Neurofibromin Structure, Functions and Regulation




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
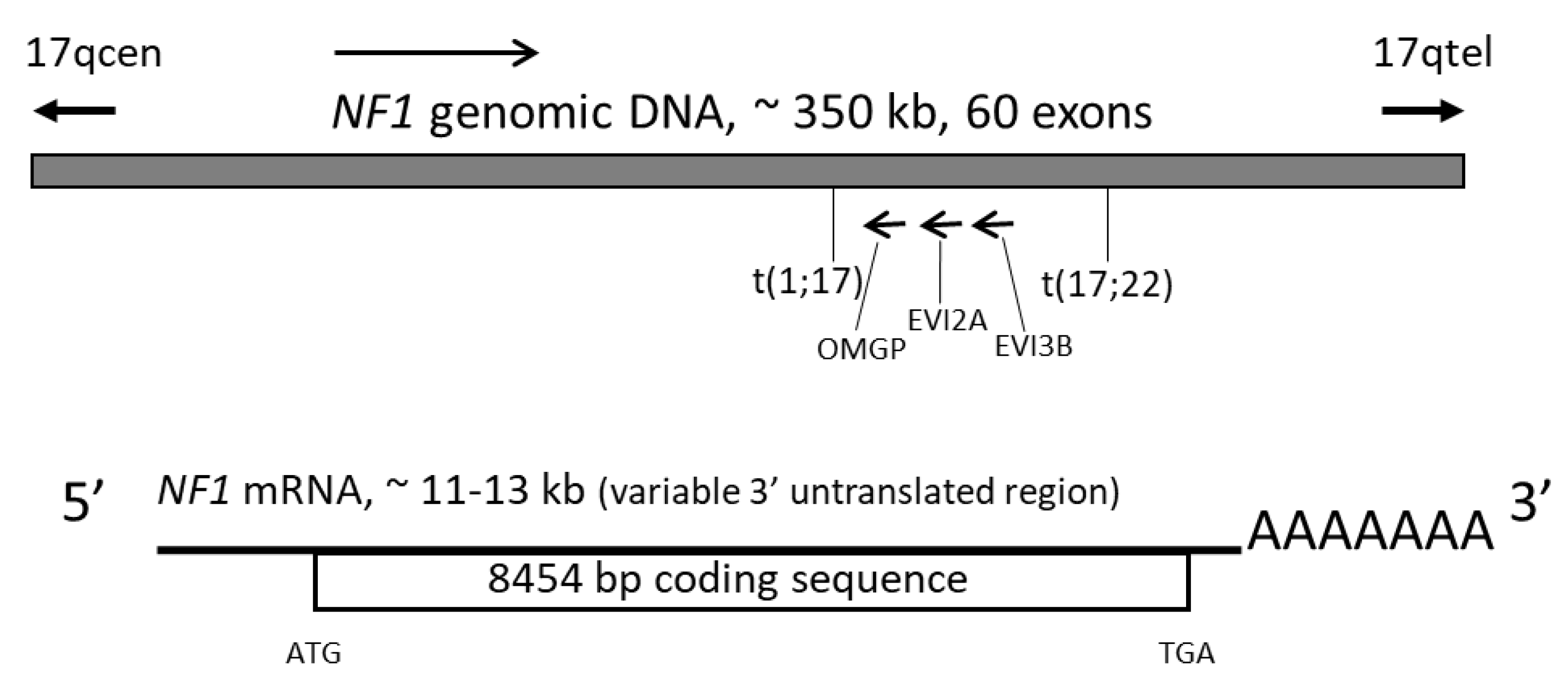
2. The NF1 Gene
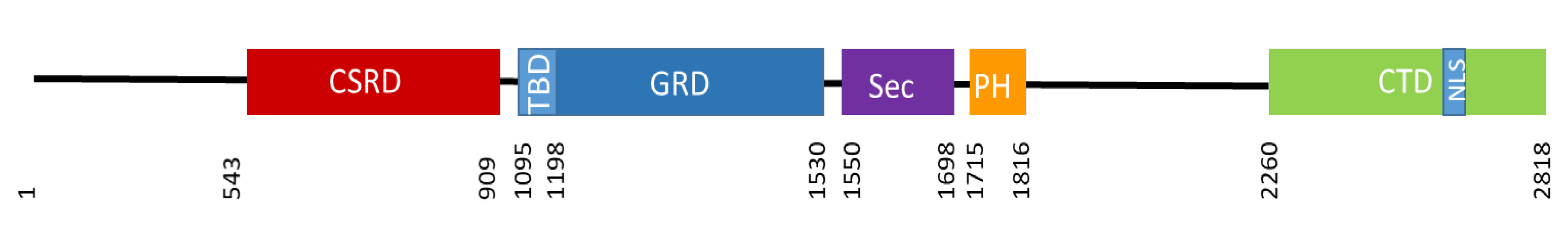
3. Neurofibromin Protein
4. Neurofibromin Isoforms
4.1. Alternative Splicing of Exon 23a
4.1.1. Type 1 and Type 2 Isoforms
4.1.2. Differences in the Function of the Two Isoforms
4.1.3. Expression of the Two Isoforms
4.2. Alternative Splicing of Exon 48a
4.3. Alternative Splicing of Exon 9a
4.4. Alternative Splicing of Exon 10a-2
4.5. Other Isoforms
5. Neurofibromin Structure and Domains
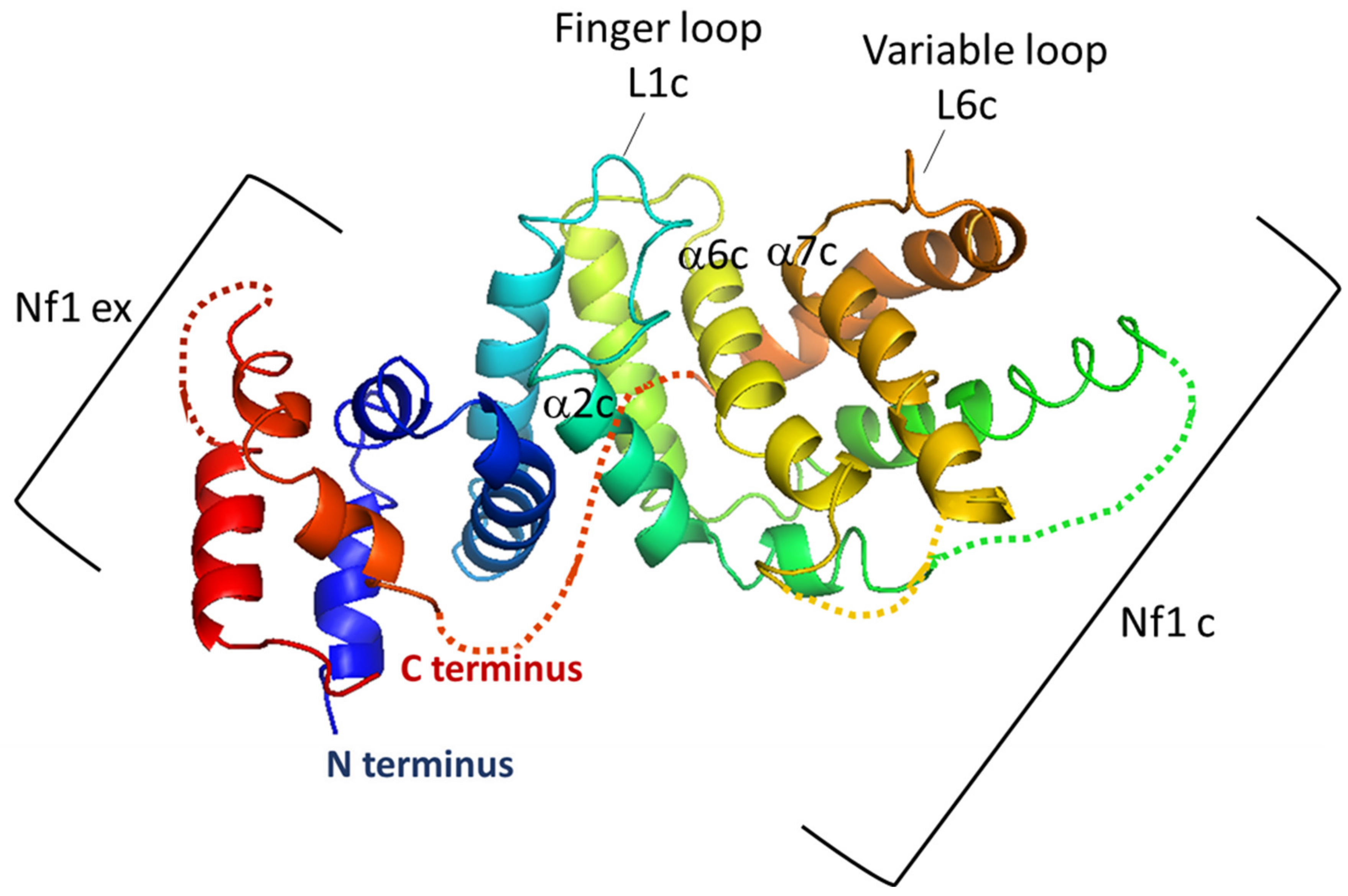
5.1. The GAP-Related Domain (GRD)
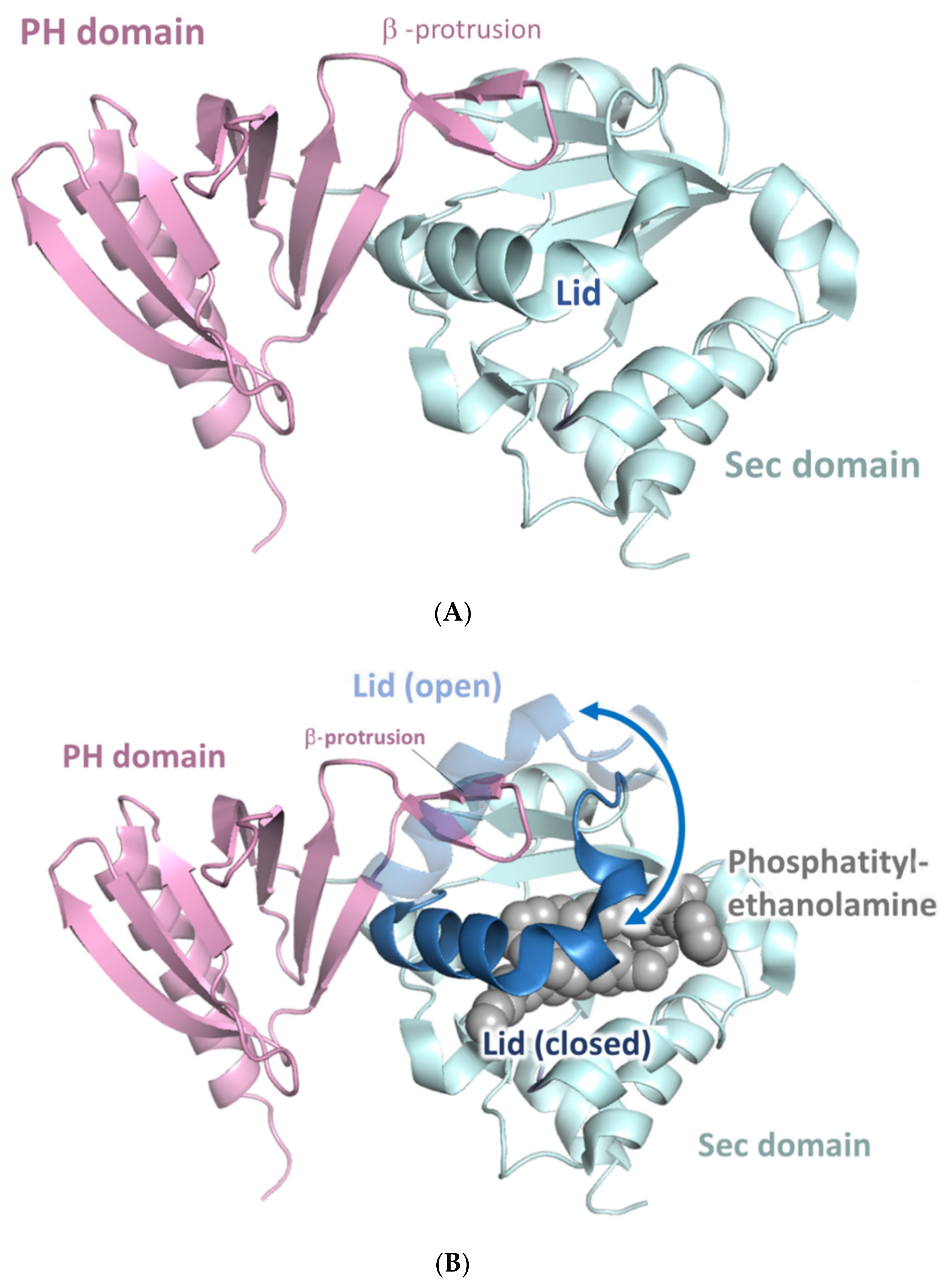
5.2. The SecPH Domain
5.3. The Cysteine-Serine Rich and C-Terminal Domains (CSRD and CTD)
5.4. Structural Data on Full-Length Neurofibromin
6. Localization
6.1. Association with Cytoskeletal Structures
6.2. Nuclear Localization
6.3. Plasma Membrane Localization
6.4. Other Reported Localization
7. Neurofibromin Functions
7.1. Ras-GAP Activity
7.1.1. Role in Cell Growth
7.1.2. Role in Learning
7.2. cAMP Regulation
7.2.1. Neurofibromin is a Positive Regulator of cAMP Levels in Various Cell Types in Both a Ras-Dependent and Ras-Independent Manner
7.2.2. Detailed Molecular Mechanisms of Neurofibromin-Mediated cAMP Regulation
7.3. Regulation of Dopamine Levels
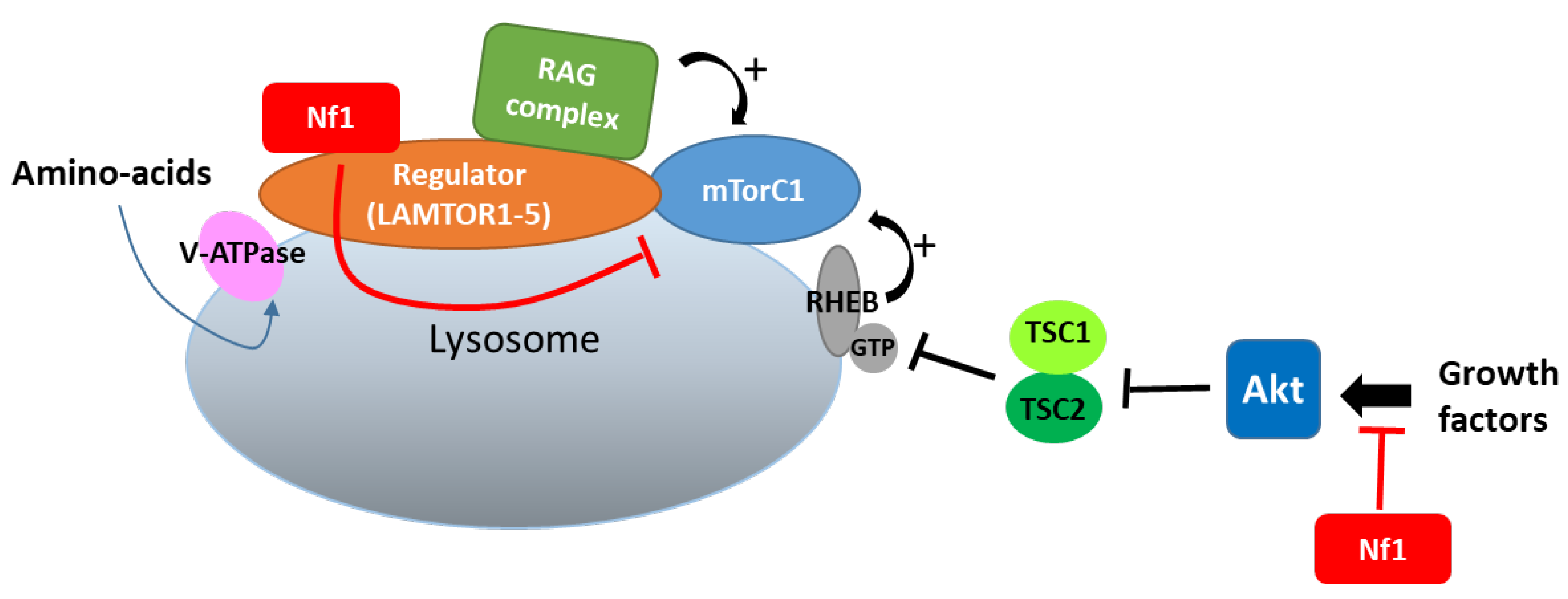
7.4. Regulation of mTOR Signaling
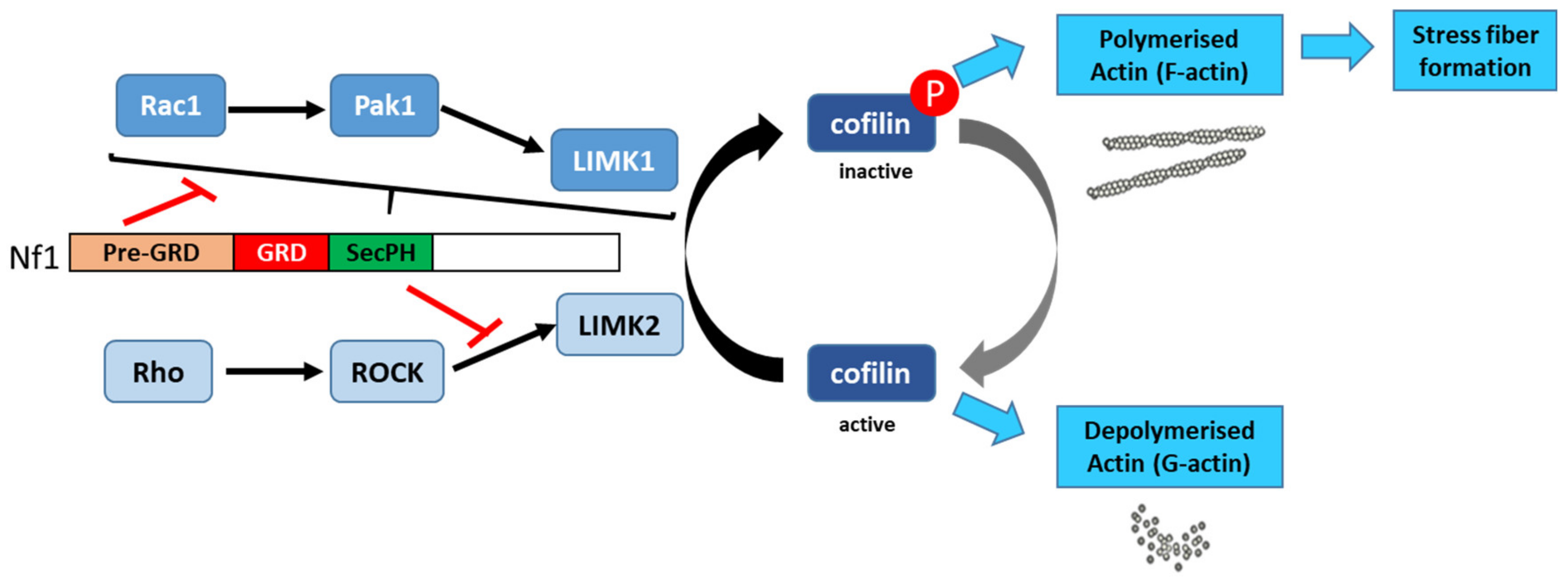
7.5. Control of Actin Cytoskeleton Organization
7.6. Microtubule-Dependent Transport in Melanocytes, Neurons, and Schwann Cells
7.7. Cell Cycle
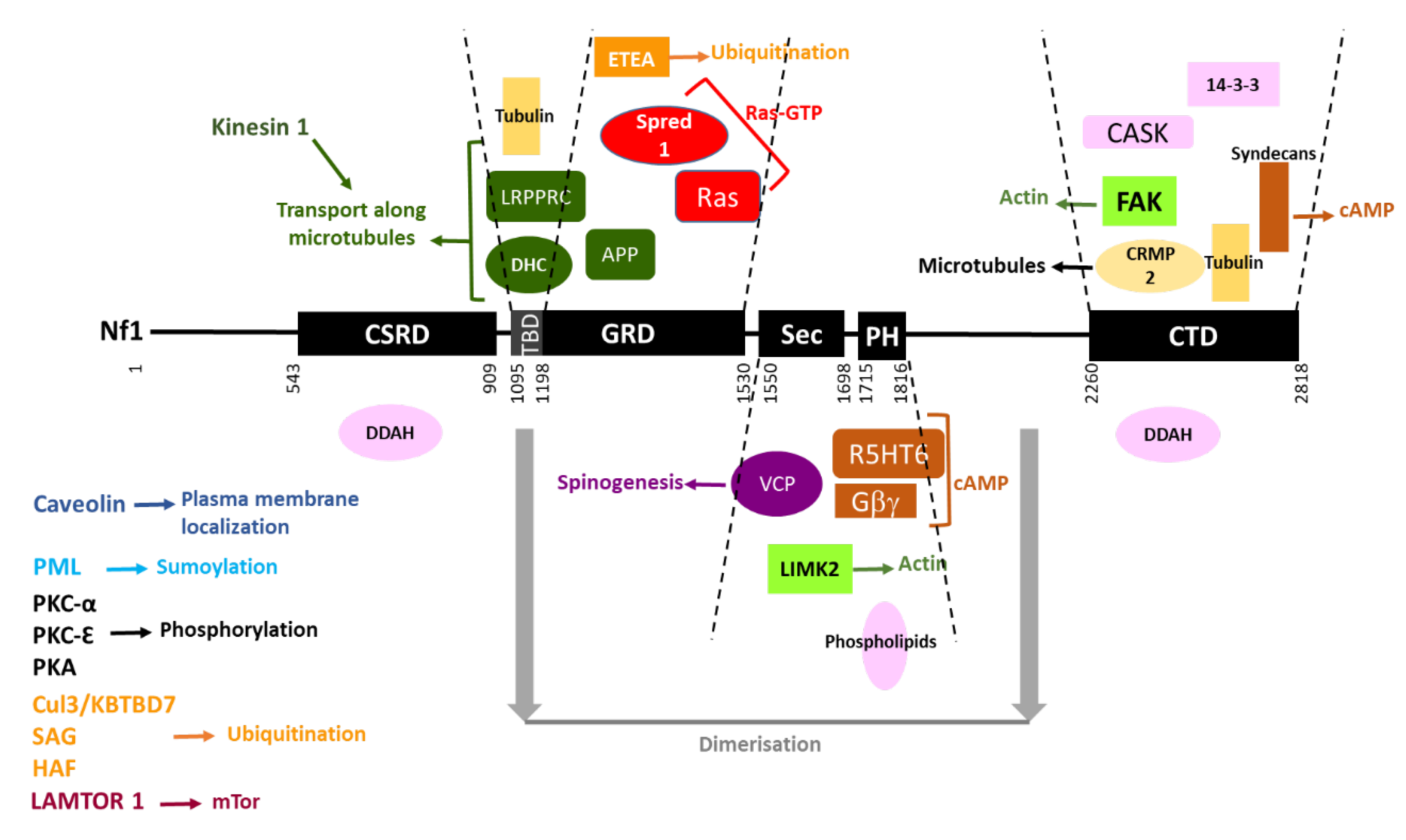
7.8. Neurofibromin Interactions
7.8.1. Partners Previously Mentioned in This Review
7.8.2. Other Partners
CRMP2
VCP
Neurofibromin Dimerization
Other Partners
8. Regulation of Neurofibromin Functions by Post-Translational Modifications (PTMs)
8.1. Phosphorylation
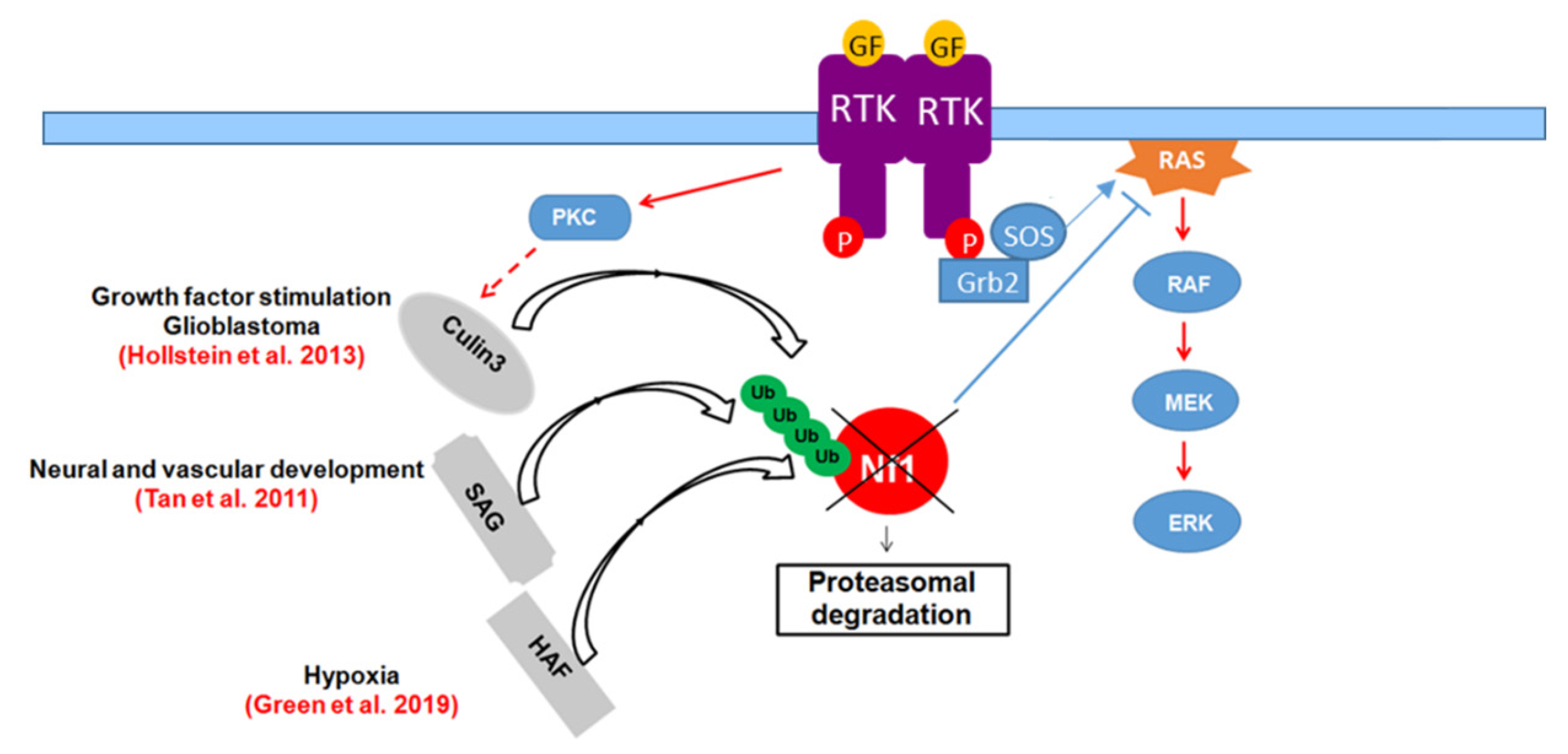
8.2. Ubiquitination
8.3. SUMOylation
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Viskochil, D.; Buchberg, A.M.; Xu, G.; Cawthon, R.M.; Stevens, J.; Wolff, R.K.; Culver, M.; Carey, J.C.; Copeland, N.G.; Jenkins, N.A.; et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990, 62, 187–192. [Google Scholar] [CrossRef]
- Wallace, M.R.; Andersen, L.B.; Fountain, J.W.; Odeh, H.M.; Viskochil, D.; Marchuk, D.A.; O’Connell, P.; White, R.; Collins, F.S. A chromosome jump crosses a translocation breakpoint in the von Recklinghausen neurofibromatosis region. Genes Chromosomes Cancer 1990, 2, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Henkemeyer, M.; Rossi, D.J.; Holmyard, D.P.; Puri, M.C.; Mbamalu, G.; Harpal, K.; Shih, T.S.; Jacks, T.; Pawson, T. Vascular system defects and neuronal apoptosis in mice lacking ras GTPase-activating protein. Nature 1995, 377, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Chen, Y.; Callens, T.; Gomes, A.; Sharp, A.; Johnson, S.; Hsiao, M.C.; Chen, Z.; Balasubramanian, M.; Barnett, C.P.; et al. Genotype-Phenotype Correlation in NF1: Evidence for a More Severe Phenotype Associated with Missense Mutations Affecting NF1 Codons 844–848. Am. J. Hum. Genet. 2018, 102, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Ferner, R.E.; Listernick, R.H.; Korf, B.R.; Wolters, P.L.; Johnson, K.J. Neurofibromatosis type 1. Nat. Rev. Dis. Prim. 2017, 3, 17004. [Google Scholar] [CrossRef]
- Philpott, C.; Tovell, H.; Frayling, I.M.; Cooper, D.N.; Upadhyaya, M. The NF1 somatic mutational landscape in sporadic human cancers. Hum. Genom. 2017, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easton, D.F.; Ponder, M.A.; Huson, S.M.; Ponder, B.A. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): Evidence for modifying genes. Am. J. Hum. Genet. 1993, 53, 305–313. [Google Scholar]
- Rieley, M.B.; Stevenson, D.A.; Viskochil, D.H.; Tinkle, B.T.; Martin, L.J.; Schorry, E.K. Variable expression of neurofibromatosis 1 in monozygotic twins. Am. J. Med Genet. Part A 2011, 155A, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.; Wright, E.; Nguyen, K.; Cannon, L.; Fain, P.; Goldgar, D.; Bishop, D.T.; Carey, J.; Baty, B.; Kivlin, J.; et al. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 1987, 236, 1100–1102. [Google Scholar] [CrossRef] [Green Version]
- Cawthon, R.M.; Weiss, R.; Xu, G.F.; Viskochil, D.; Culver, M.; Stevens, J.; Robertson, M.; Dunn, D.; Gesteland, R.; O’Connell, P.; et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell 1990, 62, 193–201. [Google Scholar] [CrossRef]
- Marchuk, D.A.; Saulino, A.M.; Tavakkol, R.; Swaroop, M.; Wallace, M.R.; Andersen, L.B.; Mitchell, A.L.; Gutmann, D.H.; Boguski, M.; Collins, F.S. cDNA cloning of the type 1 neurofibromatosis gene: Complete sequence of the NF1 gene product. Genomics 1991, 11, 931–940. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Janosch, P.; Tanji, M.; Rosenfeld, G.C.; Waymire, J.C.; Mischak, H.; Kolch, W.; Sedivy, J.M. Regulation of Raf-1 kinase activity by the 14-3-3 family of proteins. EMBO J. 1995, 14, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Cawthon, R.M.; O’Connell, P.; Buchberg, A.M.; Viskochil, D.; Weiss, R.B.; Culver, M.; Stevens, J.; Jenkins, N.A.; Copeland, N.G.; White, R. Identification and characterization of transcripts from the neurofibromatosis 1 region: The sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics 1990, 7, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Cawthon, R.M.; Andersen, L.B.; Buchberg, A.M.; Xu, G.F.; O’Connell, P.; Viskochil, D.; Weiss, R.B.; Wallace, M.R.; Marchuk, D.A.; Culver, M.; et al. cDNA sequence and genomic structure of EV12B, a gene lying within an intron of the neurofibromatosis type 1 gene. Genomics 1991, 9, 446–460. [Google Scholar] [CrossRef] [Green Version]
- Viskochil, D.; Cawthon, R.; O’Connell, P.; Xu, G.F.; Stevens, J.; Culver, M.; Carey, J.; White, R. The gene encoding the oligodendrocyte-myelin glycoprotein is embedded within the neurofibromatosis type 1 gene. Mol. Cell Biol. 1991, 11, 906–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.H.; Harper, P.S.; Upadhyaya, M. Molecular genetics of neurofibromatosis type 1 (NF1). J. Med. Genet. 1996, 33, 2–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutmann, D.H.; Collins, F.S. Neurofibromatosis type 1. Beyond positional cloning. Arch. Neurol. 1993, 50, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- Buchberg, A.M.; Cleveland, L.S.; Jenkins, N.A.; Copeland, N.G. Sequence homology shared by neurofibromatosis type-1 gene and IRA-1 and IRA-2 negative regulators of the RAS cyclic AMP pathway. Nature 1990, 347, 291–294. [Google Scholar] [CrossRef]
- Xu, G.F.; O’Connell, P.; Viskochil, D.; Cawthon, R.; Robertson, M.; Culver, M.; Dunn, D.; Stevens, J.; Gesteland, R.; White, R.; et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 1990, 62, 599–608. [Google Scholar] [CrossRef]
- Xu, G.F.; Lin, B.; Tanaka, K.; Dunn, D.; Wood, D.; Gesteland, R.; White, R.; Weiss, R.; Tamanoi, F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 1990, 63, 835–841. [Google Scholar] [CrossRef]
- Martin, G.A.; Viskochil, D.; Bollag, G.; McCabe, P.C.; Crosier, W.J.; Haubruck, H.; Conroy, L.; Clark, R.; O’Connell, P.; Cawthon, R.M.; et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63, 843–849. [Google Scholar] [CrossRef]
- Ballester, R.; Marchuk, D.; Boguski, M.; Saulino, A.; Letcher, R.; Wigler, M.; Collins, F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 1990, 63, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Kim, J.; Song, K. The C-terminal domains of human neurofibromin and its budding yeast homologs Ira1 and Ira2 regulate the metaphase to anaphase transition. Cell Cycle 2014, 13, 2780–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeClue, J.E.; Cohen, B.D.; Lowy, D.R. Identification and characterization of the neurofibromatosis type 1 protein product. Proc. Natl. Acad. Sci. USA 1991, 88, 9914–9918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daston, M.M.; Scrable, H.; Nordlund, M.; Sturbaum, A.K.; Nissen, L.M.; Ratner, N. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron 1992, 8, 415–428. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Wood, D.L.; Collins, F.S. Identification of the neurofibromatosis type 1 gene product. Proc. Natl. Acad. Sci. USA 1991, 88, 9658–9662. [Google Scholar] [CrossRef] [Green Version]
- Golovnina, K.; Blinov, A.; Chang, L.S. Evolution and origin of neurofibromin, the product of the neurofibromatosis type 1 (NF1) tumor-suppressor gene. BGRS 2006, 3142–3146. [Google Scholar]
- Bartelt-Kirbach, B.; Kaufmann, D. Insights into NF1 from Evolution; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Bernards, A.; Snijders, A.J.; Hannigan, G.E.; Murthy, A.E.; Gusella, J.F. Mouse neurofibromatosis type 1 cDNA sequence reveals high degree of conservation of both coding and non-coding mRNA segments. Hum. Mol. Genet. 1993, 2, 645–650. [Google Scholar] [CrossRef]
- White, K.A.; Swier, V.J.; Cain, J.T.; Kohlmeyer, J.L.; Meyerholz, D.K.; Tanas, M.R.; Uthoff, J.; Hammond, E.; Li, H.; Rohret, F.A.; et al. A porcine model of neurofibromatosis type 1 that mimics the human disease. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Assum, G.; Schmegner, C. NF1 Gene Evolution in Mammals; Karger AG: Basel, Switzerland, 2008; Volume 16. [Google Scholar]
- Vandenbroucke, I.; Vandesompele, J.; De Paepe, A.; Messiaen, L. Quantification of NF1 transcripts reveals novel highly expressed splice variants. FEBS Lett. 2002, 522, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Andersen, L.B.; Ballester, R.; Marchuk, D.A.; Chang, E.; Gutmann, D.H.; Saulino, A.M.; Camonis, J.; Wigler, M.; Collins, F.S. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell Biol. 1993, 13, 487–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishi, T.; Lee, P.S.; Oka, K.; Levin, V.A.; Tanase, S.; Morino, Y.; Saya, H. Differential expression of two types of the neurofibromatosis type 1 (NF1) gene transcripts related to neuronal differentiation. Oncogene 1991, 6, 1555–1559. [Google Scholar] [PubMed]
- Hinman, M.N.; Sharma, A.; Luo, G.; Lou, H. Neurofibromatosis type 1 alternative splicing is a key regulator of Ras signaling in neurons. Mol. Cell Biol. 2014, 34, 2188–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Hinman, M.N.; Guo, X.; Sharma, A.; Arakawa, H.; Luo, G.; Lou, H. Neurofibromatosis type 1 alternative splicing is a key regulator of Ras/ERK signaling and learning behaviors in mice. Hum. Mol. Genet. 2017, 26, 3797–3807. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Geist, R.T.; Wright, D.E.; Snider, W.D. Expression of the neurofibromatosis 1 (NF1) isoforms in developing and adult rat tissues. Cell Growth Differ. 1995, 6, 315–323. [Google Scholar] [PubMed]
- Gutmann, D.H.; Geist, R.T.; Rose, K.; Wright, D.E. Expression of two new protein isoforms of the neurofibromatosis type 1 gene product, neurofibromin, in muscle tissues. Dev. Dyn. 1995, 202, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Danglot, G.; Régnier, V.; Fauvet, D.; Vassal, G.; Kujas, M.; Bernheim, A. Neurofibromatosis 1 (NF1) mRNAs expressed in the central nervous system are differentially spliced in the 5’ part of the gene. Hum. Mol. Genet. 1995, 4, 915–920. [Google Scholar] [CrossRef]
- Geist, R.T.; Gutmann, D.H. Expression of a developmentally-regulated neuron-specific isoform of the neurofibromatosis 1 (NF1) gene. Neurosci. Lett. 1996, 211, 85–88. [Google Scholar] [CrossRef]
- Kaufmann, D.; Müller, R.; Kenner, O.; Leistner, W.; Hein, C.; Vogel, W.; Bartelt, B. The N-terminal splice product NF1-10a-2 of the NF1 gene codes for a transmembrane segment. Biochem. Biophys. Res. Commun. 2002, 294, 496–503. [Google Scholar] [CrossRef]
- Vandenbroucke, I.; Van Oostveldt, P.; Coene, E.; De Paepe, A.; Messiaen, L. Neurofibromin is actively transported to the nucleus. FEBS Lett. 2004, 560, 98–102. [Google Scholar] [CrossRef]
- Sherekar, M.; Han, S.W.; Ghirlando, R.; Messing, S.; Drew, M.; Rabara, D.; Waybright, T.; Juneja, P.; O’Neill, H.; Stanley, C.B.; et al. Biochemical and structural analyses reveal that the tumor suppressor neurofibromin (NF1) forms a high-affinity dimer. J. Biol. Chem. 2020, 295, 1105–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffzek, K.; Ahmadian, M.R.; Wiesmüller, L.; Kabsch, W.; Stege, P.; Schmitz, F.; Wittinghofer, A. Structural analysis of the GAP-related domain from neurofibromin and its implications. EMBO J. 1998, 17, 4313–4327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffzek, K.; Lautwein, A.; Scherer, A.; Franken, S.; Wittinghofer, A. Crystallization and preliminary X-ray crystallographic study of the Ras-GTPase-activating domain of human p120GAP. Proteins 1997, 27, 315–318. [Google Scholar] [CrossRef]
- Scheffzek, K.; Shivalingaiah, G. Ras-Specific GTPase-Activating Proteins-Structures, Mechanisms, and Interactions. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Neuwald, A.F.; Ponting, C.P. Sec14p-like domains in NF1 and Dbl-like proteins indicate lipid regulation of Ras and Rho signaling. Curr. Biol. 1999, 9, R195–R197. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, F.; D’Angelo, I.; Welti, S.; Stier, G.; Ylänne, J.; Scheffzek, K. Expression, purification and preliminary crystallographic characterization of a novel segment from the neurofibromatosis type 1 protein. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2364–2367. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, I.; Welti, S.; Bonneau, F.; Scheffzek, K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Rep. 2006, 7, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Sha, B.; Phillips, S.E.; Bankaitis, V.A.; Luo, M. Crystal structure of the Saccharomyces cerevisiae phosphatidylinositol-transfer protein. Nature 1998, 391, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Welti, S.; Fraterman, S.; D’Angelo, I.; Wilm, M.; Scheffzek, K. The sec14 homology module of neurofibromin binds cellular glycerophospholipids: Mass spectrometry and structure of a lipid complex. J. Mol. Biol. 2007, 366, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, F.; Lenherr, E.D.; Pena, V.; Hart, D.J.; Scheffzek, K. Solubility survey of fragments of the neurofibromatosis type 1 protein neurofibromin. Protein Expr. Purif. 2009, 65, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Izawa, I.; Tamaki, N.; Saya, H. Phosphorylation of neurofibromatosis type 1 gene product (neurofibromin) by cAMP-dependent protein kinase. FEBS Lett. 1996, 382, 53–59. [Google Scholar] [CrossRef] [Green Version]
- Mangoura, D.; Sun, Y.; Li, C.; Singh, D.; Gutmann, D.H.; Flores, A.; Ahmed, M.; Vallianatos, G. Phosphorylation of neurofibromin by PKC is a possible molecular switch in EGF receptor signaling in neural cells. Oncogene 2006, 25, 735–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuo, H.; Yunoue, S.; Feng, L.; Kimoto, M.; Tsuji, H.; Ono, T.; Saya, H.; Araki, N. Phosphorylation of neurofibromin by cAMP-dependent protein kinase is regulated via a cellular association of N(G),N(G)-dimethylarginine dimethylaminohydrolase. FEBS Lett. 2001, 494, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Yunoue, S.; Tokuo, H.; Ozawa, T.; Zhang, D.; Patrakitkomjorn, S.; Ichimura, T.; Saya, H.; Araki, N. PKA phosphorylation and 14-3-3 interaction regulate the function of neurofibromatosis type I tumor suppressor, neurofibromin. FEBS Lett. 2004, 557, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Koliou, X.; Fedonidis, C.; Kalpachidou, T.; Mangoura, D. Nuclear import mechanism of neurofibromin for localization on the spindle and function in chromosome congression. J. Neurochem. 2016, 136, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Patrakitkomjorn, S.; Kobayashi, D.; Morikawa, T.; Wilson, M.M.; Tsubota, N.; Irie, A.; Ozawa, T.; Aoki, M.; Arimura, N.; Kaibuchi, K.; et al. Neurofibromatosis type 1 (NF1) tumor suppressor, neurofibromin, regulates the neuronal differentiation of PC12 cells via its associating protein, CRMP-2. J. Biol. Chem. 2008, 283, 9399–9413. [Google Scholar] [CrossRef] [Green Version]
- Hsueh, Y.P.; Roberts, A.M.; Volta, M.; Sheng, M.; Roberts, R.G. Bipartite interaction between neurofibromatosis type I protein (neurofibromin) and syndecan transmembrane heparan sulfate proteoglycans. J. Neurosci. 2001, 21, 3764–3770. [Google Scholar] [CrossRef]
- Kweh, F.; Zheng, M.; Kurenova, E.; Wallace, M.; Golubovskaya, V.; Cance, W.G. Neurofibromin physically interacts with the N-terminal domain of focal adhesion kinase. Mol. Carcinog. 2009, 48, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Volta, M.; Calza, S.; Roberts, A.M.; Roberts, R.G. Characterisation of the interaction between syndecan-2, neurofibromin and CASK: Dependence of interaction on syndecan dimerization. Biochem. Biophys. Res. Commun. 2010, 391, 1216–1221. [Google Scholar] [CrossRef]
- Yang, H.W.; Hong, H.L.; Luo, W.W.; Dai, C.M.; Chen, X.Y.; Wang, L.P.; Li, Q.; Li, Z.Q.; Liu, P.Q.; Li, Z.M. mTORC2 facilitates endothelial cell senescence by suppressing Nrf2 expression via the Akt/GSK-3β/C/EBPα signaling pathway. Acta Pharmacol. Sin. 2018, 39, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; McCormick, F.; Clark, R. Characterization of full-length neurofibromin: Tubulin inhibits Ras GAP activity. EMBO J. 1993, 12, 1923–1927. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.E.; Gutmann, D.H.; Mitchell, A.; Park, S.; Boguski, M.; Jacks, T.; Wood, D.L.; Jove, R.; Collins, F.S. Neurofibromatosis type 1 gene product (neurofibromin) associates with microtubules. Somat. Cell Mol. Genet. 1993, 19, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Gutmann, D.H. Mutations in the GAP-related domain impair the ability of neurofibromin to associate with microtubules. Brain Res. 1997, 759, 149–152. [Google Scholar] [CrossRef]
- Li, C.; Cheng, Y.; Gutmann, D.A.; Mangoura, D. Differential localization of the neurofibromatosis 1 (NF1) gene product, neurofibromin, with the F-actin or microtubule cytoskeleton during differentiation of telencephalic neurons. Brain Res. Dev. Brain Res. 2001, 130, 231–248. [Google Scholar] [CrossRef]
- Koivunen, J.; Ylä-Outinen, H.; Korkiamäki, T.; Karvonen, S.L.; Pöyhönen, M.; Laato, M.; Karvonen, J.; Peltonen, S.; Peltonen, J. New function for NF1 tumor suppressor. J. Investgi. Dermatol. 2000, 114, 473–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villén, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar] [CrossRef] [Green Version]
- Nousiainen, M.; Silljé, H.H.; Sauer, G.; Nigg, E.A.; Körner, R. Phosphoproteome analysis of the human mitotic spindle. Proc. Natl. Acad. Sci. USA 2006, 103, 5391–5396. [Google Scholar] [CrossRef] [Green Version]
- Leondaritis, G.; Petrikkos, L.; Mangoura, D. Regulation of the Ras-GTPase activating protein neurofibromin by C-tail phosphorylation: Implications for protein kinase C/Ras/extracellular signal-regulated kinase 1/2 pathway signaling and neuronal differentiation. J. Neurochem. 2009, 109, 573–583. [Google Scholar] [CrossRef]
- Godin, F.; Villette, S.; Vallée, B.; Doudeau, M.; Morisset-Lopez, S.; Ardourel, M.; Hevor, T.; Pichon, C.; Bénédetti, H. A fraction of neurofibromin interacts with PML bodies in the nucleus of the CCF astrocytoma cell line. Biochem. Biophys. Res. Commun. 2012, 418, 689–694. [Google Scholar] [CrossRef]
- Stowe, I.B.; Mercado, E.L.; Stowe, T.R.; Bell, E.L.; Oses-Prieto, J.A.; Hernández, H.; Burlingame, A.L.; McCormick, F. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012, 26, 1421–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; De Schepper, S.; Fryns, J.P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.; Yusoff, P.; Wong, E.S.; Chandramouli, S.; Lao, D.H.; Fong, C.W.; Guy, G.R. The cysteine-rich sprouty translocation domain targets mitogen-activated protein kinase inhibitory proteins to phosphatidylinositol 4,5-bisphosphate in plasma membranes. Mol. Cell Biol. 2002, 22, 7953–7966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunzendorfer-Matt, T.; Mercado, E.L.; Maly, K.; McCormick, F.; Scheffzek, K. The neurofibromin recruitment factor Spred1 binds to the GAP related domain without affecting Ras inactivation. Proc. Natl. Acad. Sci. USA 2016, 113, 7497–7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Markegard, E.; Dharmaiah, S.; Urisman, A.; Drew, M.; Esposito, D.; Scheffzek, K.; Nissley, D.V.; McCormick, F.; Simanshu, D.K. Structural Insights into the SPRED1-Neurofibromin-KRAS Complex and Disruption of SPRED1-Neurofibromin Interaction by Oncogenic EGFR. Cell Rep. 2020, 32, 107909. [Google Scholar] [CrossRef] [PubMed]
- Boyanapalli, M.; Lahoud, O.B.; Messiaen, L.; Kim, B.; Anderle de Sylor, M.S.; Duckett, S.J.; Somara, S.; Mikol, D.D. Neurofibromin binds to caveolin-1 and regulates ras, FAK, and Akt. Biochem. Biophys. Res. Commun. 2006, 340, 1200–1208. [Google Scholar] [CrossRef]
- Nordlund, M.; Gu, X.; Shipley, M.T.; Ratner, N. Neurofibromin is enriched in the endoplasmic reticulum of CNS neurons. J. Neurosci. 1993, 13, 1588–1600. [Google Scholar] [CrossRef] [Green Version]
- Roudebush, M.; Slabe, T.; Sundaram, V.; Hoppel, C.L.; Golubic, M.; Stacey, D.W. Neurofibromin colocalizes with mitochondria in cultured cells. Exp. Cell Res. 1997, 236, 161–172. [Google Scholar] [CrossRef]
- De Schepper, S.; Boucneau, J.M.; Westbroek, W.; Mommaas, M.; Onderwater, J.; Messiaen, L.; Naeyaert, J.M.; Lambert, J.L. Neurofibromatosis type 1 protein and amyloid precursor protein interact in normal human melanocytes and colocalize with melanosomes. J. Investgi. Dermatol. 2006, 126, 653–659. [Google Scholar] [CrossRef] [Green Version]
- Basu, T.N.; Gutmann, D.H.; Fletcher, J.A.; Glover, T.W.; Collins, F.S.; Downward, J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature 1992, 356, 713–715. [Google Scholar] [CrossRef]
- DeClue, J.E.; Papageorge, A.G.; Fletcher, J.A.; Diehl, S.R.; Ratner, N.; Vass, W.C.; Lowy, D.R. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 1992, 69, 265–273. [Google Scholar] [CrossRef]
- Bollag, G.; McCormick, F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature 1991, 351, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, K.K.; Ingram, D.A.; Zhang, Y.; Bollag, G.; Clapp, D.W. Neurofibromin GTPase-activating protein-related domains restore normal growth in Nf1-/- cells. J. Biol. Chem. 2001, 276, 7240–7245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, B.; Gutmann, D.H. Neurofibromin regulates neural stem cell proliferation, survival, and astroglial differentiation in vitro and in vivo. J. Neurosci. 2005, 25, 5584–5594. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.F.; Yasuda, R. Neurofibromin is the major ras inactivator in dendritic spines. J. Neurosci. 2014, 34, 776–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichowski, K.; Santiago, S.; Jardim, M.; Johnson, B.W.; Jacks, T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes. Dev. 2003, 17, 449–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Y.; Vik, T.A.; Ryder, J.W.; Srour, E.F.; Jacks, T.; Shannon, K.; Clapp, D.W. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J. Exp. Med. 1998, 187, 1893–1902. [Google Scholar] [CrossRef] [Green Version]
- Hennig, A.; Markwart, R.; Wolff, K.; Schubert, K.; Cui, Y.; Prior, I.A.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Feedback activation of neurofibromin terminates growth factor-induced Ras activation. Cell Commun. Signal. 2016, 14, 5. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, B.; Li, W.; Perry, A.; Gutmann, D.H. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005, 65, 236–245. [Google Scholar]
- Widemann, B.C.; Dombi, E.; Gillespie, A.; Wolters, P.L.; Belasco, J.; Goldman, S.; Korf, B.R.; Solomon, J.; Martin, S.; Salzer, W.; et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro-Oncology 2014, 16, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.M.; Federov, N.B.; Kogan, J.H.; Murphy, G.G.; Stern, J.; Ohno, M.; Kucherlapati, R.; Jacks, T.; Silva, A.J. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002, 415, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cui, Y.; Kushner, S.A.; Brown, R.A.; Jentsch, J.D.; Frankland, P.W.; Cannon, T.D.; Silva, A.J. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol. 2005, 15, 1961–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilding, C.; McNair, K.; Stone, T.W.; Morris, B.J. Restored plasticity in a mouse model of neurofibromatosis type 1 via inhibition of hyperactive ERK and CREB. Eur. J. Neurosci. 2007, 25, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Molosh, A.I.; Johnson, P.L.; Spence, J.P.; Arendt, D.; Federici, L.M.; Bernabe, C.; Janasik, S.P.; Segu, Z.M.; Khanna, R.; Goswami, C.; et al. Social learning and amygdala disruptions in Nf1 mice are rescued by blocking p21-activated kinase. Nat. Neurosci. 2014, 17, 1583–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krab, L.C.; de Goede-Bolder, A.; Aarsen, F.K.; Pluijm, S.M.; Bouman, M.J.; van der Geest, J.N.; Lequin, M.; Catsman, C.E.; Arts, W.F.; Kushner, S.A.; et al. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: A randomized controlled trial. JAMA 2008, 300, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Vaart, T.; Plasschaert, E.; Rietman, A.B.; Renard, M.; Oostenbrink, R.; Vogels, A.; de Wit, M.C.; Descheemaeker, M.J.; Vergouwe, Y.; Catsman-Berrevoets, C.E.; et al. Simvastatin for cognitive deficits and behavioural problems in patients with neurofibromatosis type 1 (NF1-SIMCODA): A randomised, placebo-controlled trial. Lancet Neurol. 2013, 12, 1076–1083. [Google Scholar] [CrossRef]
- Payne, J.M.; Barton, B.; Ullrich, N.J.; Cantor, A.; Hearps, S.J.; Cutter, G.; Rosser, T.; Walsh, K.S.; Gioia, G.A.; Wolters, P.L.; et al. Randomized placebo-controlled study of lovastatin in children with neurofibromatosis type 1. Neurology 2016, 87, 2575–2584. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Costa, R.M.; Murphy, G.G.; Elgersma, Y.; Zhu, Y.; Gutmann, D.H.; Parada, L.F.; Mody, I.; Silva, A.J. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008, 135, 549–560. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.A.; Ratner, N.; Roberts, T.M.; Stiles, C.D. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J. Neurosci. 2001, 21, 1110–1116. [Google Scholar] [CrossRef] [Green Version]
- Warrington, N.M.; Gianino, S.M.; Jackson, E.; Goldhoff, P.; Garbow, J.R.; Piwnica-Worms, D.; Gutmann, D.H.; Rubin, J.B. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010, 70, 5717–5727. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.F.; The, I.; Hannan, F.; Bernards, A.; Zhong, Y. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 1997, 276, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Wolman, M.A.; de Groh, E.D.; McBride, S.M.; Jongens, T.A.; Granato, M.; Epstein, J.A. Modulation of cAMP and ras signaling pathways improves distinct behavioral deficits in a zebrafish model of neurofibromatosis type 1. Cell Rep. 2014, 8, 1265–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.F.; Tong, J.; Hannan, F.; Luo, L.; Zhong, Y. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature 2000, 403, 895–898. [Google Scholar] [CrossRef] [PubMed]
- The, I.; Hannigan, G.E.; Cowley, G.S.; Reginald, S.; Zhong, Y.; Gusella, J.F.; Hariharan, I.K.; Bernards, A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science 1997, 276, 791–794. [Google Scholar] [CrossRef]
- Tong, J.; Hannan, F.; Zhu, Y.; Bernards, A.; Zhong, Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat. Neurosci. 2002, 5, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, B.; Gutmann, D.H. Neurofibromatosis 1: Closing the GAP between mice and men. Curr. Opin. Genet. Dev. 2003, 13, 20–27. [Google Scholar] [CrossRef]
- Hegedus, B.; Dasgupta, B.; Shin, J.E.; Emnett, R.J.; Hart-Mahon, E.K.; Elghazi, L.; Bernal-Mizrachi, E.; Gutmann, D.H. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell 2007, 1, 443–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.A.; Gianino, S.M.; Gutmann, D.H. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci. 2010, 30, 5579–5589. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Diggs-Andrews, K.A.; Gianino, S.M.; Gutmann, D.H. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol. Cell Neurosci. 2012, 49, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Hannan, F.; Ho, I.; Tong, J.J.; Zhu, Y.; Nurnberg, P.; Zhong, Y. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum. Mol. Genet. 2006, 15, 1087–1098. [Google Scholar] [CrossRef] [Green Version]
- Deraredj Nadim, W.; Chaumont-Dubel, S.; Madouri, F.; Cobret, L.; De Tauzia, M.L.; Zajdel, P.; Bénédetti, H.; Marin, P.; Morisset-Lopez, S. Physical interaction between neurofibromin and serotonin 5-HT6 receptor promotes receptor constitutive activity. Proc. Natl. Acad. Sci. USA 2016, 113, 12310–12315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasaki, C.; Gutmann, D.H. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet. 2014, 23, 6712–6721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.L.; Lei, Y.T.; Hong, C.J.; Hsueh, Y.P. Syndecan-2 induces filopodia and dendritic spine formation via the neurofibromin-PKA-Ena/VASP pathway. J. Cell Biol. 2007, 177, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, I.; De Vries, G.H. Aberrant cAMP metabolism in NF1 malignant peripheral nerve sheath tumor cells. Neurochem. Res. 2011, 36, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Emnett, R.J.; White, C.R.; Yuede, C.M.; Conyers, S.B.; O’Malley, K.L.; Wozniak, D.F.; Gutmann, D.H. Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum. Mol. Genet. 2010, 19, 4515–4528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diggs-Andrews, K.A.; Tokuda, K.; Izumi, Y.; Zorumski, C.F.; Wozniak, D.F.; Gutmann, D.H. Dopamine deficiency underlies learning deficits in neurofibromatosis-1 mice. Ann. Neurol. 2013, 73, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mautner, V.F.; Kluwe, L.; Thakker, S.D.; Leark, R.A. Treatment of ADHD in neurofibromatosis type 1. Dev. Med. Child Neurol. 2002, 44, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Woo, A.S.; Messiaen, L.M.; Gutmann, D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015, 24, 3518–3528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pride, N.A.; Barton, B.; Hutchins, P.; Coghill, D.R.; Korgaonkar, M.S.; Hearps, S.J.C.; Rouel, M.; Malarbi, S.; North, K.N.; Payne, J.M. Effects of methylphenidate on cognition and behaviour in children with neurofibromatosis type 1: A study protocol for a randomised placebo-controlled crossover trial. BMJ Open 2018, 8, e021800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, L.P.; Muntean, B.S.; Ostrovskaya, O.; Zucca, S.; Dao, M.; Orlandi, C.; Song, C.; Xie, K.; Martemyanov, K.A. NF1-cAMP signaling dissociates cell type-specific contributions of striatal medium spiny neurons to reward valuation and motor control. PLoS Biol. 2019, 17, e3000477. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, B.; Yi, Y.; Chen, D.Y.; Weber, J.D.; Gutmann, D.H. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005, 65, 2755–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, M.; Yamamoto, H.; Setsu, N.; Kohashi, K.; Takahashi, Y.; Ishii, T.; Iida, K.; Matsumoto, Y.; Hakozaki, M.; Aoki, M.; et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2013, 19, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.A.; Cantor, A.; Korf, B.; Perentesis, J.; Gutmann, D.H.; Schorry, E.; et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas: An NF clinical trials consortium phase II study. Pediatr. Blood Cancer 2014, 61, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Weiss, B.; Widemann, B.C.; Wolters, P.; Dombi, E.; Vinks, A.; Cantor, A.; Perentesis, J.; Schorry, E.; Ullrich, N.; Gutmann, D.H.; et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas: A neurofibromatosis Clinical Trials Consortium phase II study. Neuro-Oncology 2015, 17, 596–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widemann, B.C.; Lu, Y.; Reinke, D.; Okuno, S.H.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Hirbe, A.C.; Kim, A.; et al. Targeting Sporadic and Neurofibromatosis Type 1 (NF1) Related Refractory Malignant Peripheral Nerve Sheath Tumors (MPNST) in a Phase II Study of Everolimus in Combination with Bevacizumab (SARC016). Sarcoma 2019, 2019, 7656747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullrich, N.J.; Prabhu, S.P.; Reddy, A.T.; Fisher, M.J.; Packer, R.; Goldman, S.; Robison, N.J.; Gutmann, D.H.; Viskochil, D.H.; Allen, J.C.; et al. A phase II study of continuous oral mTOR inhibitor everolimus for recurrent, radiographic-progressive neurofibromatosis type 1-associated pediatric low-grade glioma: A Neurofibromatosis Clinical Trials Consortium study. Neuro-Oncology 2020, 22, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, M.; Choi, J.M.; Kim, B.J.; Zhou, M.T.; Chen, Z.; Jain, A.N.; Jung, S.Y.; Yuan, J.; Wang, W.; et al. Clustered, Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9-coupled Affinity Purification/Mass Spectrometry Analysis Revealed a Novel Role of Neurofibromin in mTOR Signaling. Mol. Cell Proteom. 2017, 16, 594–607. [Google Scholar] [CrossRef] [Green Version]
- Nada, S.; Mori, S.; Takahashi, Y.; Okada, M. p18/LAMTOR1: A late endosome/lysosome-specific anchor protein for the mTORC1/MAPK signaling pathway. Methods Enzymol. 2014, 535, 249–263. [Google Scholar] [CrossRef]
- Xie, K.; Colgan, L.A.; Dao, M.T.; Muntean, B.S.; Sutton, L.P.; Orlandi, C.; Boye, S.L.; Boye, S.E.; Shih, C.C.; Li, Y.; et al. NF1 Is a Direct G Protein Effector Essential for Opioid Signaling to Ras in the Striatum. Curr. Biol. 2016, 26, 2992–3003. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.W.; Olson, M.F. LIM kinases: Function, regulation and association with human disease. J. Mol. Med. 2007, 85, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, T.; Araki, N.; Yunoue, S.; Tokuo, H.; Feng, L.; Patrakitkomjorn, S.; Hara, T.; Ichikawa, Y.; Matsumoto, K.; Fujii, K.; et al. The neurofibromatosis type 1 gene product neurofibromin enhances cell motility by regulating actin filament dynamics via the Rho-ROCK-LIMK2-cofilin pathway. J. Biol. Chem. 2005, 280, 39524–39533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallée, B.; Doudeau, M.; Godin, F.; Gombault, A.; Tchalikian, A.; de Tauzia, M.L.; Bénédetti, H. Nf1 RasGAP inhibition of LIMK2 mediates a new cross-talk between Ras and Rho pathways. PLoS ONE 2012, 7, e47283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starinsky-Elbaz, S.; Faigenbloom, L.; Friedman, E.; Stein, R.; Kloog, Y. The pre-GAP-related domain of neurofibromin regulates cell migration through the LIM kinase/cofilin pathway. Mol. Cell Neurosci. 2009, 42, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, M.A.; Speicher, D.W.; Shiekhattar, R. The motor protein kinesin-1 links neurofibromin and merlin in a common cellular pathway of neurofibromatosis. J. Biol. Chem. 2002, 277, 36909–36912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, A.; Stokin, G.B.; Yang, Z.; Xia, C.H.; Goldstein, L.S. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 2000, 28, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Arun, V.; Worrell, L.; Wiley, J.C.; Kaplan, D.R.; Guha, A. Neurofibromin interacts with the cytoplasmic Dynein Heavy Chain 1 in melanosomes of human melanocytes. FEBS Lett. 2013, 587, 1466–1473. [Google Scholar] [CrossRef] [Green Version]
- Donarum, E.A.; Halperin, R.F.; Stephan, D.A.; Narayanan, V. Cognitive dysfunction in Nf1 knock-out mice may result from altered vesicular trafficking of APP/DRD3 complex. BMC Neurosci. 2006, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Arun, V.; Wiley, J.C.; Kaur, H.; Kaplan, D.R.; Guha, A. A novel neurofibromin (NF1) interaction with the leucine-rich pentatricopeptide repeat motif-containing protein links neurofibromatosis type 1 and the French Canadian variant of Leigh’s syndrome in a common molecular complex. J. Neurosci. Res. 2013, 91, 494–505. [Google Scholar] [CrossRef]
- Wang, W.; van Niekerk, E.; Willis, D.E.; Twiss, J.L. RNA transport and localized protein synthesis in neurological disorders and neural repair. Dev. Neurobiol. 2007, 67, 1166–1182. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Hsueh, Y.P. Neurofibromin interacts with CRMP-2 and CRMP-4 in rat brain. Biochem. Biophys. Res. Commun. 2008, 369, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Moutal, A.; Yang, X.; Li, W.; Gilbraith, K.B.; Luo, S.; Cai, S.; François-Moutal, L.; Chew, L.A.; Yeon, S.K.; Bellampalli, S.S.; et al. CRISPR/Cas9 editing of Nf1 gene identifies CRMP2 as a therapeutic target in neurofibromatosis type 1-related pain that is reversed by (S)-Lacosamide. Pain 2017, 158, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Shih, Y.T.; Chen, C.Y.; Chao, H.W.; Lee, M.J.; Hsueh, Y.P. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Investig. 2011, 121, 4820–4837. [Google Scholar] [CrossRef] [PubMed]
- Carnes, R.M.; Kesterson, R.A.; Korf, B.R.; Mobley, J.A.; Wallis, D. Affinity Purification of NF1 Protein-Protein Interactors Identifies Keratins and Neurofibromin Itself as Binding Partners. Genes 2019, 10, 650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnes, R.M.; Mobley, J.A.; Crossman, D.K.; Liu, H.; Korf, B.R.; Kesterson, R.A.; Wallis, D. Multi-Omics Profiling for NF1 Target Discovery in Neurofibromin (NF1) Deficient Cells. Proteomics 2019, 19, e1800334. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 2006, 22, 159–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGillicuddy, L.T.; Fromm, J.A.; Hollstein, P.E.; Kubek, S.; Beroukhim, R.; De Raedt, T.; Johnson, B.W.; Williams, S.M.; Nghiemphu, P.; Liau, L.M.; et al. Proteasomal and genetic inactivation of the NF1 tumor suppressor in gliomagenesis. Cancer Cell 2009, 16, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phan, V.T.; Ding, V.W.; Li, F.; Chalkley, R.J.; Burlingame, A.; McCormick, F. The RasGAP proteins Ira2 and neurofibromin are negatively regulated by Gpb1 in yeast and ETEA in humans. Mol. Cell Biol. 2010, 30, 2264–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Zhao, Y.; Kim, S.J.; Liu, M.; Jia, L.; Saunders, T.L.; Zhu, Y.; Sun, Y. SAG/RBX2/ROC2 E3 ubiquitin ligase is essential for vascular and neural development by targeting NF1 for degradation. Dev. Cell 2011, 21, 1062–1076. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.A.; Beggs, A.H. Kelch proteins: Emerging roles in skeletal muscle development and diseases. Skelet. Muscle 2014, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollstein, P.E.; Cichowski, K. Identifying the Ubiquitin Ligase complex that regulates the NF1 tumor suppressor and Ras. Cancer Discov. 2013, 3, 880–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, Y.S.; Sargis, T.; Reichert, E.C.; Rudasi, E.; Fuja, D.; Jonasch, E.; Koh, M.Y. Hypoxia-Associated Factor (HAF) Mediates Neurofibromin Ubiquitination and Degradation Leading to Ras-ERK Pathway Activation in Hypoxia. Mol. Cancer Res. 2019, 17, 1220–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Thé, H.; Le Bras, M.; Lallemand-Breitenbach, V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J. Cell Biol. 2012, 198, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Lallemand-Breitenbach, V.; de Thé, H. PML nuclear bodies. Cold Spring Harb Perspect. Biol. 2010, 2, a000661. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; de Thé, H.; Lallemand-Breitenbach, V. PML nuclear bodies: Assembly and oxidative stress-sensitive sumoylation. Nucleus 2014, 5, 499–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauclair, G.; Bridier-Nahmias, A.; Zagury, J.F.; Saïb, A.; Zamborlini, A. JASSA: A comprehensive tool for prediction of SUMOylation sites and SIMs. Bioinformatics 2015, 31, 3483–3491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, J.; Eifler, K.; Sigurðsson, J.O.; Cuijpers, S.A.; Hendriks, I.A.; Verlaan-de Vries, M.; Kelstrup, C.D.; Francavilla, C.; Medema, R.H.; Olsen, J.V.; et al. Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol. Cell 2014, 53, 1053–1066. [Google Scholar] [CrossRef] [Green Version]
- Hendriks, I.A.; Lyon, D.; Young, C.; Jensen, L.J.; Vertegaal, A.C.; Nielsen, M.L. Site-specific mapping of the human SUMO proteome reveals co-modification with phosphorylation. Nat. Struct. Mol. Biol. 2017, 24, 325–336. [Google Scholar] [CrossRef]
- Gross, A.M.; Dombi, E.; Widemann, B.C. Current status of MEK inhibitors in the treatment of plexiform neurofibromas. Childs Nerv. Syst. 2020. [Google Scholar] [CrossRef]
- Wallis, D.; Li, K.; Lui, H.; Hu, K.; Chen, M.J.; Li, J.; Kang, J.; Das, S.; Korf, B.R.; Kesterson, R.A. Neurofibromin (NF1) genetic variant structure-function analyses using a full-length mouse cDNA. Hum. Mutat. 2018, 39, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Morrison, H. Construction of cloning-friendly minigenes for mammalian expression of full-length human NF1 isoforms. Hum. Mutat. 2019, 40, 187–192. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. https://doi.org/10.3390/cells9112365
Bergoug M, Doudeau M, Godin F, Mosrin C, Vallée B, Bénédetti H. Neurofibromin Structure, Functions and Regulation. Cells. 2020; 9(11):2365. https://doi.org/10.3390/cells9112365
Chicago/Turabian StyleBergoug, Mohammed, Michel Doudeau, Fabienne Godin, Christine Mosrin, Béatrice Vallée, and Hélène Bénédetti. 2020. "Neurofibromin Structure, Functions and Regulation" Cells 9, no. 11: 2365. https://doi.org/10.3390/cells9112365
APA StyleBergoug, M., Doudeau, M., Godin, F., Mosrin, C., Vallée, B., & Bénédetti, H. (2020). Neurofibromin Structure, Functions and Regulation. Cells, 9(11), 2365. https://doi.org/10.3390/cells9112365