–3202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagadish, N.; Rana, R.; Mishra, D.; Kumar, M.; Suri, A. Sperm associated antigen 9 (SPAG9): A new member of c-Jun NH2 -terminal kinase (JNK) interacting protein exclusively expressed in testis. Keio J. Med. 2005, 54, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelkar, N.; Standen, C.L.; Davis, R.J. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol. Cell. Biol. 2005, 25, 2733–2743. [Google Scholar] [CrossRef] [Green Version]
- Verhey, K.J.; Meyer, D.; Deehan, R.; Blenis, J.; Schnapp, B.J.; Rapoport, T.A.; Margolis, B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 2001, 152, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, S.; Yasukawa, T.; Homma, Y.; Ito, Y.; Niikura, T.; Hiraki, T.; Hirai, S.; Ohno, S.; Kita, Y.; Kawasumi, M.; et al. c-Jun N-terminal kinase (JNK)-interacting protein-1b/islet-brain-1 scaffolds Alzheimer’s amyloid precursor protein with JNK. J. Neurosci. 2001, 21, 6597–6607. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, J.; Whitmarsh, A.J.; Cavanagh, J.; Sharma, M.; Davis, R.J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Biol. 1999, 19, 7245–7254. [Google Scholar] [CrossRef] [Green Version]
- Drerup, C.M.; Nechiporuk, A.V. JNK-interacting protein 3 mediates the retrograde transport of activated c-Jun N-terminal kinase and lysosomes. PLoS Genetics. 2013, 9, e1003303. [Google Scholar] [CrossRef] [Green Version]
- Kelkar, N.; Gupta, S.; Dickens, M.; Davis, R.J. Interaction of a mitogen-activated protein kinase signaling module with the neuronal protein JIP3. Mol. Cell. Biol. 2000, 20, 1030–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonny, C.; Oberson, A.; Negri, S.; Sauser, C.; Schorderet, D.F. Cell-permeable peptide inhibitors of JNK: Novel blockers of beta-cell death. Diabetes 2001, 50, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsello, T.; Clarke, P.G.H.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef]
- Pellet, J.B.; Haefliger, J.A.; Staple, J.K.; Widmann, C.; Welker, E.; Hirling, H.; Bonny, C.; Nicod, P.; Catsicas, S.; Waeber, G.; et al. Spatial, temporal and subcellular localization of islet-brain 1 (IB1), a homologue of JIP-1, in mouse brain. Eur. J. Neurosci. 2000, 12, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhou, L.; Del Villar, K.; Ghanevati, M.; Tashjian, V.; Miller, C.A. JIP1 regulates neuronal apoptosis in response to stress. Brain Res. Mol. Brain Res. 2005, 134, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Borsello, T.; Centeno, C.; Riederer, I.M.; Haefliger, J.-A.; Riederer, B.M. Phosphorylation-dependent dimerization and subcellular localization of islet-brain 1/c-Jun N-terminal kinase-interacting protein 1. J. Neurosci. Res. 2007, 85, 3632–3641. [Google Scholar] [CrossRef] [PubMed]
- Penela, P.; Ribas, C.; Mayor, F. Mechanisms of regulation of the expression and function of G protein-coupled receptor kinases. Cell. Signal. 2003, 15, 973–981. [Google Scholar] [CrossRef]
- Goodman, O.B.; Krupnick, J.G.; Santini, F.; Gurevich, V.V.; Penn, R.B.; Gagnon, A.W.; Keen, J.H.; Benovic, J.L. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996, 383, 447–450. [Google Scholar] [CrossRef]
- Srivastava, A.; Gupta, B.; Gupta, C.; Shukla, A.K. Emerging Functional Divergence of β-Arrestin Isoforms in GPCR Function. Trends Endocrinol. Metab. 2015, 26, 628–642. [Google Scholar] [CrossRef]
- Zurkovsky, L.; Sedaghat, K.; Ahmed, M.R.; Gurevich, V.V.; Gurevich, E.V. Arrestin-2 and arrestin-3 differentially modulate locomotor responses and sensitization to amphetamine. Neuropharmacology 2017, 121, 20–29. [Google Scholar] [CrossRef]
- Gurevich, E.V.; Gurevich, V.V. Arrestins: Ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006, 7, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 72 McDonald, P.H.; Chow, C.W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, E.A.; Collins, M.K. Dynamic interaction between the dual specificity phosphatase MKP7 and the JNK3 scaffold protein beta-arrestin 2. J. Biol. Chem. 2005, 280, 25651–25658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Ben-Shalom, R.; Ahn, M.; Liptak, A.T.; van Rijn, R.M.; Whistler, J.L.; Bender, K.J. β-Arrestin-Dependent Dopaminergic Regulation of Calcium Channel Activity in the Axon Initial Segment. Cell Rep. 2016, 16, 1518–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontrello, C.G.; Sun, M.-Y.; Lin, A.; Fiacco, T.A.; DeFea, K.A.; Ethell, I.M. Cofilin under control of β-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc. Natl. Acad. Sci. USA 2012, 109, E442–E451. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Xie, R.-G.; Gao, Y.-J.; Xu, Z.-Z.; Zhao, L.-X.; Bang, S.; Berta, T.; Park, C.-K.; Lay, M.; Chen, W.; et al. β-arrestin-2 regulates NMDA receptor function in spinal lamina II neurons and duration of persistent pain. Nat. Commun 2016, 7, 12531. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, S.S.G. Receptor tyrosine kinase transactivation: Fine-tuning synaptic transmission. Trends Neurosci. 2003, 26, 119–122. [Google Scholar] [CrossRef]
- Gao, H.; Sun, Y.; Wu, Y.; Luan, B.; Wang, Y.; Qu, B.; Pei, G. Identification of beta-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-kappaB pathways. Mol. Cell 2004, 14, 303–317. [Google Scholar] [CrossRef]
- Feng, X.; Wu, C.-Y.; Burton, F.H.; Loh, H.H.; Wei, L.-N. β-arrestin protects neurons by mediating endogenous opioid arrest of inflammatory microglia. Cell Death Differ. 2014, 21, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Jaeschke, A.; Karasarides, M.; Ventura, J.-J.; Ehrhardt, A.; Zhang, C.; Flavell, R.A.; Shokat, K.M.; Davis, R.J. JNK2 is a positive regulator of the cJun transcription factor. Mol. Cell 2006, 23, 899–911. [Google Scholar] [CrossRef]
- Kuan, C.Y.; Yang, D.D.; Samanta Roy, D.R.; Davis, R.J.; Rakic, P.; Flavell, R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999, 22, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.D.; Kuan, C.Y.; Whitmarsh, A.J.; Rincón, M.; Zheng, T.S.; Davis, R.J.; Rakic, P.; Flavell, R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature 1997, 389, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. [Google Scholar] [CrossRef] [PubMed]
- Brecht, S.; Kirchhof, R.; Chromik, A.; Willesen, M.; Nicolaus, T.; Raivich, G.; Wessig, J.; Waetzig, V.; Goetz, M.; Claussen, M.; et al. Specific pathophysiological functions of JNK isoforms in the brain. Eur. J. Neurosci. 2005, 21, 363–377. [Google Scholar] [CrossRef]
- Kunde, S.-A.; Rademacher, N.; Tzschach, A.; Wiedersberg, E.; Ullmann, R.; Kalscheuer, V.M.; Shoichet, S.A. Characterisation of de novo MAPK10/JNK3 truncation mutations associated with cognitive disorders in two unrelated patients. Hum. Genet. 2013, 132, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baptista, J.; Mercer, C.; Prigmore, E.; Gribble, S.M.; Carter, N.P.; Maloney, V.; Thomas, N.S.; Jacobs, P.A.; Crolla, J.A. Breakpoint mapping and array CGH in translocations: Comparison of a phenotypically normal and an abnormal cohort. Am. J. Hum. Genet. 2008, 82, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Shoichet, S.A.; Duprez, L.; Hagens, O.; Waetzig, V.; Menzel, C.; Herdegen, T.; Schweiger, S.; Dan, B.; Vamos, E.; Ropers, H.-H.; et al. Truncation of the CNS-expressed JNK3 in a patient with a severe developmental epileptic encephalopathy. Hum. Genet. 2006, 118, 559–567. [Google Scholar] [CrossRef]
- Waetzig, V.; Zhao, Y.; Herdegen, T. The bright side of JNKs-Multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog. Neurobiol. 2006, 80, 84–97. [Google Scholar] [CrossRef]
- Eminel, S.; Roemer, L.; Waetzig, V.; Herdegen, T. c-Jun N-terminal kinases trigger both degeneration and neurite outgrowth in primary hippocampal and cortical neurons. J. Neurochem. 2008, 104, 957–969. [Google Scholar] [CrossRef]
- Bevilaqua, L.R.M.; Kerr, D.S.; Medina, J.H.; Izquierdo, I.; Cammarota, M. Inhibition of hippocampal Jun N-terminal kinase enhances short-term memory but blocks long-term memory formation and retrieval of an inhibitory avoidance task. Eur. J. Neurosci. 2003, 17, 897–902. [Google Scholar] [CrossRef]
- Lin, W.; Wang, S.; Yang, Z.; Lin, J.; Ke, Q.; Lan, W.; Shi, J.; Wu, S.; Cai, B. Heme Oxygenase-1 Inhibits Neuronal Apoptosis in Spinal Cord Injury through Down-Regulation of Cdc42-MLK3-MKK7-JNK3 Axis. J. Neurotrauma 2017, 34, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Resnick, L.; Fennell, M. Targeting JNK3 for the treatment of neurodegenerative disorders. Drug Discov. Today 2004, 9, 932–939. [Google Scholar] [CrossRef]
- Antoniou, X.; Falconi, M.; Di Marino, D.; Borsello, T. JNK3 as a therapeutic target for neurodegenerative diseases. J. Alzheimers Dis. 2011, 24, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Sclip, A.; Arnaboldi, A.; Colombo, I.; Veglianese, P.; Colombo, L.; Messa, M.; Mancini, S.; Cimini, S.; Morelli, F.; Antoniou, X.; et al. Soluble Aβ oligomer-induced synaptopathy: C-Jun N-terminal kinase’s role. J. Mol. Cell Biol. 2013, 5, 277–279. [Google Scholar] [CrossRef]
- Sclip, A.; Antoniou, X.; Colombo, A.; Camici, G.G.; Pozzi, L.; Cardinetti, D.; Feligioni, M.; Veglianese, P.; Bahlmann, F.H.; Cervo, L.; et al. c-Jun N-terminal kinase regulates soluble Aβ oligomers and cognitive impairment in AD mouse model. J. Biol. Chem. 2011, 286, 43871–43880. [Google Scholar] [CrossRef] [Green Version]
- Sclip, A.; Tozzi, A.; Abaza, A.; Cardinetti, D.; Colombo, I.; Calabresi, P.; Salmona, M.; Welker, E.; Borsello, T. c-Jun N-terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis. 2014, 5, e1019. [Google Scholar] [CrossRef]
- Buccarello, L.; Borsello, T. The Tat-Aβ1-6A2V(D) peptide against AD synaptopathy. Oncotarget 2017, 8, 10773–10774. [Google Scholar] [CrossRef] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef]
- Sun, B.; Halabisky, B.; Zhou, Y.; Palop, J.J.; Yu, G.; Mucke, L.; Gan, L. Imbalance between GABAergic and Glutamatergic Transmission Impairs Adult Neurogenesis in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 2009, 5, 624–633. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Levy, J.M.; Hou, A.; Winters, C.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Nicoll, R.A.; Reese, T.S. PSD-95 family MAGUKs are essential for anchoring AMPA and NMDA receptor complexes at the postsynaptic density. Proc. Natl. Acad. Sci. USA 2015, 112, E6983–E6992. [Google Scholar] [CrossRef] [Green Version]
- Cimini, S.; Sclip, A.; Mancini, S.; Colombo, L.; Messa, M.; Cagnotto, A.; Di Fede, G.; Tagliavini, F.; Salmona, M.; Borsello, T. The cell-permeable Aβ1-6A2VTAT(D) peptide reverts synaptopathy induced by Aβ1-42wt. Neurobiol. Dis. 2016, 89, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Musi, C.A.; Agrò, G.; Buccarello, L.; Camuso, S.; Borsello, T. JNK signaling activation in the Ube3a maternal deficient mouse model: Its specific inhibition prevents post-synaptic protein-enriched fraction alterations and cognitive deficits in Angelman Syndrome model. Neurobiol. Dis. 2020, 140, 104812. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Futai, K.; Jo, J.; Hayashi, Y.; Cho, K.; Sheng, M. Synaptic accumulation of PSD-95 and synaptic function regulated by phosphorylation of serine-295 of PSD-95. Neuron 2007, 56, 488–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Gong, J.; Ma, J.; Zhang, T.; Li, Y.; Lan, T.; Guo, P.; Qi, S. Targeting the Dvl-1/β-arrestin2/JNK3 interaction disrupts Wnt5a-JNK3 signaling and protects hippocampal CA1 neurons during cerebral ischemia reperfusion. Neuropharmacology 2018, 135, 11–21. [Google Scholar] [CrossRef]
- Ge, X.-H.; Zhu, G.-J.; Geng, D.-Q.; Zhang, H.-Z.; He, J.-M.; Guo, A.-Z.; Ma, L.-L.; Yu, D.-H. Metformin protects the brain against ischemia/reperfusion injury through PI3K/Akt1/JNK3 signaling pathways in rats. Physiol. Behav. 2017, 170, 115–123. [Google Scholar] [CrossRef]
- Wen, X.-R.; Fu, Y.-Y.; Liu, H.-Z.; Wu, J.; Shao, X.-P.; Zhang, X.-B.; Tang, M.; Shi, Y.; Ma, K.; Zhang, F.; et al. Neuroprotection of Sevoflurane Against Ischemia/Reperfusion-Induced Brain Injury Through Inhibiting JNK3/Caspase-3 by Enhancing Akt Signaling Pathway. Mol. Neurobiol. 2016, 53, 1661–1671. [Google Scholar] [CrossRef]
- Qi, S.-H.; Zhao, H.; Gong, J.-J.; Sun, F.-M.; Yue, J.; Guan, Q.-H.; Wang, M. Neuroprotection of paclitaxel against cerebral ischemia/reperfusion-induced brain injury through JNK3 signaling pathway. J. Recept. Signal. Transduct. Res. 2011, 31, 402–407. [Google Scholar] [CrossRef]
- Song, Y.-J.; Dai, C.-X.; Li, M.; Cui, M.-M.; Ding, X.; Zhao, X.-F.; Wang, C.-L.; Li, Z.-L.; Guo, M.-Y.; Fu, Y.-Y.; et al. The potential role of HO-1 in regulating the MLK3-MKK7-JNK3 module scaffolded by JIP1 during cerebral ischemia/reperfusion in rats. Behav. Brain Res. 2019, 359, 528–535. [Google Scholar] [CrossRef]
- Guan, Q.-H.; Pei, D.-S.; Zong, Y.-Y.; Xu, T.-L.; Zhang, G.-Y. Neuroprotection against ischemic brain injury by a small peptide inhibitor of c-Jun N-terminal kinase (JNK) via nuclear and non-nuclear pathways. Neuroscience 2006, 139, 609–627. [Google Scholar] [CrossRef]
- Pirianov, G.; Brywe, K.G.; Mallard, C.; Edwards, A.D.; Flavell, R.A.; Hagberg, H.; Mehmet, H. Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury. J. Cereb. Blood Flow Metab. 2007, 27, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Carulla, P.; Bribián, A.; Rangel, A.; Gavín, R.; Ferrer, I.; Caelles, C.; Del Río, J.A.; Llorens, F. Neuroprotective role of PrPC against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by GluR6/7-PSD-95 binding. Mol. Biol. Cell 2011, 22, 3041–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhang, C.-W.; Zhou, Y.; Wong, W.Q.; Lee, L.C.; Ong, W.Y.; Yoon, S.O.; Hong, W.; Fu, X.-Y.; Soong, T.W.; et al. APP upregulation contributes to retinal ganglion cell degeneration via JNK3. Cell Death Differ. 2018, 25, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.A.; Harder, J.M.; Fornarola, L.B.; Freeman, R.S.; Clark, A.F.; Pang, I.-H.; John, S.W.M.; Libby, R.T. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol. Dis. 2012, 46, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.-S.; Abel, G.; Klintworth, H.; Flavell, R.A.; Xia, Z. JNK3 mediates paraquat- and rotenone-induced dopaminergic neuron death. J. Neuropathol. Exp. Neurol. 2010, 69, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Wang, G.; Yang, H.-Q.; Hong, Z.; Xiao, Q.; Ren, R.-J.; Zhou, H.-Y.; Bai, L.; Chen, S.-D. K252a prevents nigral dopaminergic cell death induced by 6-OHDA through inhibition of both MLK3/JNK3 and ASK1/JNK3 signaling pathways. Mol. Pharm. 2007. [Google Scholar] [CrossRef]
- Pan, J.; Xiao, Q.; Sheng, C.-Y.; Hong, Z.; Yang, H.-Q.; Wang, G.; Ding, J.-Q.; Chen, S.-D. Blockade of the translocation and activation of c-Jun N-terminal kinase 3 (JNK3) attenuates dopaminergic neuronal damage in mouse model of Parkinson’s disease. Neurochem. Int. 2009, 54, 418–425. [Google Scholar] [CrossRef]
- Hunot, S.; Vila, M.; Teismann, P.; Davis, R.J.; Hirsch, E.C.; Przedborski, S.; Rakic, P.; Flavell, R.A. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 665–670. [Google Scholar] [CrossRef] [Green Version]
- Nagai, H.; Noguchi, T.; Takeda, K.; Ichijo, H. Pathophysiological roles of ASK1-MAP kinase signaling pathways. J. Biochem. Mol. Biol. 2007, 40, 1–6. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef]
- Garcia, M.; Vanhoutte, P.; Pages, C.; Besson, M.-J.; Brouillet, E.; Caboche, J. The Mitochondrial Toxin 3-Nitropropionic Acid Induces Striatal Neurodegeneration via a c-Jun N-Terminal Kinase/c-Jun Module. J. Neurosci. 2002, 22, 2174–2184. [Google Scholar] [CrossRef] [Green Version]
- Junyent, F.; De Lemos, L.; Verdaguer, E.; Pallàs, M.; Folch, J.; Camins, A.; Auladell, C.; Beas-Zárate, C. Lack of Jun-N-terminal kinase 3 (JNK3) does not protect against neurodegeneration induced by 3-nitropropionic acid. Neuropathol. Appl. Neurobiol. 2012, 38, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.A.; You, Y.-M.; Pollema, S.L.; Kaminska, A.; Liu, K.; Yoshioka, K.; Björkblom, B.; Coffey, E.T.; Bagnato, C.; Han, D.; et al. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009, 12, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourmaud, S.; Paquet, C.; Dumurgier, J.; Pace, C.; Bouras, C.; Gray, F.; Laplanche, J.-L.; Meurs, E.F.; Mouton-Liger, F.; Hugon, J. Increased levels of cerebrospinal fluid JNK3 associated with amyloid pathology: Links to cognitive decline. J. Psychiatry Neurosci 2015, 40, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Standen, C.L.; Brownlees, J.; Grierson, A.J.; Kesavapany, S.; Lau, K.F.; McLoughlin, D.M.; Miller, C.C. Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer’s disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3). J. Neurochem. 2001, 76, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Scheinfeld, M.H.; Ghersi, E.; Davies, P.; D’Adamio, L. Amyloid beta protein precursor is phosphorylated by JNK-1 independent of, yet facilitated by, JNK-interacting protein (JIP)-1. J. Biol. Chem. 2003, 278, 42058–42063. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.-S.; Kao, S.-C.; Lemere, C.A.; Xia, W.; Tseng, H.-C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.-H. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef]
- Kimberly, W.T.; Zheng, J.B.; Town, T.; Flavell, R.A.; Selkoe, D.J. Physiological regulation of the beta-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. J. Neurosci. 2005, 25, 5533–5543. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Tatebayashi, Y.; Akagi, T.; Chui, D.-H.; Murayama, M.; Miyasaka, T.; Planel, E.; Tanemura, K.; Sun, X.; Hashikawa, T.; et al. Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem. 2002, 277, 42060–42065. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Fei, Z.; Luo, S.; Wang, H. MiR-335-5p Inhibits β-Amyloid (Aβ) Accumulation to Attenuate Cognitive Deficits Through Targeting c-jun-N-terminal Kinase 3 in Alzheimer’s Disease. Curr. Neurovasc. Res. 2020, 17, 93–101. [Google Scholar] [CrossRef]
- Xu, Y.; Hou, X.-Y.; Liu, Y.; Zong, Y.-Y. Different protection of K252a and N-acetyl-L-cysteine against amyloid-beta peptide-induced cortical neuron apoptosis involving inhibition of MLK3-MKK7-JNK3 signal cascades. J. Neurosci. Res. 2009, 87, 918–927. [Google Scholar] [CrossRef]
- McGuire, J.L.; Depasquale, E.A.; Funk, A.J.; O’Donnovan, S.M.; Hasselfeld, K.; Marwaha, S.; Hammond, J.H.; Hartounian, V.; Meador-Woodruff, J.H.; Meller, J.; et al. Abnormalities of signal transduction networks in chronic schizophrenia. NPJ Schizophr 2017, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Funk, A.J.; McCullumsmith, E.R.; Haroutunian, V.; Meador-Woodruff, J.M. Abnormal Activity of the MAPK and cAMP-Associated Signaling Pathways in Frontal Cortical Areas in Postmortem Brain in Schizophrenia. Neuropsychopharmacology 2012, 37, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, J.; Chen, X.; Cai, B.; Chen, G. A logical relationship for schizophrenia, bipolar, and major depressive disorder. Part 4: Evidence from chromosome 4 high-density association screen. J. Comp. Neurol. 2019, 527, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Openshaw, R.L.; Kwon, J.; McColl, A.; Penninger, J.M.; Cavanagh, J.; Pratt, J.A.; Morris, B.J. JNK signalling mediates aspects of maternal immune activation: Importance of maternal genotype in relation to schizophrenia risk. J. Neuroinflamm. 2019, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Watts, M.E.; Rubin, L.L. MAP4K4 Activation Mediates Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Cell Rep. 2019, 26, 1143–1156.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genabai, N.K.; Ahmad, S.; Zhang, Z.; Jiang, X.; Gabaldon, C.A.; Gangwani, L. Genetic inhibition of JNK3 ameliorates spinal muscular atrophy. Human Molec. Gen. 2015, 24, 6986–7004. [Google Scholar] [CrossRef] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Z.-Y.; Kawasaki, Y.; Tan, P.-H.; Wen, Y.-R.; Huang, J.; Ji, R.-R. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav. Immun. 2007, 21, 642–651. [Google Scholar] [CrossRef] [Green Version]
- Gourmaud, S.; Thomas, P.; Thomasseau, S.; Tible, M.; Abadie, C.; Paquet, C.; Hugon, J. Brimapitide Reduced Neuronal Stress Markers and Cognitive Deficits in 5XFAD Transgenic Mice. J. Alzheimers Dis. 2018, 63, 665–674. [Google Scholar] [CrossRef]
- Orejana, L.; Barros-Miñones, L.; Aguirre, N.; Puerta, E. Implication of JNK pathway on tau pathology and cognitive decline in a senescence-accelerated mouse model. Exp. Gerontol. 2013, 48, 565–571. [Google Scholar] [CrossRef]
- Schellino, R.; Boido, M.; Borsello, T.; Vercelli, A. Pharmacological c-Jun NH2-Terminal Kinase (JNK) Pathway Inhibition Reduces Severity of Spinal Muscular Atrophy Disease in Mice. Front. Mol. Neurosci. 2018, 11, 308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staecker, H.; Jokovic, G.; Karpishchenko, S.; Kienle-Gogolok, A.; Krzyzaniak, A.; Lin, C.-D.; Navratil, P.; Tzvetkov, V.; Wright, N.; Meyer, T. Efficacy and Safety of AM-111 in the Treatment of Acute Unilateral Sudden Deafness-A Double-blind, Randomized, Placebo-controlled Phase 3 Study. Otol. Neurotol. 2019, 40, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Suckfuell, M.; Lisowska, G.; Domka, W.; Kabacinska, A.; Morawski, K.; Bodlaj, R.; Klimak, P.; Kostrica, R.; Meyer, T. Efficacy and safety of AM-111 in the treatment of acute sensorineural hearing loss: A double-blind, randomized, placebo-controlled phase II study. Otol. Neurotol. 2014, 35, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Chiquet, C.; Aptel, F.; Creuzot-Garcher, C.; Berrod, J.-P.; Kodjikian, L.; Massin, P.; Deloche, C.; Perino, J.; Kirwan, B.-A.; de Brouwer, S.; et al. Postoperative Ocular Inflammation: A Single Subconjunctival Injection of XG-102 Compared to Dexamethasone Drops in a Randomized Trial. Am. J. Ophthalmol. 2017, 174, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Relja, B.; Weber, R.; Maraslioglu, M.; Wagner, N.; Borsello, T.; Jobin, C.; Marzi, I.; Lehnert, M. Differential Relevance of NF-κB and JNK in the Pathophysiology of Hemorrhage/Resususcitation-Induced Liver Injury after Chronic Ethanol Feeding. PLoS ONE 2015, 10, e0137875. [Google Scholar] [CrossRef] [Green Version]
- Repici, M.; Chen, X.; Morel, M.-P.; Doulazmi, M.; Sclip, A.; Cannaya, V.; Veglianese, P.; Kraftsik, R.; Mariani, J.; Borsello, T.; et al. Specific inhibition of the JNK pathway promotes locomotor recovery and neuroprotection after mouse spinal cord injury. Neurobiol. Dis. 2012, 46, 710–721. [Google Scholar] [CrossRef]
- Tran, H.T.; Sanchez, L.; Brody, D.L. Inhibition of JNK by a peptide inhibitor reduces traumatic brain injury-induced tauopathy in transgenic mice. J. Neuropathol. Exp. Neurol. 2012, 71, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Ortolano, F.; Colombo, A.; Zanier, E.R.; Sclip, A.; Longhi, L.; Perego, C.; Stocchetti, N.; Borsello, T.; De Simoni, M.G. c-Jun N-terminal kinase pathway activation in human and experimental cerebral contusion. J. Neuropathol. Exp. Neurol. 2009, 68, 964–971. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-K.; Yeap, Y.Y.C.; Bogoyevitch, M.A. The JNK1/JNK3 interactome--contributions by the JNK3 unique N-terminus and JNK common docking site residues. Biochem. Biophys. Res. Commun. 2014, 453, 576–581. [Google Scholar] [CrossRef]
- Dou, X.; Huang, H.; Li, Y.; Jiang, L.; Wang, Y.; Jin, H.; Jiao, N.; Zhang, L.; Zhang, L.; Liu, Z. Multistage Screening Reveals 3-Substituted Indolin-2-one Derivatives as Novel and Isoform-Selective c-Jun N-terminal Kinase 3 (JNK3) Inhibitors: Implications to Drug Discovery for Potential Treatment of Neurodegenerative Diseases. J. Med. Chem. 2019, 62, 6645–6664. [Google Scholar] [CrossRef]
- Rajan, R.K.; Ramanathan, M. Identification and neuroprotective evaluation of a potential c-Jun N-terminal kinase 3 inhibitor through structure-based virtual screening and in-vitro assay. J. Comput. Aided Mol. Des. 2020, 34, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Schneider, L.S.; Mangialasche, F.; Andreasen, N.; Feldman, H.; Giacobini, E.; Jones, R.; Mantua, V.; Mecocci, P.; Pani, L.; Winblad, B.; et al. Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J. Intern. Med. 2014, 275, 251–283. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-L.; Gao, C.-M. c-Jun N-terminal kinase 3 signalling serves a potential role as a biomarker for determining the pathogenesis of Parkinson’s disease. Mol. Med. Rep. 2018, 17, 3255–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musi, C.A.; Agrò, G.; Santarella, F.; Iervasi, E.; Borsello, T. JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases. Cells 2020, 9, 2190. https://doi.org/10.3390/cells9102190
Musi CA, Agrò G, Santarella F, Iervasi E, Borsello T. JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases. Cells. 2020; 9(10):2190. https://doi.org/10.3390/cells9102190
Chicago/Turabian StyleMusi, Clara Alice, Graziella Agrò, Francesco Santarella, Erika Iervasi, and Tiziana Borsello. 2020. "JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases" Cells 9, no. 10: 2190. https://doi.org/10.3390/cells9102190
APA StyleMusi, C. A., Agrò, G., Santarella, F., Iervasi, E., & Borsello, T. (2020). JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases. Cells, 9(10), 2190. https://doi.org/10.3390/cells9102190