Perlecan Facilitates Neuronal Nitric Oxide Synthase Delocalization in Denervation-Induced Muscle Atrophy

 ,
,  ,
,  and
and 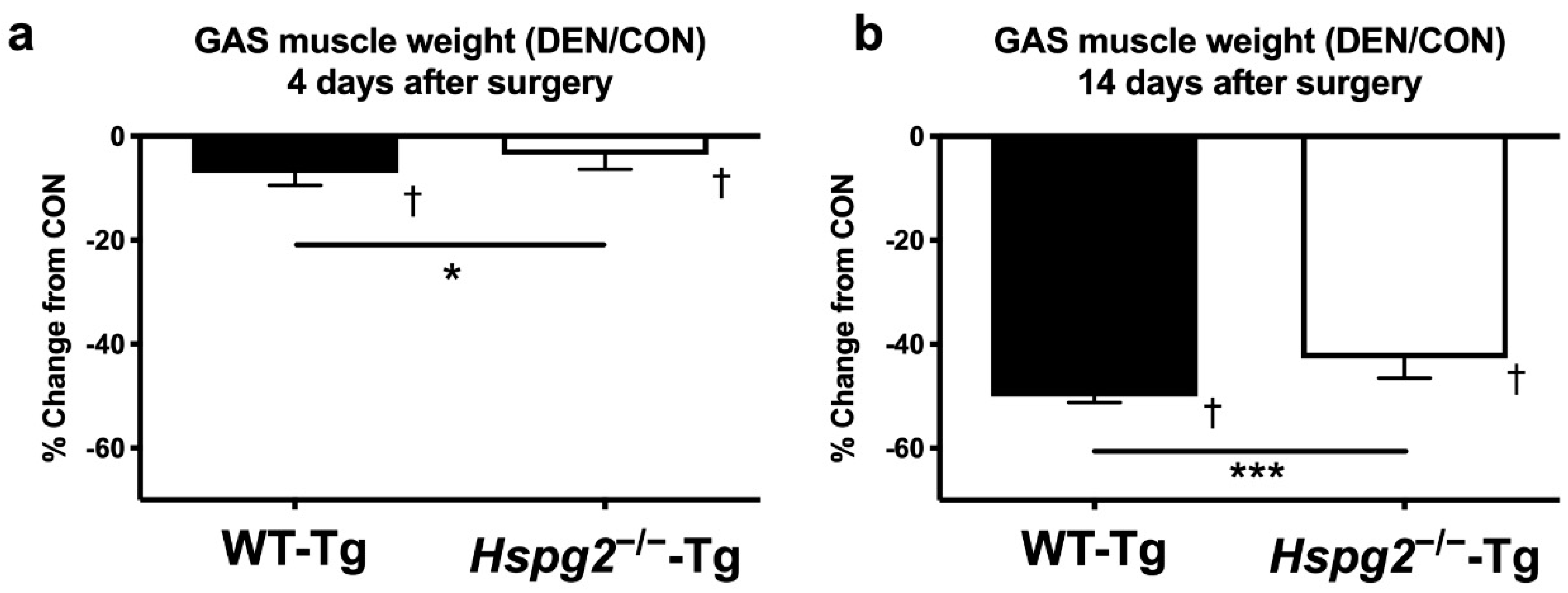{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Denervation Surgery
2.3. Muscle Sampling
2.4. Immunofluorescence Microscopy
2.5. Total Protein Extraction
2.6. Western Blotting
2.7. Statistical Analyses
3. Results
3.1. Changes in Muscle Weight after Denervation Surgery
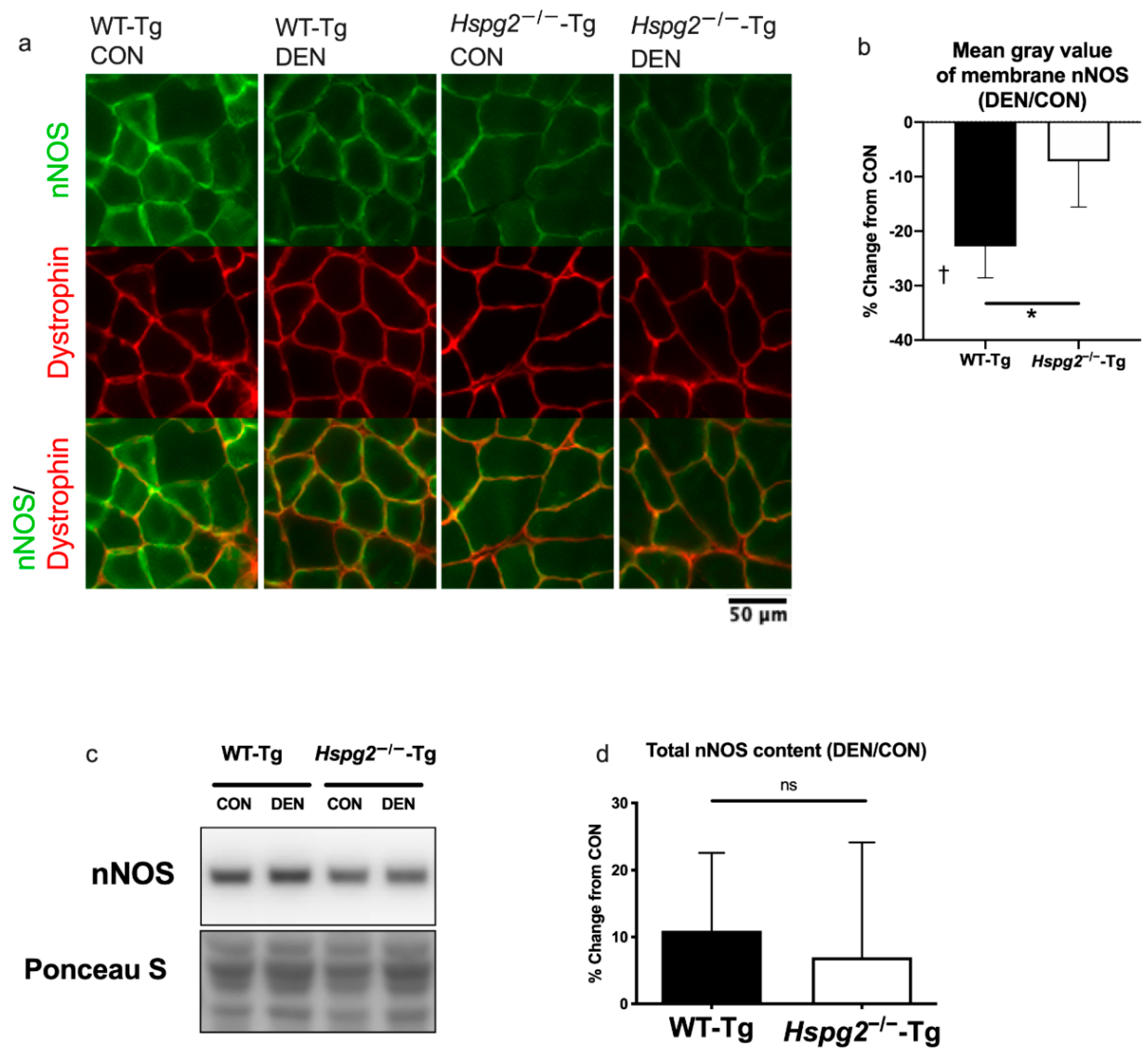
3.2. Changes in Membrane nNOS Expression
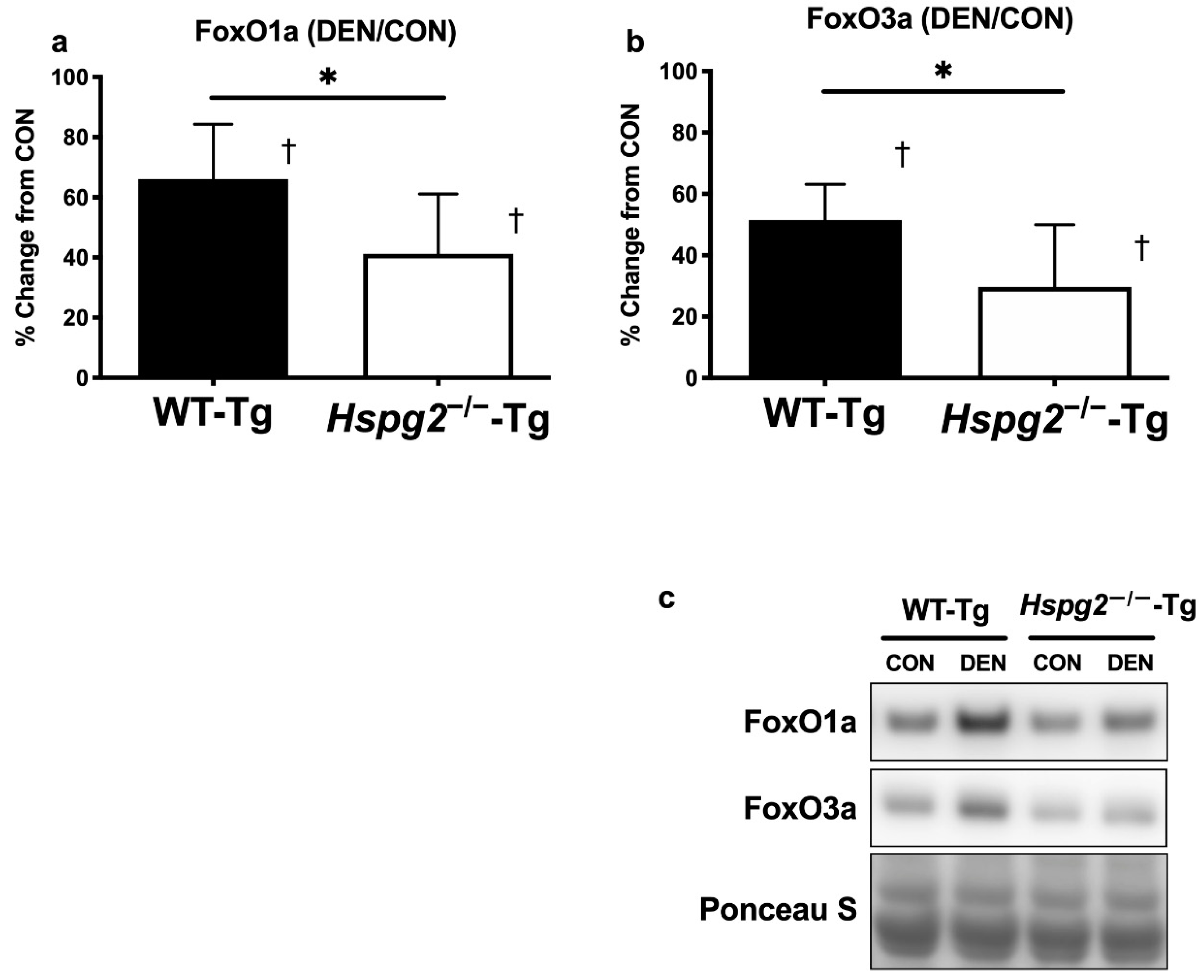
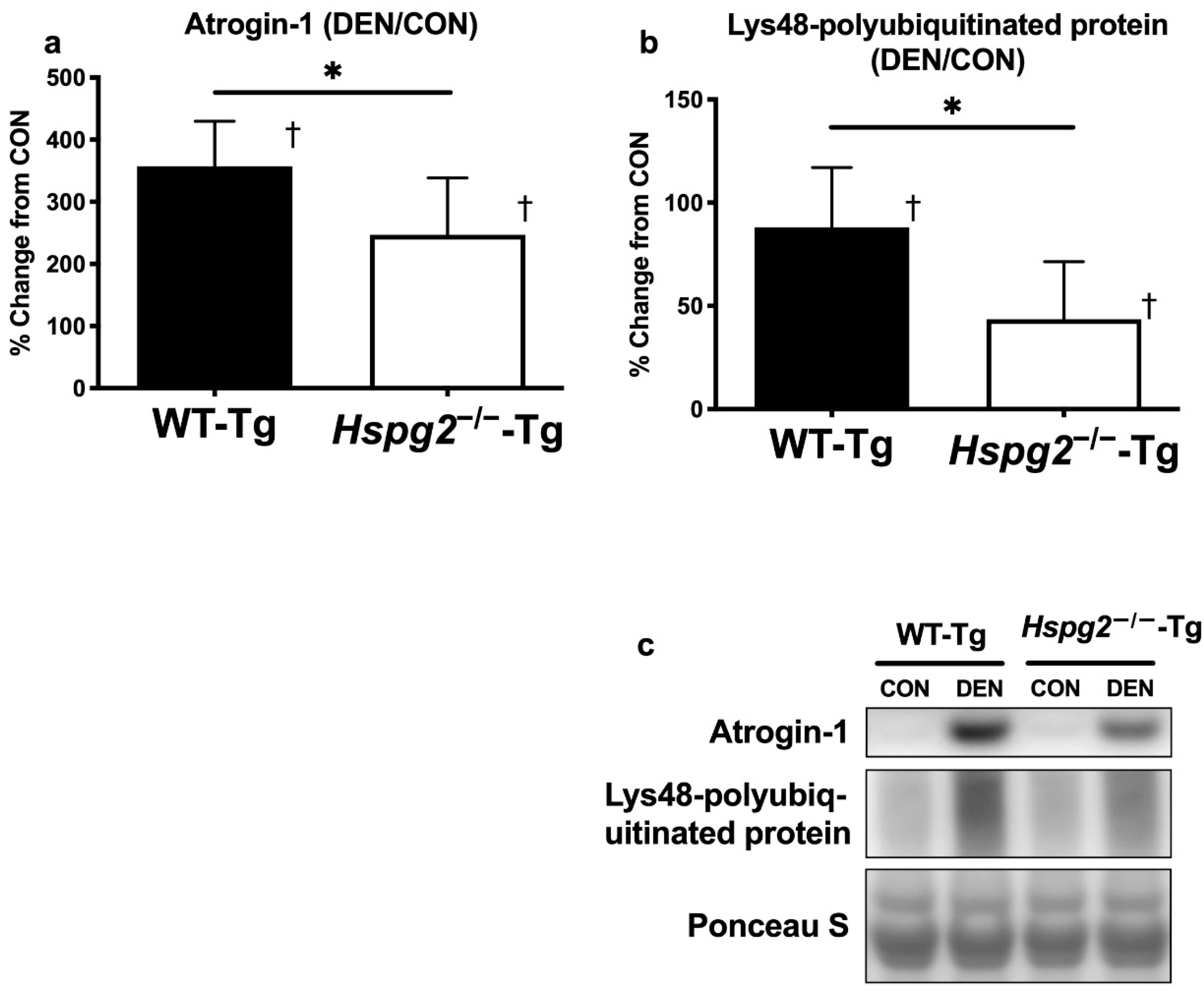
3.3. FoxO Signaling and Ubiquitin—Proteasome Signaling Activation
4. Discussion
4.1. Perlecan Promotes Skeletal Muscle Atrophy Following Denervation
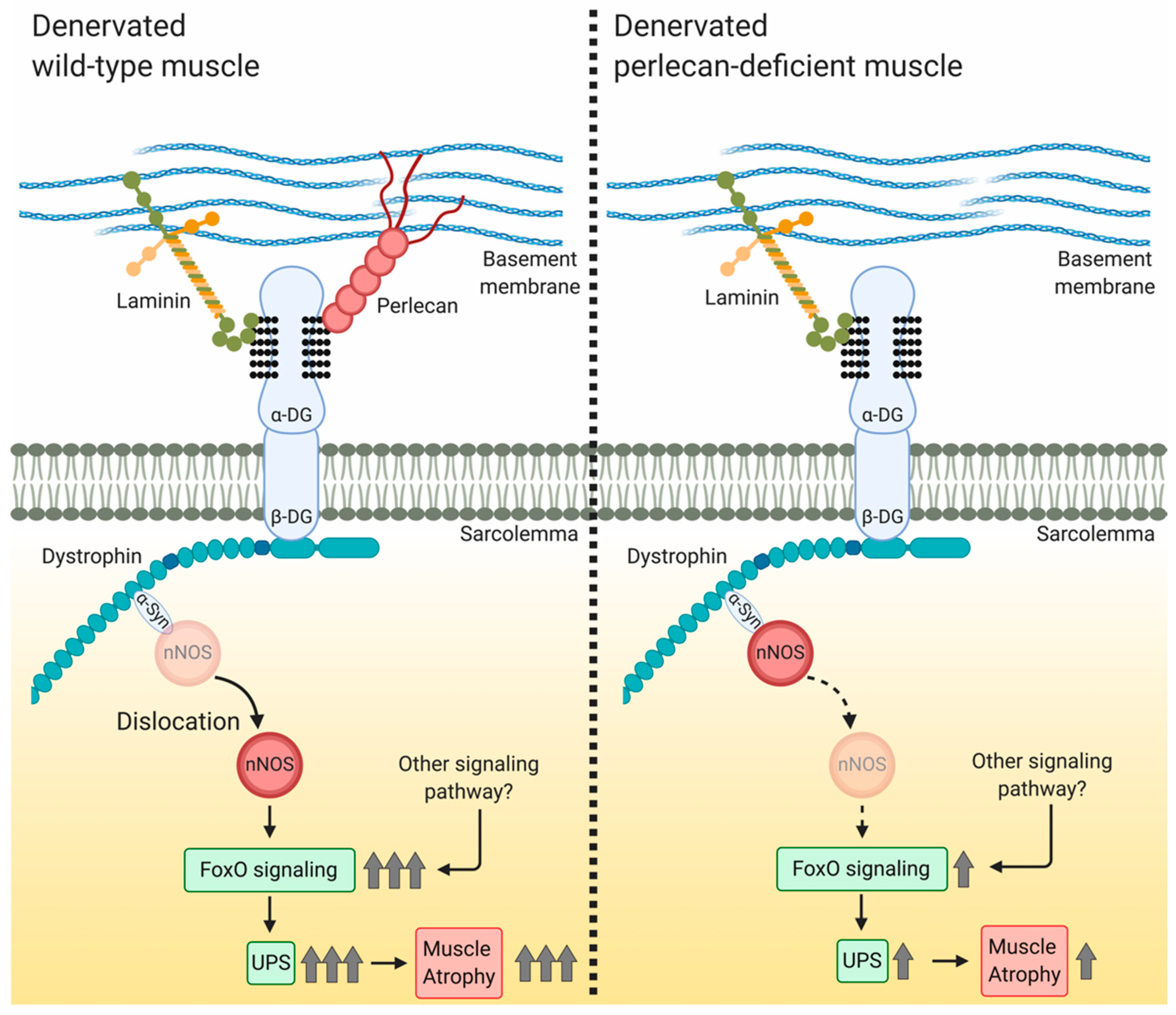
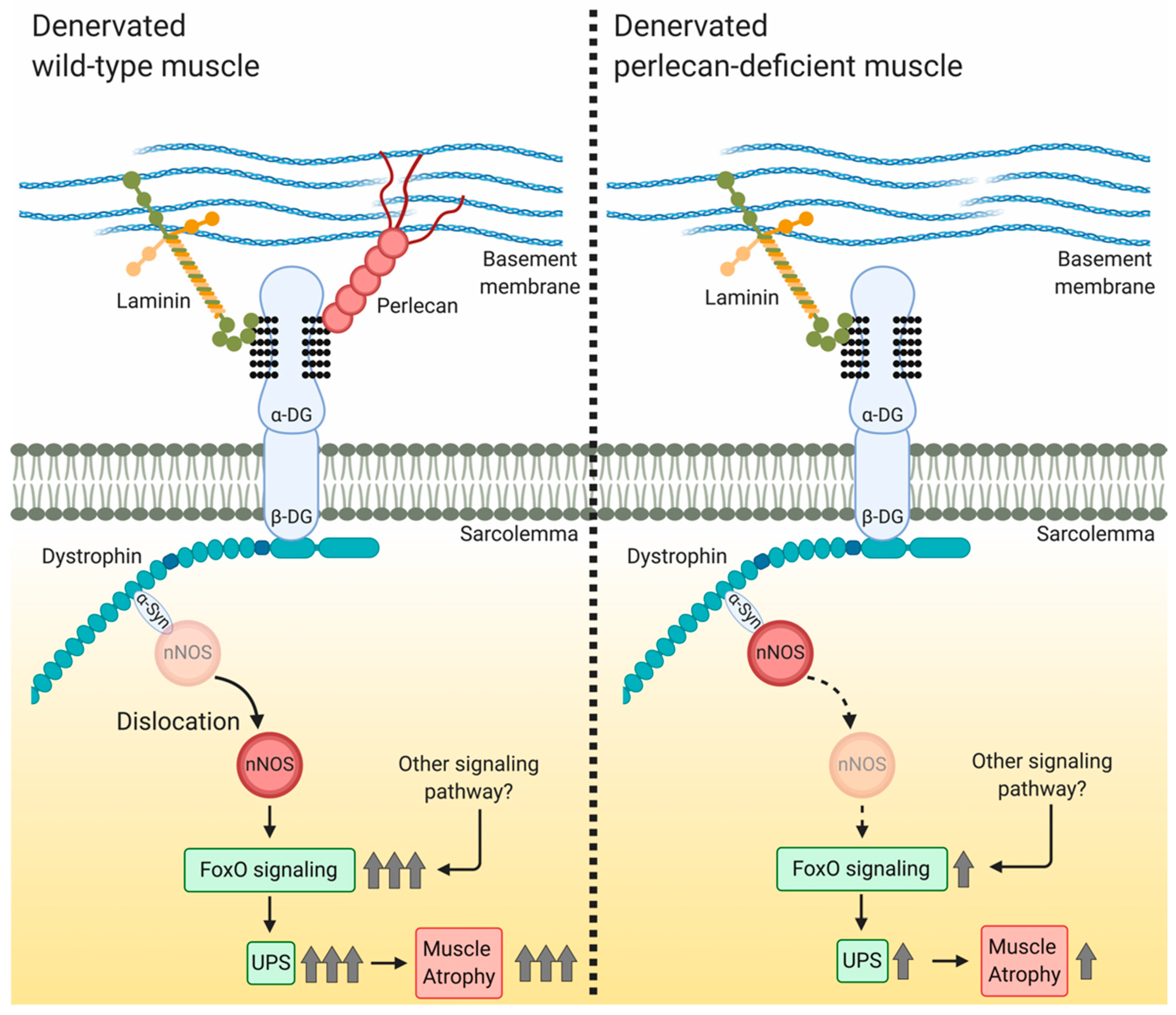
4.2. Perlecan Facilitates nNOS Delocalization, FoxO Signaling, and Ubiquitin—Proteasome System Activation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, A.; Sharif-Naeini, R.; Folgering, J.R.H.; Bichet, D.; Duprat, F.; Honoré, E. Canonical TRP Channels and Mechanotransduction: From Physiology to Disease States. Pflug. Arch. Eur. J. Physiol. 2010, 460, 571–581. [Google Scholar] [CrossRef]
- Smith, L.W.; Smith, J.D.; Criswell, D.S. Involvement of Nitric Oxide Synthase in Skeletal Muscle Adaptation to Chronic Overload. J. Appl. Physiol. 2002, 92, 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokri, B.; Engel, A.G. Duchenne Dystrophy: Electron Microscopic Findings Pointing to a Basic or Early Abnormality in the Plasma Membrane of the Muscle Fiber. Neurology 1975, 25, 1111–1120. [Google Scholar] [PubMed]
- Pestronk, A.; Parhad, I.M.; Drachman, D.B.; Price, D.L. Membrane Myopathy: Morphological Similarities to Duchenne Muscular Dystrophy. Muscle Nerve 1982, 5, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and Extracellular Matrix Homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [Green Version]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric Oxide Synthase Complexed with Dystrophin and Absent from Skeletal Muscle Sarcolemma in Duchenne Muscular Dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Kumar, A.; Khandelwal, N.; Malya, R.; Reid, M.B.; Boriek, A.M. Loss of Dystrophin Causes Aberrant Mechanotransduction in Skeletal Muscle Fibers. FASEB J. 2004, 18, 102–113. [Google Scholar] [CrossRef]
- Wu, M.; Fannin, J.; Rice, K.M.; Wang, B.; Blough, E.R. Effect of Aging on Cellular Mechanotransduction. Ageing Res. Rev. 2011, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Edgerton, V.R.; Roy, R.R.; Allen, D.L.; Monti, R.J. Adaptations in Skeletal Muscle Disuse or Decreased-Use Atrophy. Am. J. Phys. Med. Rehabil. 2002, 81 (Suppl. 11), S127–S147. [Google Scholar] [CrossRef]
- Lawler, J.M.; Song, W.; Kwak, H.B. Differential Response of Heat Shock Proteins to Hindlimb Unloading and Reloading in the Soleus. Muscle Nerve 2006, 33, 200–207. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo Transcription Factors Induce the Atrophy-Related Ubiquitin Ligase Atrogin-1 and Cause Skeletal Muscle Atrophy. Cell 2004, 117, 399–412. [Google Scholar] [PubMed] [Green Version]
- Bodine, S.C.; Baehr, L.M. Skeletal Muscle Atrophy and the E3 Ubiquitin Ligases MuRF1 and MAFbx/Atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, A.D.; Zhang, N.Y.; Xu, P.; Han, K.J.; Noone, S.; Peng, J.; Liu, C.W. The Lysine 48 and Lysine 63 Ubiquitin Conjugates Are Processed Differently by the 26 S Proteasome. J. Biol. Chem. 2009, 284, 35485–35494. [Google Scholar] [CrossRef] [Green Version]
- Foletta, V.C.; White, L.J.; Larsen, A.E.; Léger, B.; Russell, A.P. The Role and Regulation of MAFbx/Atrogin-1 and MuRFl in Skeletal Muscle Atrophy. Pflugers Arch. Eur. J. Physiol. 2011, 461, 325–335. [Google Scholar] [CrossRef]
- Kamei, Y.; Miura, S.; Suzuki, M.; Kai, Y.; Mizukami, J.; Taniguchi, T.; Mochida, K.; Hata, T.; Matsuda, J.; Aburatani, H.; et al. Skeletal Muscle FOXO1 (FKHR) Transgenic Mice Have Less Skeletal Muscle Mass, down-Regulated Type I (Slow Twitch/Red Muscle) Fiber Genes, and Impaired Glycemic Control. J. Biol. Chem. 2004, 279, 41114–41123. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Motohashi, N.; Uezumi, A.; Fukada, S.; Yoshimura, T.; Itoyama, Y.; Aoki, M.; Miyagoe-Suzuki, Y.; Takeda, S. NO Production Results in Suspension-Induced Muscle Atrophy through Dislocation of Neuronal NOS. J. Clin. Investig. 2007, 117, 2468–2476. [Google Scholar] [CrossRef] [Green Version]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of Nitric Oxide Synthase with the Postsynaptic Density Protein PSD-95 and A1-Syntrophin Mediated by PDZ Domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Kameya, S.; Miyagoe, Y.; Nonaka, I.; Ikemoto, T.; Endo, M.; Hanaoka, K.; Nabeshima, Y.I.; Takeda, S. A1-Syntrophin Gene Disruption Results in the Absence of Neuronal-Type Nitric-Oxide Synthase at the Sarcolemma but Does Not Induce Muscle Degeneration. J. Biol. Chem. 1999, 274, 2193–2200. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Thomas, G.D.; Yue, Y.; Yang, H.T.; Li, D.; Long, C.; Judge, L.; Bostick, B.; Chamberlain, J.S.; Terjung, R.L.; et al. Dystrophins Carrying Spectrin-like Repeats 16 and 17 Anchor NNOS to the Sarcolemma and Enhance Exercise Performance in a Mouse Model of Muscular Dystrophy. J. Clin. Investig. 2009, 119, 624–635. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Zhao, J.; Yue, Y.; Duan, D. A2 and A3 Helices of Dystrophin R16 and R17 Frame a Microdomain in the A1 Helix of Dystrophin R17 for Neuronal NOS Binding. Proc. Natl. Acad. Sci. USA 2013, 110, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.E.; Odom, G.L.; Kim, M.J.; Chamberlain, J.S.; Froehner, S.C. Syntrophin Binds Directly to Multiple Spectrin-like Repeats in Dystrophin and Mediates Binding of NNOS to Repeats 16–17. Hum. Mol. Genet. 2018, 27, 2978–2985. [Google Scholar] [CrossRef] [PubMed]
- Froehner, S.C.; Reed, S.M.; Anderson, K.N.; Huang, P.L.; Percival, J.M. Loss of NNOS Inhibits Compensatory Muscle Hypertrophy and Exacerbates Inflammation and Eccentric Contraction-Induced Damage in Mdx Mice. Hum. Mol. Genet. 2015, 24, 492–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.M.; Rader, E.P.; Crawford, R.W.; Iyengar, N.K.; Thedens, D.R.; Faulkner, J.A.; Parikh, S.V.; Weiss, R.M.; Chamberlain, J.S.; Moore, S.A.; et al. Sarcolemma-Localized NNOS Is Required to Maintain Activity after Mild Exercise. Nature 2008, 456, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finanger Hedderick, E.L.; Simmers, J.L.; Soleimani, A.; Andres-Mateos, E.; Marx, R.; Files, D.C.; King, L.; Crawford, T.O.; Corse, A.M.; Cohn, R.D. Loss of Sarcolemmal NNOS Is Common in Acquired and Inherited Neuromuscular Disorders. Neurology 2011, 76, 960–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellman, J.E.; DeRuisseau, K.C.; Betters, J.L.; Lira, V.A.; Soltow, Q.A.; Selsby, J.T.; Criswell, D.S. In Vivo Inhibition of Nitric Oxide Synthase Impairs Upregulation of Contractile Protein MRNA in Overloaded Plantaris Muscle. J. Appl. Physiol. 2006, 100, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamler, J.S.; Meissner, G. Physiology of Nitric Oxide in Skeletal Muscle. Physiol Rev. 2001, 81, 209–237. [Google Scholar]
- Ito, N.; Ruegg, U.T.; Kudo, A.; Miyagoe-Suzuki, Y.; Takeda, S. Activation of Calcium Signaling through Trpv1 by NNOS and Peroxynitrite as a Key Trigger of Skeletal Muscle Hypertrophy. Nat. Med. 2013, 19, 101–106. [Google Scholar] [CrossRef]
- Crosbie, R.H.; Barresi, R.; Campbell, K.P. Loss of Sarcolemma NNOS in Sarcoglycan-Deficient Muscle. FASEB J. 2002, 16, 1786–1791. [Google Scholar] [CrossRef] [Green Version]
- Côté, P.D.; Moukhles, H.; Carbonetto, S. Dystroglycan Is Not Required for Localization of Dystrophin, Syntrophin, and Neuronal Nitric-Oxide Synthase at the Sarcolemma but Regulates Integrin A7B Expression and Caveolin-3 Distribution. J. Biol. Chem. 2002, 277, 4672–4679. [Google Scholar] [CrossRef] [Green Version]
- Mercado, M.L.; Amenta, A.R.; Hagiwara, H.; Rafii, M.S.; Lechner, B.E.; Owens, R.T.; McQuillan, D.J.; Froehner, S.C.; Fallon, J.R. Biglycan Regulates the Expression and Sarcolemmal Localization of Dystrobrevin, Syntrophin, and NNOS. FASEB J. 2006, 20, 1724–1726. [Google Scholar] [CrossRef]
- Mercado, M.L.; Amenta, A.R.; Hagiwara, H.; Rafii, M.S.; Owens, R.T.; Mcquillan, D.J.; Froehner, S.C.; Justin, R. Biglycan Targets Dystrobrevin, Syntrophin and NNOS to the Muscle Cell Membrane. FASEB J. 2011, 20, 1724–1726. [Google Scholar] [CrossRef]
- Kanagawa, M.; Michele, D.E.; Satz, J.S.; Barresi, R.; Kusano, H.; Sasaki, T.; Timpl, R.; Henry, M.D.; Campbell, K.P. Disruption of Perlecan Binding and Matrix Assembly by Post-Translational or Genetic Disruption of Dystroglycan Function. FEBS Lett. 2005, 579, 4792–4796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bello, V.; Moreau, N.; Sirour, C.; Hidalgo, M.; Buisson, N.; Darribère, T. The Dystroglycan: Nestled in an Adhesome during Embryonic Development. Dev. Biol. 2015, 401, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Talts, J.F.; Andac, Z.; Göhring, W.; Brancaccio, A.; Timpl, R. Binding of the G Domains of Laminin A1 and A2 Chains and Perlecan to Heparin, Sulfatides, α-Dystroglycan and Several Extracellular Matrix Proteins. EMBO J. 1999, 18, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ichikawa, N.; Kosaki, K.; Yamada, Y.; Sasaki, T.; Sakai, L.Y.; Kurosawa, H.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan Deficiency Causes Muscle Hypertrophy, a Decrease in Myostatin Expression, and Changes in Muscle Fiber Composition. Matrix Biol. 2010, 29, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Xu, Z.; Furuya, N.; Nonaka, R.; Yamada, Y.; Arikawa-Hirasawa, E. Perlecan Inhibits Autophagy to Maintain Muscle Homeostasis in Mouse Soleus Muscle. Matrix Biol. 2015, 48, 26–35. [Google Scholar] [CrossRef]
- Yamashita, Y.; Nakada, S.; Yoshihara, T.; Nara, T.; Furuya, N.; Miida, T.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan, a Heparan Sulfate Proteoglycan, Regulates Systemic Metabolism with Dynamic Changes in Adipose Tissue and Skeletal Muscle. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Nonaka, R.; Iesaki, T.; de Vega, S.; Daida, H.; Okada, T.; Sasaki, T.; Arikawa-Hirasawa, E. Perlecan Deficiency Causes Endothelial Dysfunction by Reducing the Expression of Endothelial Nitric Oxide Synthase. Physiol. Rep. 2015, 3, e12272. [Google Scholar] [CrossRef]
- Arikawa-hirasawa, E.; Watanabe, H.; Takami, H.; Hassell, J.R.; Yamada, Y. Perlecan Is Essential for Cartilage and Cephalic Development. Nat. Genet. 1999, 23, 354–358. [Google Scholar] [CrossRef]
- Costell, M.; Gustafsson, E.; Aszódi, A.; Mörgelin, M.; Bloch, W.; Hunziker, E.; Addicks, K.; Timpl, R.; Fässler, R. Perlecan Maintains the Integrity of Cartilage and Some Basement Membranes. J. Cell Biol. 1999, 147, 1109–1122. [Google Scholar]
- Tsumaki, N.; Tanaka, K.; Arikawa-Hirasawa, E.; Nakase, T.; Kimura, T.; Terrig Thomas, J.; Ochi, T.; Luyten, F.P.; Yamada, Y. Role of CDMP-1 in Skeletal Morphogenesis: Promotion of Mesenchymal Cell Recruitment and Chondrocyte Differentiation. J. Cell Biol. 1999, 144, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Romero-calvo, I.; Ocón, B.; Martínez-moya, P.; Suárez, M.D.; Zarzuelo, A.; Martínez-augustin, O.; De Medina, F.S. Reversible Ponceau Staining as a Loading Control Alternative to Actin in Western Blots. Anal. Biochem. 2010, 401, 318–320. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of Autophagy and the Ubiquitin-Proteasome System by the FoxO Transcriptional Network during Muscle Atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, R.V. Perlecan: A Gem of a Proteoglycan. Matrix Biol. 1994, 14, 203–208. [Google Scholar] [CrossRef]
- Gubbiotti, M.A.; Neill, T.; Iozzo, R.V. A Current View of Perlecan in Physiology and Pathology: A Mosaic of Functions. Matrix Biol. 2017, 57–58, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Brandan, E.; Gutierrez, J. Role of Skeletal Muscle Proteoglycans during Myogenesis. Matrix Biol. 2013, 32, 289–297. [Google Scholar] [CrossRef]
- Brandan, E.; Maldonado, M.; Garrido, J.; Inestrosa, N.C. Anchorage of Collagen-Tailed Acetylcholinesterase to the Extracellular Matrix Is Mediated by Heparan Sulfate Proteoglycans. J. Cell Biol. 1985, 101, 985–992. [Google Scholar] [CrossRef] [Green Version]
- Arikawa-Hirasawa, E.; Rossi, S.G.; Rotundo, R.L.; Yamada, Y. Absence of Acetylcholinesterase at the Neuromuscular Junctions of Perlecan-Null Mice. Nat. Neurosci. 2002, 5, 119–123. [Google Scholar] [CrossRef]
- Villar, M.J.; Hassell, J.R.; Brandan, E. Interaction of Skeletal Muscle Cells with Collagen Type IV Is Mediated by Perlecan Associated with the Cell Surface. J. Cell Biochem. 1999, 75, 665–674. [Google Scholar] [PubMed]
- Henriquez, J.P.; Casar, J.C.; Fuentealba, L.; Carey, D.J.; Brandan, E. Extracellular Matrix Histone H1 Binds to Perlecan, Is Present in Regenerating Skeletal Muscle and Stimulates Myoblast Proliferation. J. Cell Sci. 2002, 115, 2041–2051. [Google Scholar]
- Echaniz-Laguna, A.; Rene, F.; Marcel, C.; Bangratz, M.; Fontaine, B.; Loeffler, J.P.; Nicole, S. Electrophysiological Studies in a Mouse Model of Schwartz-Jampel Syndrome Demonstrate Muscle Fiber Hyperactivity of Peripheral Nerve Origin. Muscle Nerve 2009, 40, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Bangratz, M.; Sarrazin, N.; Devaux, J.; Zambroni, D.; Echaniz-Laguna, A.; René, F.; Borio, D.; Davoine, C.S.; Fontaine, B.; Feltri, M.L.; et al. A Mouse Model of Schwartz-Jampel Syndrome Reveals Myelinating Schwann Cell Dysfunction with Persistent Axonal Depolarization in Vitro and Distal Peripheral Nerve Hyperexcitability When Perlecan Is Lacking. Am. J. Pathol. 2012, 180, 2040–2055. [Google Scholar] [CrossRef] [Green Version]
- Colombelli, C.; Palmisano, M.; Eshed-Eisenbach, Y.; Zambroni, D.; Pavoni, E.; Ferri, C.; Saccucci, S.; Nicole, S.; Soininen, R.; McKee, K.K.; et al. Perlecan Is Recruited by Dystroglycan to Nodes of Ranvier and Binds the Clustering Molecule Gliomedin. J. Cell Biol. 2015, 208, 313–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirks, M.L.; Wall, B.T.; Snijders, T.; Ottenbros, C.L.P.; Verdijk, L.B.; Van Loon, L.J.C. Neuromuscular Electrical Stimulation Prevents Muscle Disuse Atrophy during Leg Immobilization in Humans. Acta Physiol. 2014, 210, 628–641. [Google Scholar] [CrossRef]
- Vitadello, M.; Germinario, E.; Ravara, B.; Libera, L.D.; Danieli-Betto, D.; Gorza, L. Curcumin Counteracts Loss of Force and Atrophy of Hindlimb Unloaded Rat Soleus by Hampering Neuronal Nitric Oxide Synthase Untethering from Sarcolemma. J. Physiol. 2014, 592, 2637–2652. [Google Scholar] [CrossRef]
- Vitadello, M.; Gherardini, J.; Gorza, L. The Stress Protein/Chaperone Grp94 Counteracts Muscle Disuse Atrophy by Stabilizing Subsarcolemmal Neuronal Nitric Oxide Synthase. Antioxid. Redox Signal. 2014, 20, 2479–2496. [Google Scholar] [CrossRef] [Green Version]
- Lawler, J.M.; Kunst, M.; Hord, J.M.; Lee, Y.; Joshi, K.; Botchlett, R.E.; Ramirez, A.; Martinez, D.A. EUK-134 Ameliorates NNOSμ Translocation and Skeletal Muscle Fiber Atrophy during Short-Term Mechanical Unloading. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 306, R470–R482. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Therapeutic Strategies to Address Neuronal Nitric Oxide Synthase Deficiency and the Loss of Nitric Oxide Bioavailability in Duchenne Muscular Dystrophy. Orphanet J. Rare Dis. 2017, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Le, S.; Yu, M.; Hovan, L.; Zhao, Z.; Ervasti, J.; Yan, J. Dystrophin As a Molecular Shock Absorber. ACS Nano 2018, 12, 12140–12148. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.W.; Helmke, B.P.; Blackman, B.R.; Schwartz, M.A. Mechanisms of Mechanotransduction. Dev. Cell 2006, 10, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klapholz, B.; Brown, N.H. Talin—The Master of Integrin Adhesions. J. Cell Sci. 2017, 130, 2435–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor-Pareja, J.C.; Xu, T. Shaping Cells and Organs in Drosophila by Opposing Roles of Fat Body-Secreted Collagen IV and Perlecan. Dev. Cell 2011, 21, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayadev, R.; Sherwood, D.R. Basement Membranes. Curr. Biol. 2017, 27, R207–R211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crest, J.; Diz-Muñoz, A.; Chen, D.Y.; Fletcher, D.A.; Bilder, D. Organ Sculpting by Patterned Extracellular Matrix Stiffness. eLife 2017, 6, e24958. [Google Scholar] [CrossRef]
- Ramos-Lewis, W.; Page-McCaw, A. Basement Membrane Mechanics Shape Development: Lessons from the Fly. Matrix Biol. 2019, 75–76, 72–81. [Google Scholar] [CrossRef]
- Bix, G.; Iozzo, R.V. Novel Interactions of Perlecan: Unraveling Perlecan’s Role in Angiogenesis. Microsc. Res. Tech. 2008, 71, 339–348. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakada, S.; Yamashita, Y.; Machida, S.; Miyagoe-Suzuki, Y.; Arikawa-Hirasawa, E. Perlecan Facilitates Neuronal Nitric Oxide Synthase Delocalization in Denervation-Induced Muscle Atrophy. Cells 2020, 9, 2524. https://doi.org/10.3390/cells9112524
Nakada S, Yamashita Y, Machida S, Miyagoe-Suzuki Y, Arikawa-Hirasawa E. Perlecan Facilitates Neuronal Nitric Oxide Synthase Delocalization in Denervation-Induced Muscle Atrophy. Cells. 2020; 9(11):2524. https://doi.org/10.3390/cells9112524
Chicago/Turabian StyleNakada, Satoshi, Yuri Yamashita, Shuichi Machida, Yuko Miyagoe-Suzuki, and Eri Arikawa-Hirasawa. 2020. "Perlecan Facilitates Neuronal Nitric Oxide Synthase Delocalization in Denervation-Induced Muscle Atrophy" Cells 9, no. 11: 2524. https://doi.org/10.3390/cells9112524
APA StyleNakada, S., Yamashita, Y., Machida, S., Miyagoe-Suzuki, Y., & Arikawa-Hirasawa, E. (2020). Perlecan Facilitates Neuronal Nitric Oxide Synthase Delocalization in Denervation-Induced Muscle Atrophy. Cells, 9(11), 2524. https://doi.org/10.3390/cells9112524