Leukocytes, Systemic Inflammation and Immunopathology in Acute-on-Chronic Liver Failure


{kind=link}
{kind=link}
Abstract
:1. Acute-on-Chronic Liver Failure (ACLF)
2. Systemic Inflammation and Immunopathology Are Major Drivers of ACLF
3. Immunosuppression Is a Common Feature in ACLF
4. Portal Hypertension, Endothelial Activation and the Interplay with the Innate Immune System
5. Immunometabolism Also Plays a Critical Role in ACLF
6. Cells of the Innate Immune System: Role in ACLF
6.1. Mononuclear Phagocytes
6.2. Neutrophils
6.3. Macrophages
7. Mediators of Inflammation in ACLF
7.1. Cytokines
7.2. Chemokines
7.3. Growth Factors
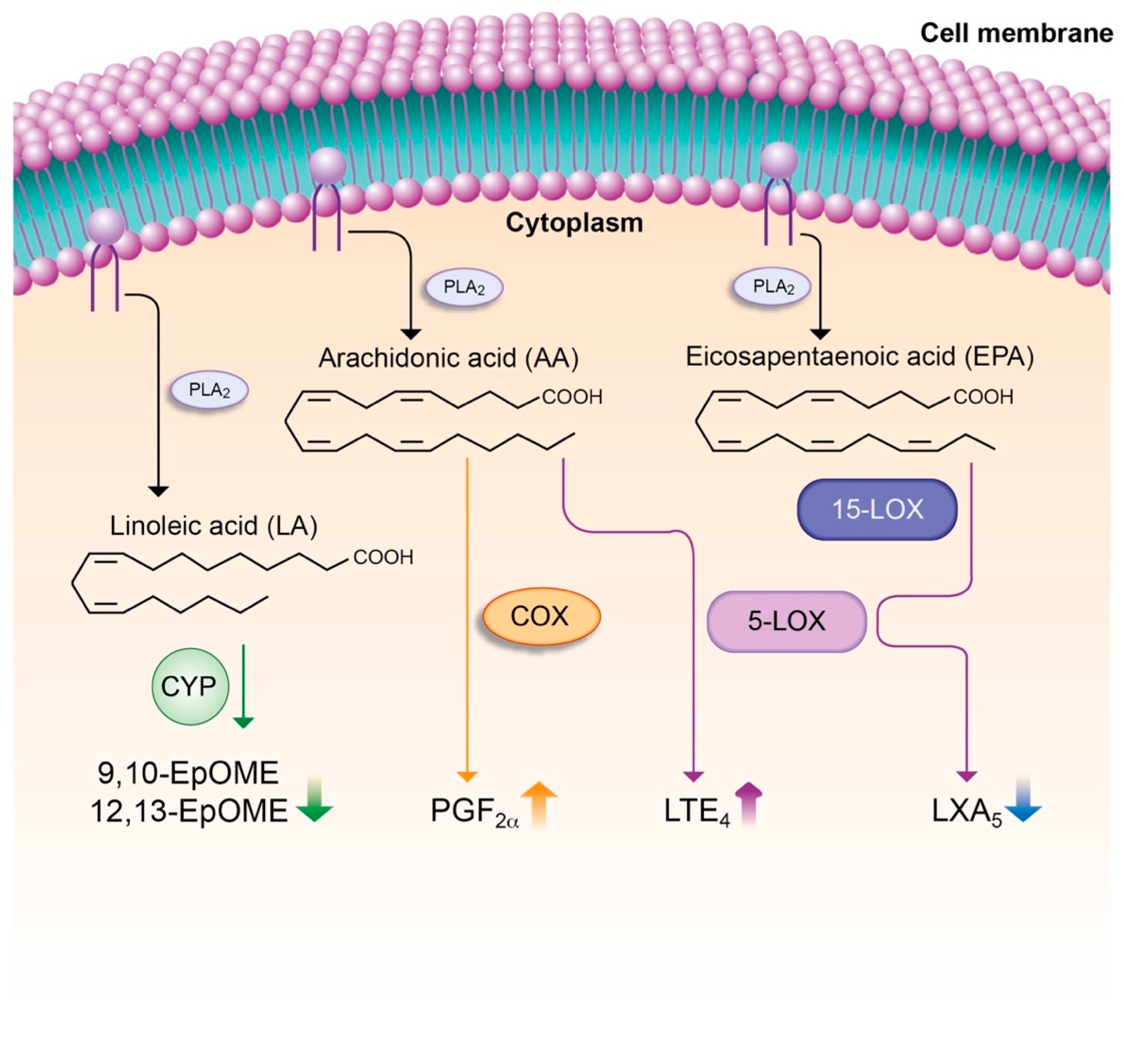
7.4. Lipid Mediators
8. Therapeutic Approaches to Limit Systemic Inflammation in ACLF
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moreau, R.; Jalan, R.; Gines, P.; Pavesi, M.; Angeli, P.; Cordoba, J.; Durand, F.; Gustot, T.; Saliba, F.; Domenicali, M.; et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 2013, 144, 1426–1437. [Google Scholar] [CrossRef]
- Arroyo, V.; Moreau, R.; Kamath, P.S.; Jalan, R.; Ginès, P.; Nevens, F.; Fernández, J.; To, U.; García-Tsao, G.; Schnabl, B. Acute-on-chronic liver failure in cirrhosis. Nat. Rev. Dis. Prim. 2016, 2, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, V.; Moreau, R.; Jalan, R. Acute-on-Chronic Liver Failure. N. Engl. J. Med. 2020, 382, 2137–2145. [Google Scholar] [CrossRef]
- Clària, J.; Stauber, R.E.; Coenraad, M.J.; Moreau, R.; Jalan, R.; Pavesi, M.; Amorós, À.; Titos, E.; Alcaraz-Quiles, J.; Oettl, K.; et al. Systemic inflammation in decompensated cirrhosis: Characterization and role in acute-on-chronic liver failure. Hepatology 2016, 64, 1249–1264. [Google Scholar] [CrossRef] [Green Version]
- Trebicka, J.; Amoros, A.; Pitarch, C.; Titos, E.; Alcaraz-quiles, J.; Schierwagen, R.; Deulofeu, C.; Fernandez-Gomez, J.; Piano, S.; Caraceni, P.; et al. Addressing Profiles of Systemic Inflammation Across the Different Clinical Phenotypes of Acutely Decompensated Cirrhosis. Front. Immunol. 2019, 10, 476. [Google Scholar] [CrossRef] [Green Version]
- Van Der Poll, T.; Van De Veerdonk, F.L.; Scicluna, B.P.; Netea, M.G. The immunopathology of sepsis and potencial therapeutic targets. Nat. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef]
- Fernández, J.; Acevedo, J.; Wiest, R.; Gustot, T.; Amoros, A.; Deulofeu, C.; Reverter, E.; Martínez, J.; Saliba, F.; Jalan, R.; et al. Bacterial and fungal infections in acute-on-chronic liver failure: Prevalence, characteristics and impact on prognosis. Gut 2018, 67, 1870–1880. [Google Scholar] [CrossRef]
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Stärkel, P.; Turner, J.R.; Ho, S.B.; Schnabl, B. Dysbiosis-induced intestinal inflammation activates TNFRI and mediates alcoholic liver disease in mice. Hepatology 2015, 61, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizawa, S.; Brar, G.; Tsukamoto, H. Cell Death and Liver Disease. Gut Liver 2020, 14, 20–29. [Google Scholar] [CrossRef] [Green Version]
- Schaapman, J.J.; Amoros, À.; Van Der Reijden, J.J.; Laleman, W.; Zeuzem, S.; Bañares, R.; Jalan, R.; Arroyo, V.; Clària, J.; Verspaget, H.W.; et al. Genetic variants of innate immunity receptors are associated with mortality in cirrhotic patients with bacterial infection. Liver Int. 2020, 40, 646–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaraz-Quiles, J.; Titos, E.; Casulleras, M.; Pavesi, M.; López-Vicario, C.; Rius, B.; Lopategi, A.; de Gottardi, A.; Graziadei, I.; Gronbaek, H.; et al. Polymorphisms in the IL-1 gene cluster influence systemic inflammation in patients at risk for acute-on-chronic liver failure. Hepatology 2017, 65, 216–220. [Google Scholar] [CrossRef] [PubMed]
- De Meo, A.N.; Andersen, B.R.; English, D.K.; Peterson, J. Defective Chemotaxis Associated with a Serum Inhibitor in Cirrhotic Patients. N. Engl. J. Med. 1972, 286, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lu, Q.; Zhu, M.; Huang, C.; Yu, K.; Huang, Y.; Zhao, X.; Luo, X.-G.; Zheng, J.-M. Lower level of complement component C3 and C3a in the plasma means poor outcome in the patients with hepatitis B virus related acute-on-chronic liver failure. BMC Gastroenterol. 2020, 20, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, M.; Maggioli, C.; Zaccherini, G. Human albumin in the management of complications of liver cirrhosis. Crit. Care 2012, 16, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homann, C.; Varming, K.; Hogasen, K.; Mollnes, T.E.; Graudal, N.; Thomsen, A.C.; Garred, P. Acquired C3 deficiency in patients with alcoholic cirrhosis predisposes to infection and increased mortality. Gut 1997, 40, 544–549. [Google Scholar] [CrossRef]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Bernsmeier, C.; Pop, O.T.; Singanayagam, A.; Triantafyllou, E.; Patel, V.C.; Weston, C.J.; Curbishley, S.; Sadiq, F.; Vergis, N.; Khamri, W.; et al. Patients With Acute-on-Chronic Liver Failure Have Increased Numbers of Regulatory Immune Cells Expressing the Receptor Tyrosine Kinase MERTK. Gastroenterology 2015, 148, 603–615. [Google Scholar] [CrossRef]
- Bernsmeier, C.; Triantafyllou, E.; Brenig, R.; Lebosse, F.J.; Singanayagam, A.; Patel, V.C.; Pop, O.T.; Khamri, W.; Nathwani, R.; Tidswell, R.; et al. CD14+ CD15− HLA-DR− myeloid-derived suppressor cells impair antimicrobial responses in patients with acute-on-chronic liver failure. Gut 2018, 67, 1155–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, A.J.; Fullerton, J.N.; Massey, K.A.; Auld, G.; Sewell, G.; James, S.; Newson, J.; Karra, E.; Winstanley, A.; Alazawi, W.; et al. Immunosuppression in acutely decompensated cirrhosis is mediated by prostaglandin E2. Nat. Med. 2014, 20, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Xing, T.; Li, L.; Cao, H.; Huang, J. Altered immune function of monocytes in different stages of patients with acute on chronic liver failure. Clin. Exp. Immunol. 2006, 147, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Trebicka, J.; Fernandez, J.; Papp, M.; Caraceni, P.; Laleman, W.; Gambino, C.; Giovo, I.; Uschner, F.E.; Jimenez, C.; Mookerjee, R.; et al. The PREDICT study uncovers three clinical courses of acutely decompensated cirrhosis that have distinct pathophysiology. J. Hepatol. 2020, 73, 842–854. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Albillos, A.; Barden, G.E.; West, A.B. Bacterial translocation in acute and chronic portal hypertension. Hepatology 1993, 17, 1081–1085. [Google Scholar] [CrossRef]
- Lozano-ruiz, B.; Bachiller, V.; García-martínez, I.; Zapater, P.; Gómez-hurtado, I.; Moratalla, A.; Giménez, P.; Bellot, P.; Francés, R.; Such, J.; et al. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J. Hepatol. 2015, 62, 64–71. [Google Scholar] [CrossRef]
- Michelena, J.; Altamirano, J.; Abraldes, J.G.; Affò, S.; Morales-Ibanez, O.; Sancho-Bru, P.; Dominguez, M.; García-Pagán, J.C.; Fernández, J.; Arroyo, V.; et al. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology 2015, 62, 762–772. [Google Scholar] [CrossRef]
- Berres, M.; Asmacher, S.; Lehmann, J.; Jansen, C.; Görtzen, J.; Klein, S.; Meyer, C.; Strunk, H.M.; Fimmers, R.; Tacke, F.; et al. CXCL9 is a prognostic marker in patients with liver cirrhosis receiving transjugular intrahepatic portosystemic shunt. J. Hepatol. 2015, 62, 332–339. [Google Scholar] [CrossRef]
- Lehmann, J.M.; Claus, K.; Jansen, C.; Pohlmann, A.; Schierwagen, R.; Meyer, C.; Thomas, D.; Manekeller, S.; Clària, J.; Strassburg, C.P.; et al. Circulating CXCL10 in cirrhotic portal hypertension might reflect systemic inflammation and predict ACLF and mortality. Liver Int. 2018, 38, 875–884. [Google Scholar] [CrossRef]
- Levi, M.; Keller, T.T.; van Gorp, E.; ten Cate, H. Infection and inflammation and the coagulation system. Cardiovasc. Res. 2003, 60, 26–39. [Google Scholar] [CrossRef]
- Coughlin, S.R. Thrombin signalling and protease-activated receptors. Nature 2000, 407, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Plessier, A.; Denninger, M.-H.; Consigny, Y.; Pessione, F.; Francoz, C.; Durand, F.; Francque, S.; Bezeaud, A.; Chauvelot-Moachon, L.; Lebrec, D.; et al. Coagulation disorders in patients with cirrhosis and severe sepsis. Liver Int. 2003, 23, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, A.S.; Parada, X.V.; Ortiz, G.A.; Long, J.; Krinsky, S.; Zhao, S.; Fuchs, B.C.; Sojoodi, M.; Zhang, D.; Karumanchi, S.A.; et al. Serum Angiopoietin-2 Predicts Mortality and Kidney Outcomes in Decompensated Cirrhosis. Hepatology 2019, 69, 729–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solé, C.; Solà, E.; Morales-ruiz, M.; Fernàndez, G.; Huelin, P.; Graupera, I.; Moreira, R.; de Prada, G.; Ariza, X.; Pose, E.; et al. Characterization of Inflammatory Response in Acute-on-Chronic Liver Failure and Relationship with Prognosis. Sci. Rep. 2016, 6, 32341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amalakuhan, B.; Habib, S.A.; Mangat, M.; Reyes, L.F.; Rodriguez, A.H.; Hinojosa, C.A.; Soni, N.J.; Gilley, R.P.; Bustamante, C.A.; Anzueto, A.; et al. Endothelial adhesion molecules and multiple organ failure in patients with severe sepsis. Cytokine 2016, 88, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Gauley, J.; Pisetsky, D.S. The release of microparticles by RAW 264.7 macrophage cells stimulated with TLR ligands. J. Leukoc. Biol. 2010, 87, 1115–1123. [Google Scholar] [CrossRef]
- Rautou, P.; Bresson, J.; Sainte-Marie, Y.; Vion, A.; Paradis, V.; Renard, J.M.; Devue, C.; Heymes, C.; Letteron, P.; Elkrief, L.; et al. Abnormal Plasma Microparticles Impair Vasoconstrictor Responses in Patients with Cirrhosis. Gastroenterology 2012, 143, 166–176. [Google Scholar] [CrossRef]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. Molecular Cell Biology, 4th ed.; W.H. Free: New York, NY, USA, 2000; ISBN 0-7167-3136-3. [Google Scholar]
- Wang, A.; Luan, H.; Medzhitov, R. An Evolutionary Perspective on Immunometabolism. Science 2019, 363, 6423. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-schughart, L.A.; Mueller-klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef]
- Ratter, J.M.; Rooijackers, H.M.M.; Hooiveld, G.J.; Hijmans, A.G.M.; de Galan, B.E.; Tack, C.; Stienstra, R. In vitro and in vivo Effects of Lactate on Metabolism and Cytokine Production of Human Primary PBMCs and Monocytes. Front. Immunol. 2018, 9, 2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, R.; Clària, J.; Aguilar, F.; Fenaille, F.; Lozano, J.J.; Junot, C.; Colsch, B.; Caraceni, P.; Trebicka, J.; Pavesi, M.; et al. Blood metabolomics uncovers inflammation-associated mitochondrial dysfunction as a potential mechanism underlying ACLF. J. Hepatol. 2020, 72, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Bellance, N.; Damm, A.; Bing, H.; Zhu, Z.; Yovchev, M.I.; Sehgal, V.; Moss, T.J.; Oertel, M.; Pipinos, I.I.; et al. A switch in the source of ATP production and a loss in capacity to perform glycolysis are hallmarks of hepatocyte failure in advance liver disease. J. Hepatol. 2014, 60, 1203–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C. Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaccherini, G.; Aguilar, F.; Caraceni, P.; Clària, J.; Lozano, J.; Fenaille, F. Addressing the role of amino acids in systemic inflammatory responses and organ failures in patients with ACLF. J. Hepatol. 2020, in press. [Google Scholar] [CrossRef]
- Van Wyngene, L.; Vandewalle, J.; Libert, C. Reprogramming of basic metabolic pathways in microbial sepsis: Therapeutic targets at last? EMBO Mol. Med. 2018, 10, 1–18. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Reddy, K.R.; Leary, J.G.O.; Vargas, H.E.; Lai, J.C.; Kamath, P.S.; Tandon, P.; Wong, F.; Subramanian, R.M.; Thuluvath, P.; et al. Serum Levels of Metabolites Produced by Intestinal Microbes and Lipid Moieties Independently Associated With Acute-on-Chronic Liver Failure and Death in Patients With Cirrhosis. Gastroenterology 2020, 159, 1715–1730. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef]
- Weiss, E.; de la Grange, P.; Defaye, M.; Lozano, J.J.; Aguilar, F.; Hedge, P.; Jolly, A.; Moga, L.; Baweja, S.; Agarwal, B.; et al. RNA Identification of Dysregulated Blood Immune Cells Playing a Pathophysiological Role in Critically Ill Patients With ACLF. Front. Immunol. 2020, in press. [Google Scholar]
- Liu, F.; Duan, X.; Wan, Z.; Zang, H.; You, S.; Yang, R.; Liu, H.; Li, D.; Li, J.; Zhang, Y.; et al. Lower number and decreased function of natural killer cells in hepatitis B virus related acute-on-chronic liver failure. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 605–613. [Google Scholar] [CrossRef]
- Stephan, F.; Yang, K.; Tankovic, J.; Soussy, C.-J.; Dhonneur, G.; Duvaldestin, P.; Brochard, L.; Brun-Buisson, C.; Harf, A.; Delclaux, C. Impairment of polymorphonuclear neutrophil functions precedes nosocomial infections in critically ill patients. Crit. Care Med. 2002, 30, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Vergis, N.; Khamri, W.; Beale, K.; Sadiq, F.; Aletrari, M.O.; Moore, C.; Atkinson, S.R.; Bernsmeier, C.; Possamai, L.A.; Petts, G.; et al. Defective monocyte oxidative burst predicts infection in alcoholic hepatitis and is associated with reduced expression of NADPH oxidase. Gut 2017, 66, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albillos, A.; Lario, M.; Álvarez-Mon, M. Cirrhosis-associated immune dysfunction: Distinctive features and clinical relevance. J. Hepatol. 2014, 61, 1385–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasmuth, H.E.; Kunz, D.; Yagmur, E.; Timmer-strangho, A.; Vidacek, D.; Siewert, E.; Bach, J.; Geier, A.; Purucker, E.A.; Gressner, A.M.; et al. Patients with acute on chronic liver failure display ′sepsis-like′ immune paralysis. J. Hepatol. 2005, 42, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de la Hera, A.; Reyes, E.; Monserrat, J.; Muñoz, L.; Nieto, M.; Prieto, A.; Sanz, E.; Alvarez-Mon, M. Tumour necrosis factor-alpha expression by activated monocytes and altered T-cell homeostasis in ascitic alcoholic cirrhosis: Amelioration with norfloxacin. J. Hepatol. 2004, 40, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Korf, H.; Plessis, J.; Van Pelt, J.; De Groote, S.; Cassiman, D.; Verbeke, L.; Ghesquière, B.; Fendt, S.; Bird, M.J.; Talebi, A.; et al. Inhibition of glutamine synthetase in monocytes from patients with acute-on-chronic liver failure resuscitates their antibacterial and inflammatory capacity. Gut 2019, 68, 1872–1883. [Google Scholar] [CrossRef]
- Sampath, P.; Moideen, K.; Ranganathan, U.D.; Bethunaickan, R. Monocyte Subsets: Phenotypes and Function in Tuberculosis Infection. Front. Immunol. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Wu, W.; Yang, Y.; Yang, Q.; Song, G.; Wu, Y.; Wei, L.; Chen, Z. Decreased Tim-3 expression is associated with functional abnormalities of monocytes in decompensated cirrhosis without overt bacterial infection. J. Hepatol. 2015, 63, 60–67. [Google Scholar] [CrossRef]
- Berry, P.A.; Mcphail, M.J.W.; Davies, E.T.; Wendon, J.A.; Vergani, D. Severity of the compensatory anti-inflammatory response determined by monocyte HLA-DR expression may assist outcome prediction in cirrhosis. Intensive Care Med. 2011, 37, 453–460. [Google Scholar] [CrossRef]
- Kusaba, N.; Kumashiro, R.; Ogata, H.; Sata, M.; Tanikawa, K. In Vitro Study of Neutrophil Apoptosis in Liver Cirrhosis. Intern. Med. 1998, 37, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Khanam, A.; Trehanpati, N.; Riese, P.; Rastogi, A.; Guzman, C.A.; Sarin, S.K. Blockade of Neutrophil’s Chemokine Receptors CXCR1/2 Abrogate Liver Damage in Acute-on-Chronic Liver Failure. Front. Immunol. 2017, 8, 464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Jeng, W.; Ho, Y.; Teng, W.; Hsieh, Y.; Chen, W.-T.; Lin, H.-H.; Sheen, I.-S.; Lin, C.-Y. Increased EMR2 expression on neutrophils correlates with disease severity and predicts overall mortality in cirrhotic patients. Sci. Rep. 2016, 6, 38250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussif, A.; Rolas, L.; Weiss, E.; Bouriche, H.; Moreau, R.; Périanin, A. Impaired intracellular signaling, myeloperoxidase release and bactericidal activity of neutrophils from patients with alcoholic cirrhosis. J. Hepatol. 2016, 64, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Fiuza, C.; Salcedo, M.; Clemente, G.; Tellado, J.M. Granulocyte Colony-Stimulating Factor Improves Deficient In Vitro Neutrophil Transendothelial Migration in Patients with Advanced Liver Disease. Clin. Diagn. Lab. Immunol. 2002, 9, 433–439. [Google Scholar] [CrossRef] [Green Version]
- Artru, F.; Bou Saleh, M.; Maggiotto, F.; Lassailly, G.; Ningarhari, M.; Demaret, J.; Ntandja-Wandji, L.-C.; Pais de Barros, J.-P.J.L.; Drumez, E.; Helou, D.G.; et al. IL-33/ST2 pathway regulates neutrophil migration and predicts outcome in patients with severe alcoholic hepatitis. J. Hepatol. 2020, 72, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Rajkovic, I.A.; Williams, R. Abnormalities of Neutrophil Phagocytosis, Intracellular Killing and Metabolic Activity in Alcoholic Cirrhosis and Hepatitis. Hepatology 1986, 6, 252–262. [Google Scholar] [CrossRef]
- Rolas, L.; Boussif, A.; Weiss, E.; Lettéron, P.; Haddad, O.; El-benna, J.; Rautou, P.; Moreau, R.; Périanin, A. NADPH oxidase depletion in neutrophils from patients with cirrhosis and restoration via toll-like receptor 7/8 activation. Gut 2018, 67, 1505–1516. [Google Scholar] [CrossRef]
- Garfia, C.; García-Ruiz, I.; Solís-Herruzo, J.A. Deficient phospholipase C activity in blood polimorphonuclear neutrophils from patients with liver cirrhosis. J. Hepatol. 2004, 40, 749–756. [Google Scholar] [CrossRef]
- Mookerjee, R.P.; Stadlbauer, V.; Lidder, S.; Wright, G.A.K.; Hodges, S.J.; Davies, N.A.; Jalan, R. Neutrophil Dysfunction in Alcoholic Hepatitis Superimposed on Cirrhosis is Reversible and Predicts the Outcome. Hepatology 2007, 46, 831–840. [Google Scholar] [CrossRef]
- Taylor, N.J.; Vijay, G.K.M.; Abeles, R.D.; Auzinger, G.; Bernal, W.; Ma, Y.; Wendon, J.A.; Shawcross, D.L. The severity of circulating neutrophil dysfunction in patients with cirrhosis is associated with 90-day and 1-year mortality. Aliment. Pharmacol. Ther. 2014, 40, 705–715. [Google Scholar] [CrossRef]
- Moreau, N.; Wittebole, X.; Fleury, Y.; Forget, P.; Laterre, P.-F.; Castanares-Zapatero, D. Neutrophil-to-Lymphocyte Ratio Predicts Death in Acute-on-Chronic Liver Failure Patients Admitted to the Intensive Care Unit: A Retrospective Cohort Study. Shock 2018, 49, 385–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grønbæk, H.; Rødgaard-Hansen, S.; Aagaard, N.K.; Arroyo, V.; Moestrup, S.K.; Garcia, E.; Solà, E.; Domenicali, M.; Piano, S.; Vilstrup, H.; et al. Macrophage activation markers predict mortality in patients with liver cirrhosis without or with acute-on-chronic liver failure (ACLF). J. Hepatol. 2016, 64, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriadis, T.; Georgios Pissas, V.L.; Stefanidis, I.; Lawson, B.R. Toll-like receptors and their role in renal pathologies. Inflamm Allergy Drug Targets 2012, 11, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Clària, J.; Arroyo, V.; Moreau, R. The Acute-on-Chronic Liver Failure Syndrome, or When the Innate Immune System Goes Astray. J. Immunol. 2016, 197, 3755–3761. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C.; Kushner, I. Acute-Phase Proteins and Other Systemic Responses to Inflammation. N. Engl. J. Med. 1999, 340, 448–454. [Google Scholar] [CrossRef]
- Borish, L.C.; Steinke, J.W. 2. Cytokines and chemokines. J. Allergy Clin. Immunol. 2003, 2, 460–475. [Google Scholar] [CrossRef]
- Byl, B.; Roucloux, I.; Crusiaux, A.; Dupont, E.; Devière, J. Tumor necrosis factor α and interleukin 6 plasma levels in infected cirrhotic patients. Gastroenterology 1993, 104, 1492–1497. [Google Scholar] [CrossRef]
- Albillos, A.; de la Hera, A.; González, M.; Moya, J.-L.; Calleja, J.-L.; Monserrat, J.; Ruiz-del-Arbol, L.; Alvarez-Mon, M. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology 2003, 37, 208–217. [Google Scholar] [CrossRef]
- Navasa, M.; Follo, A.; Filella, X.; Jiménez, W.; Francitorra, A.; Planas, R.; Rimola, A.; Arroyo, V.; Rodés, J. Tumor Necrosis Factor and Interleukin-6 in Spontaneous Bacterial Peritonitis in Cirrhosis: Relationship With the Development of Renal Impairment and Mortality. Hepatology 1998, 27, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-arras, D.; Rose-john, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy 2016, 8, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Genovese, M.C.; Kremer, J.M.; van Vollenhoven, R.F.; Alten, R.; Scali, J.J.; Kelman, A.; Dimonaco, S.; Brockwell, L. Transaminase Levels and Hepatic Events During Tocilizumab Treatment. Pooled Analysis of Long-Term Clinical Trial Safety Data in Rheumatoid Arthritis. Arthritis Rheumatol. 2017, 69, 1751–1761. [Google Scholar] [CrossRef] [Green Version]
- Boettler, T.; Newsome, P.N.; Mondelli, M.U.; Maticic, M.; Cordero, E.; Cornberg, M.; Berg, T. Care of patients with liver disease during the COVID-19 pandemic: EASL-ESCMID position paper. J. Hepatol. Reports 2020, 2, 100113. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. The Chemokine Superfamily Revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef]
- Simonetto, D.; Shah, V.; Kamath, P. Improving survival in ACLF: Growing evidence for use of G-CSF. Hepatol. Int. 2017, 11, 473–475. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Guo, R.; Ming, D.; Deng, Y.; Su, M.; Lin, C.; Li, J.; Lin, Z.; Su, Z. The Transforming Growth Factor β1/Interleukin-31 Pathway Is Upregulated in Patients with Hepatitis B Virus-Related Acute-on-Chronic Liver Failure and Is Associated with Disease Severity and Survival. Clin. Vaccine Immunol. 2015, 22, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 13–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buczynski, M.W.; Dumlao, D.S.; Dennis, E.A. An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009, 50, 1015–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clària, J.; Romano, M. Pharmacological intervention of cyclooxygenase-2 and 5-lipoxygenase pathways. Impact on inflammation and cancer. Curr. Pharm. Des. 2005, 11, 3431–3447. [Google Scholar] [CrossRef] [PubMed]
- Stables, M.J.; Gilroy, D.W. Old and new generation lipid mediators in acute inflammation and resolution. Prog. Lipid Res. 2011, 50, 35–51. [Google Scholar] [CrossRef]
- Claria, J.; Serhan, C.N. Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc. Natl. Acad. Sci. USA 1995, 92, 9475–9479. [Google Scholar] [CrossRef] [Green Version]
- López-vicario, C.; Rius, B.; Alcaraz-quiles, J.; García-alonso, V.; Lopategi, A.; Titos, E.; Clària, J. Pro-resolving mediators produced from EPA and DHA: Overview of the pathways involved and their mechanisms in metabolic syndrome and related liver diseases. Eur. J. Pharmacol. 2016, 785, 133–143. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [Green Version]
- China, L.; Maini, A.; Skene, S.S.; Shabir, Z.; Sylvestre, Y.; Colas, R.A.; Ly, L.; Salles, N.B.; Belloti, V.; Dalli, J.; et al. Albumin Counteracts Immune-Suppressive Effects of Lipid Mediators in Patients With Advanced Liver Disease. Clin. Gastroenterol. Hepatol. 2018, 16, 738–747. [Google Scholar] [CrossRef] [Green Version]
- Becares, N.; Härmälä, S.; Colas, R.A.; Maini, A.A.; Bennet, K.; Skene, S.S.; Shabir, Z.; Dalli, J.; O’Brien, A. Immune Regulatory Mediators in Plasma from Patients With Acute Decompensation Are Associated With 3-Month Mortality. Clin. Gastroenterol. Hepatol. 2020, 18, 1207–1215. [Google Scholar] [CrossRef] [Green Version]
- López-Vicario, C.; Checa, A.; Urdangarin, A.; Aguilar, F.; Alcaraz-Quiles, J.; Caraceni, P.; Amorós, A.; Pavesi, M.; Gómez-Cabrero, D.; Trebicka, J.; et al. Targeted lipidomics reveals extensive changes in circulating lipid mediators in patients with acutely decompensated cirrhosis. J. Hepatol. 2020, 73, 817–828. [Google Scholar] [CrossRef]
- Sort, P.; Navasa, M.; Arroyo, V.; Aldeguer, X.; Planas, R.; Ruiz-del-Arbol, L.; Castells, L.; Vargas, V.; Soriano, G.; Guevara, M.; et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N. Engl. J. Med. 1999, 403–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardi, M.; Angeli, P.; Claria, J.; Moreau, R.; Gines, P.; Jalan, R.; Caraceni, P.; Fernandez, J.; Gerbes, A.L.; Brien, A.J.O.; et al. Albumin in decompensated cirrhosis: New concepts and perspectives. Gut 2020, 69, 1127–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caraceni, P.; Riggio, O.; Angeli, P.; Alessandria, C.; Neri, S.; Foschi, F.G.; Levantesi, F.; Airoldi, A.; Boccia, S.; Svegliati-Baroni, G.; et al. Long-term albumin administration in decompensated cirrhosis (ANSWER): An open-label randomised trial. Lancet 2018, 391, 2417–2429. [Google Scholar] [CrossRef]
- Fernández, J.; Clària, J.; Amorós, A.; Aguilar, F.; Castro, M.; Casulleras, M.; Acevedo, J.; Duran-Güell, M.; Nuñez, L.; Costa, M.; et al. Effects of Albumin Treatment on Systemic and Portal Hemodynamics and Systemic Inflammation in Patients With Decompensated Cirrhosis. Gastroenterology 2019, 157, 149–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bañares, R.; Ibáñez-samaniego, L.; Torner, J.M.; Pavesi, M.; Olmedo, C.; Catalina, M.V.; Albillos, A.; Larsen, F.S.; Nevens, F.; Hassanein, T.; et al. Meta-analysis of individual patient data of albumin dialysis in acute-on-chronic liver failure: Focus on treatment intensity. Therap. Adv. Gastroenterol. 2019, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Niewinski, G.; Raszeja-Wyszomirska, J.; Hrenczuk, M.; Rozga, A.; Malkowski, P.; Rozga, J. Intermittent high-flux albumin dialysis with continuous venovenous hemodialysis for acute-on-chronic liver failure and acute kidney injury. Artif. Organs 2020, 44, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhang, Z.; Cheng, Q.; Chen, G.; Li, W.; Ma, K.; Guo, W.; Luo, X.; Chen, T.; Ning, Q. Plasma perfusion combined with plasma exchange in chronic hepatitis B-related acute-on-chronic liver failure patients. Hepatol. Int. 2020, 14, 491–502. [Google Scholar] [CrossRef]
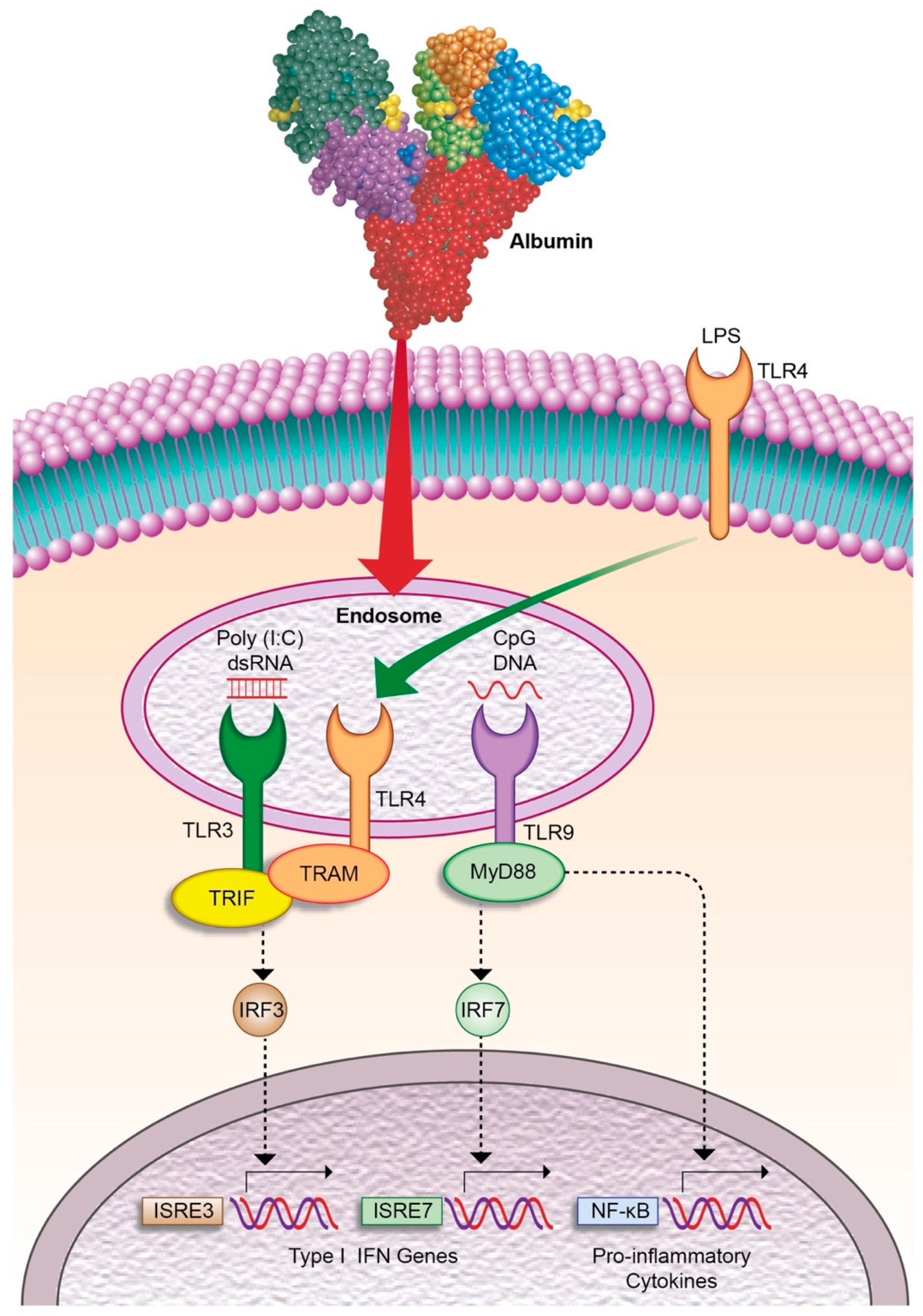
- Casulleras, M.; Flores-Costa, R.; Duran-Güell, M.; Alcaraz-quiles, J.; Sanz, S.; Titos, E.; López-vicario, C.; Fernández, J.; Horrillo, R.; Costa, M.; et al. Albumin internalizes and inhibits endosomal TLR signaling in leukocytes from patients with decompensated cirrhosis. Sci. Transl. Med. 2020, 12, eaax5135. [Google Scholar] [CrossRef]
- Garg, V.; Garg, H.; Khan, A.; Trehanpati, N.; Kumar, A.; Sharma, B.C.; Sakhuja, P.; Sarin, S.K. Granulocyte Colony–Stimulating Factor Mobilizes CD34+ Cells and Improves Survival of Patients With Acute-on-Chronic Liver Failure. Gastroenterology 2012, 142, 505–512. [Google Scholar] [CrossRef]
- Duan, X.; Liu, F.; Tong, J.; Yang, H.; Chen, J.; Liu, X.; Mao, Y.; Xin, S.-J.; Hu, J.-H. Granulocyte-colony stimulating factor therapy improves survival in patients with hepatitis B virus-associated acute-on-chronic liver failure. World J. Gastroenterol. 2013, 19, 1104–1110. [Google Scholar] [CrossRef]
- Saha, B.K.; Al Mahtab, M.; Akbar, S.M.F.; Noor-E-Alam, S.M.; Al Mamun, A.; Hossain, S.M.S.; Alam, M.A.; Moben, A.L.; Khondaker, F.A.; Chowdhury, F.I.; et al. Therapeutic implications of granulocyte colony stimulating factor in patients with acute-on-chronic liver failure: Increased survival and containment of liver damage. Hepatol. Int. 2017, 11, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, C.; Brosteanu, O.; Tenckhoff, H.; Splith, K.; Schmelzle, M.; Berg, T. Granulocyte colony stimulating factor (G-CSF) to treat acute-on-chronic liver failure: A multicenter randomized trial (GRAFT STUDY). J. Hepatol. 2015, 62, S847. [Google Scholar] [CrossRef]
- Sharma, M.; Kulkarni, A.; Sasikala, M.; Kumar, P.; Jaggaiahgari, S.; Pondugala, K.; Jaishetwar, G.; Darisetty, S.; Jagtap, N.; Gupta, R.; et al. Long-term Outcome of Autologous Hematopoietic Stem Cell Infusion in Cirrhosis: Waning Effect over Time. J. Clin. Transl. Hepatol. 2020, 9, 1–6. [Google Scholar] [CrossRef]
- Engelmann, C.; Sheikh, M.; Sharma, S.; Kondo, T.; Loeffler-Wirth, H.; Zheng, Y.B.; Novelli, S.; Hall, A.; Kerbert, A.J.C.; Macnaughtan, J.; et al. Toll-like receptor 4 is a therapeutic target for prevention and treatment of liver failure. J. Hepatol. 2020, 73, 102–112. [Google Scholar] [CrossRef]
- Xiang, X.; Feng, D.; Hwang, S.; Ren, T.; Wang, X.; Trojnar, E.; Matyas, C.; Mo, R.; Shang, D.; He, Y.; et al. Interleukin-22 ameliorates acute-on-chronic liver failure by reprogramming impaired regeneration pathways in mice. J. Hepatol. 2020, 72, 736–745. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, Z.; Xu, R.; Lin, H.; Fu, J.; Zou, Z.; Zhang, A.; Shi, J.; Chen, L.; Lv, S.; et al. Human Mesenchymal Stem Cell Transfusion Is Safe and Improves Liver Function in Acute-on-Chronic Liver Failure Patients. Stem. Cells Transl. Med. 2012, 1, 725–731. [Google Scholar] [CrossRef]
- Lin, B.-L.; Chen, J.-F.; Qiu, W.-H.; Wang, K.-W.; Xie, D.-Y.; Chen, X.-Y.; Liu, Q.-L.; Peng, L.; Li, J.-G.; Mei, Y.-Y.; et al. Allogeneic Bone Marrow–Derived Mesenchymal Stromal Cells for Hepatitis B Virus–Related Acute-on-Chronic Liver Failure: A Randomized Controlled Trial. Hepatology 2017, 66, 209–219. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casulleras, M.; Zhang, I.W.; López-Vicario, C.; Clària, J. Leukocytes, Systemic Inflammation and Immunopathology in Acute-on-Chronic Liver Failure. Cells 2020, 9, 2632. https://doi.org/10.3390/cells9122632
Casulleras M, Zhang IW, López-Vicario C, Clària J. Leukocytes, Systemic Inflammation and Immunopathology in Acute-on-Chronic Liver Failure. Cells. 2020; 9(12):2632. https://doi.org/10.3390/cells9122632
Chicago/Turabian StyleCasulleras, Mireia, Ingrid W. Zhang, Cristina López-Vicario, and Joan Clària. 2020. "Leukocytes, Systemic Inflammation and Immunopathology in Acute-on-Chronic Liver Failure" Cells 9, no. 12: 2632. https://doi.org/10.3390/cells9122632
APA StyleCasulleras, M., Zhang, I. W., López-Vicario, C., & Clària, J. (2020). Leukocytes, Systemic Inflammation and Immunopathology in Acute-on-Chronic Liver Failure. Cells, 9(12), 2632. https://doi.org/10.3390/cells9122632