NOTCH1 Signaling in Head and Neck Squamous Cell Carcinoma

 ,
, 
Abstract
:1. Introduction
2. NOTCH1 Encodes for a Conserved Receptor Regulating Transcription and Cell Fate
3. NOTCH1 Mutations in HNSCC
3.1. Initial Identification of NOTCH1 Mutations
3.2. Confirmation of NOTCH1 Mutations in HPV-Negative and -Positive HNSCC
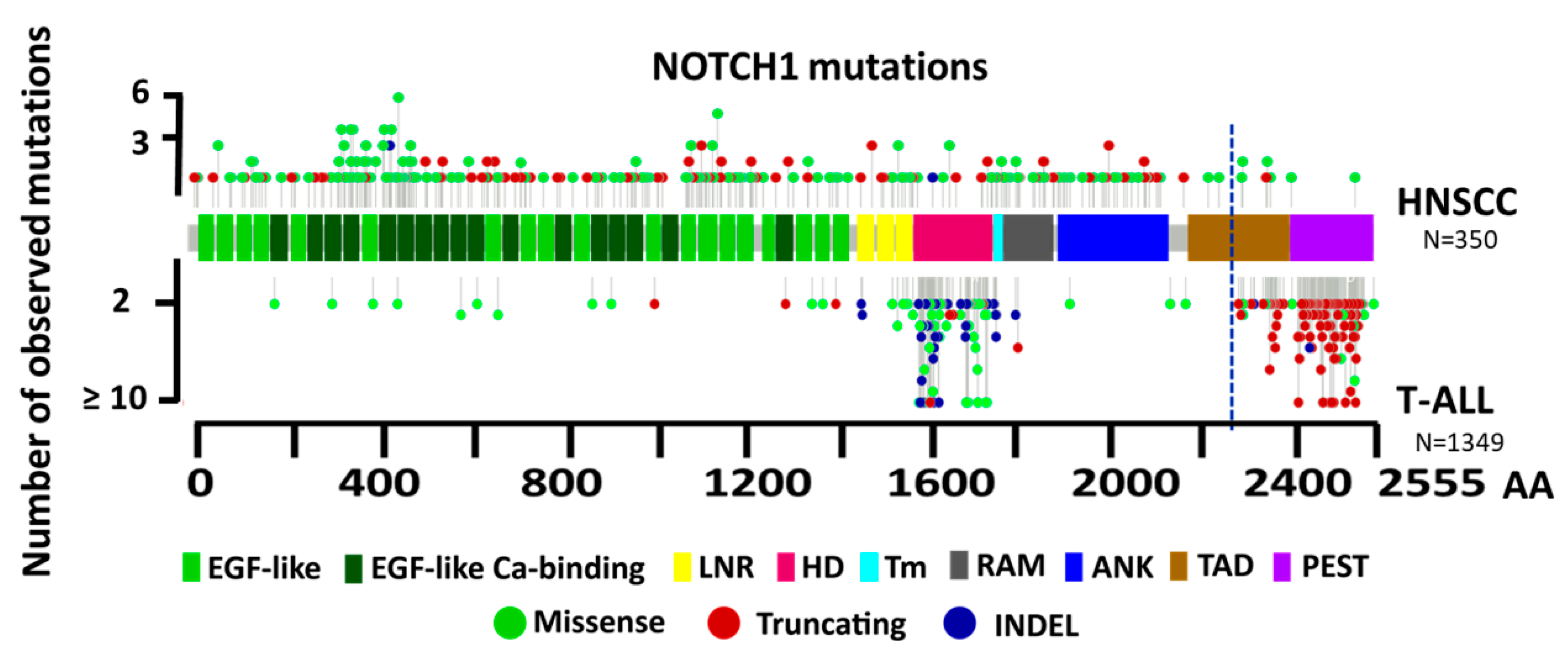
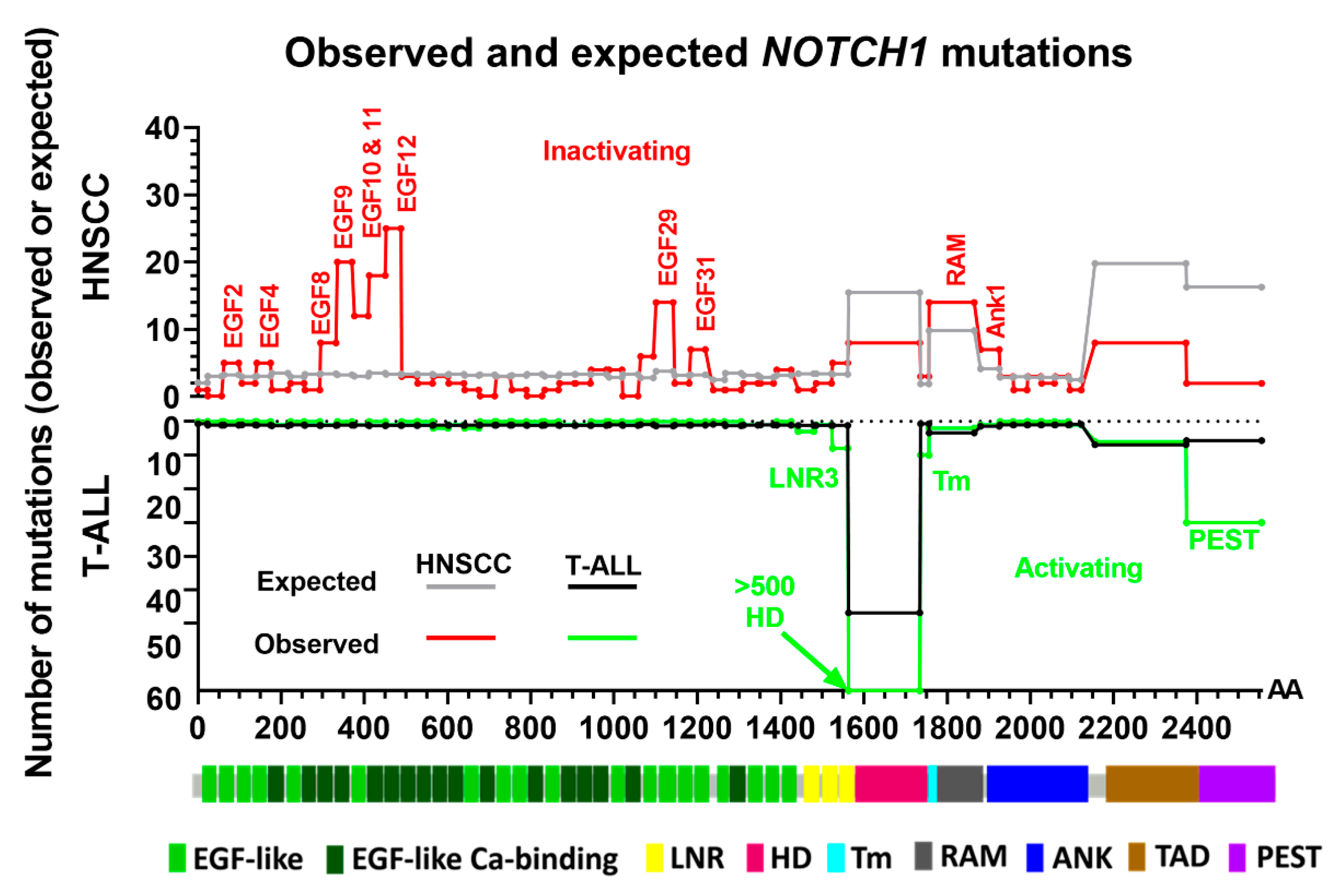
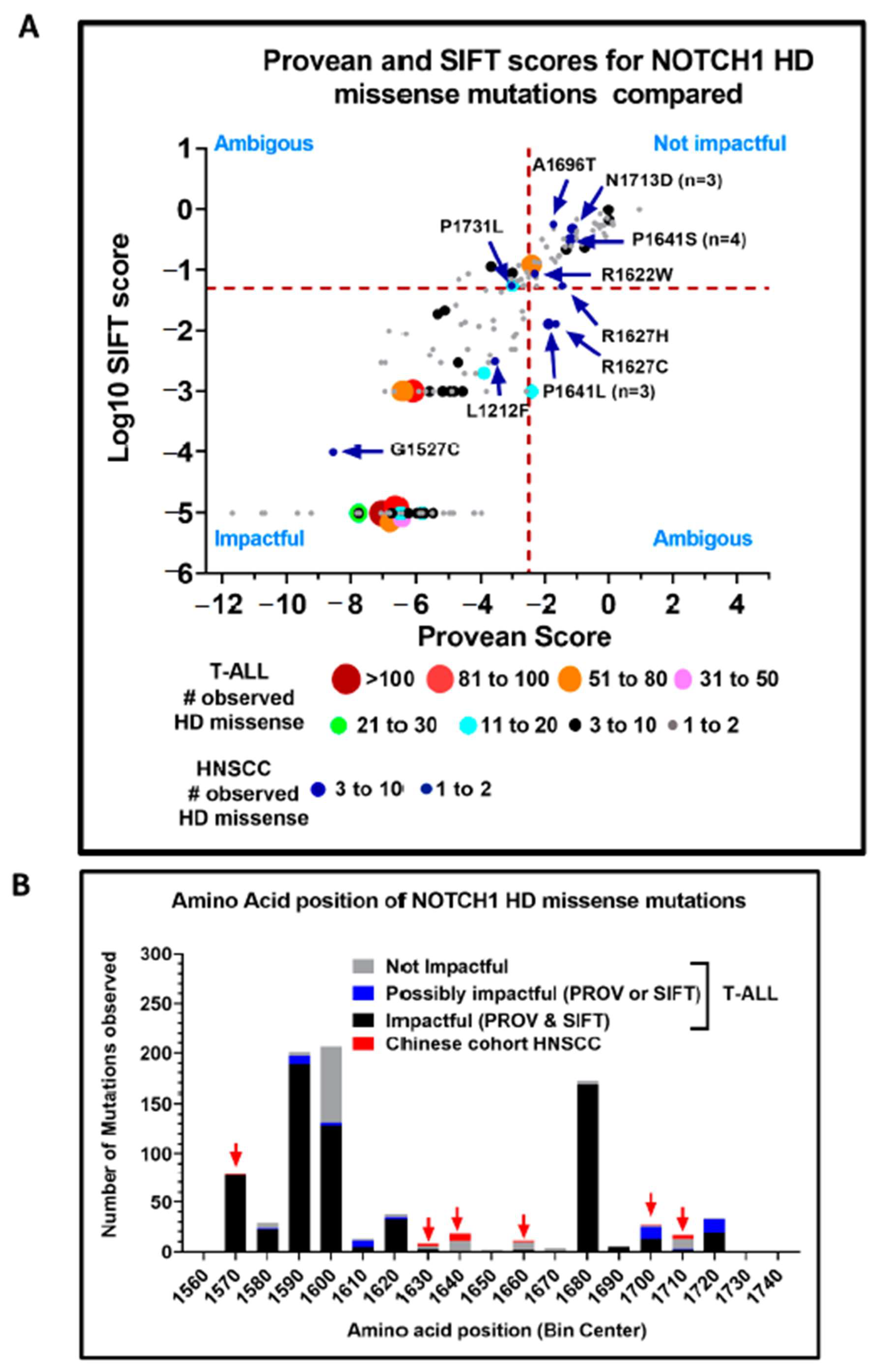
3.3. Structural Characterization of NOTCH1 Mutations
3.4. Evidence of Activating NOTCH1 Mutations in HNSCC
4. NOTCH1 Signaling in HNSCCs
4.1. HNSCC Cell Line Models for Studying NOTCH1
4.2. Restoration of NOTCH1 Signaling Alters Growth of NOTCH1 Mutant HNSCC Cell Lines
4.3. NOTCH1 Pathway in HNSCCs Harboring wt NOTCH1.
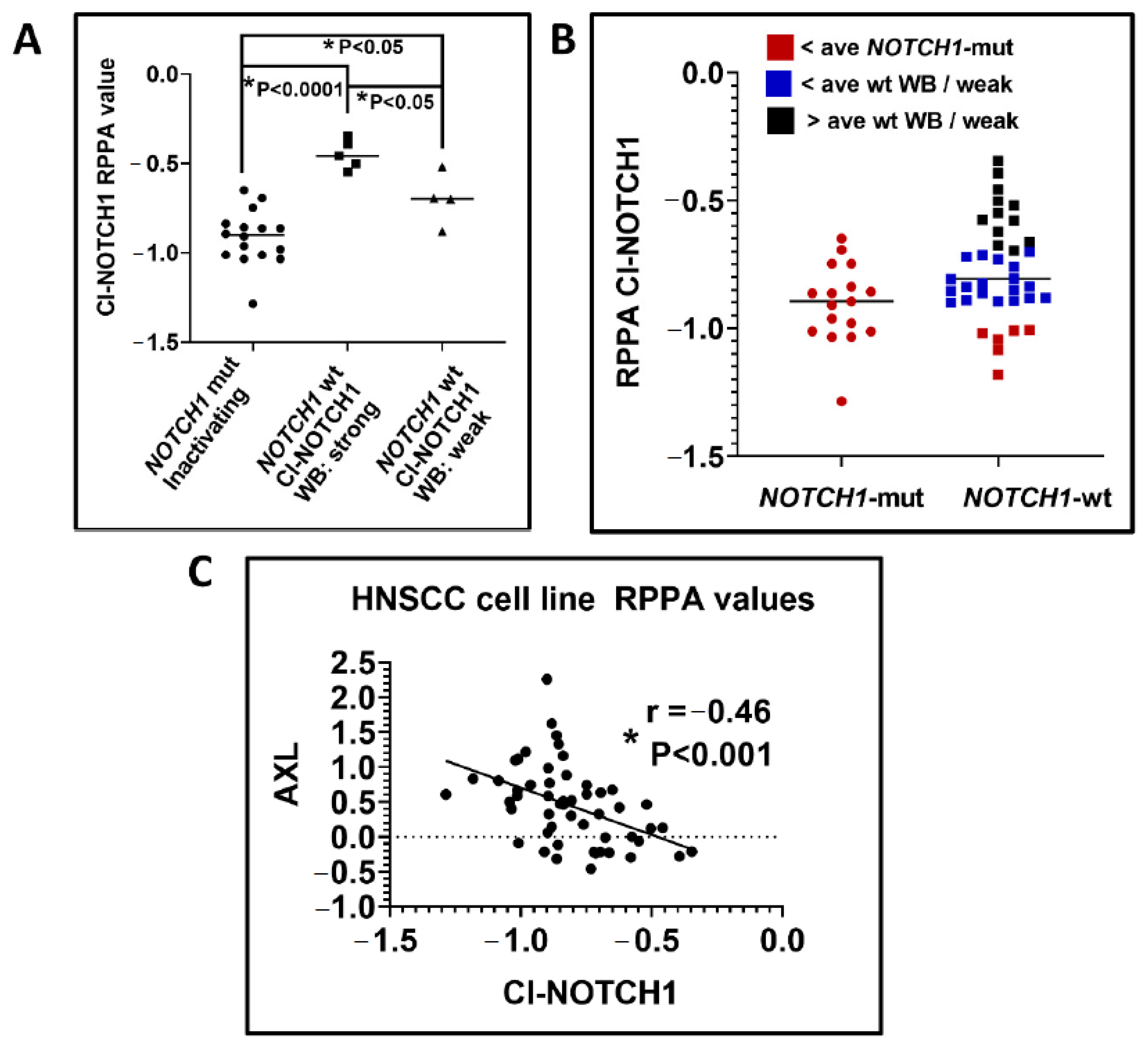
4.4. Evidence That NOTCH1 Is Oncogenic in a Subset of HNSCCs
4.5. NOTCH1 Pathway Activation in Clinical HNSCC Specimens
4.6. Epithelial to Mesenchymal Transition and NOTCH1 Signaling
5. NOTCH1 Mutations in Other SCCs
6. The Prognostic Role of NOTCH Signaling and NOTCH1 Mutations in HNSCC
7. Targeting NOTCH1 Mutant HNSCC
7.1. PI3K Inhibitors
7.2. Chemotherapy
7.3. Immunotherapy
8. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Torabi, S.J.; Yarbrough, W.G.; Mehra, S.; Osborn, H.A.; Judson, B. Association of Human Papillomavirus Status at Head and Neck Carcinoma Subsites with Overall Survival. JAMA Otolaryngol. Head Neck Surg. 2018, 144, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; Mahipal, A.; Mehra, R.; Tahara, M.; Berger, R.; Eder, J.P.; Burtness, B.; Lee, S.H.; et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients with Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results from the Phase Ib KEYNOTE-012 Expansion Cohort. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef] [PubMed]
- Bauml, J.; Seiwert, T.Y.; Pfister, D.G.; Worden, F.; Liu, S.V.; Gilbert, J.; Saba, N.F.; Weiss, J.; Wirth, L.; Sukari, A.; et al. Pembrolizumab for Platinum- and Cetuximab-Refractory Head and Neck Cancer: Results from a Single-Arm, Phase II Study. J. Clin. Oncol. 2017, 35, 1542–1549. [Google Scholar] [CrossRef]
- Harrington, K.J.; Ferris, R.L.; Blumenschein, G., Jr.; Colevas, A.D.; Fayette, J.; Licitra, L.; Kasper, S.; Even, C.; Vokes, E.E.; Worden, F.; et al. Nivolumab versus standard, single-agent therapy of investigator’s choice in recurrent or metastatic squamous cell carcinoma of the head and neck (CheckMate 141): Health-related quality-of-life results from a randomised, phase 3 trial. Lancet Oncol. 2017, 18, 1104–1115. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulieres, D.; Tahara, M.; de Castro, G., Jr.; Psyrri, A.; Baste, N.; Neupane, P.; Bratland, A.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Mehra, R.; Seiwert, T.Y.; Gupta, S.; Weiss, J.; Gluck, I.; Eder, J.P.; Burtness, B.; Tahara, M.; Keam, B.; Kang, H.; et al. Efficacy and safety of pembrolizumab in recurrent/metastatic head and neck squamous cell carcinoma: Pooled analyses after long-term follow-up in KEYNOTE-012. Br. J. Cancer 2018, 119, 153–159. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; De Castro, G., Jr.; Psyrri, A.; Baste Rotllan, N.; Neupane, P.C.; Bratland, Å.; et al. KEYNOTE-048: Phase III study of first-line pembrolizumab (P) for recurrent/metastatic head and neck squamous cell carcinoma (R/M HNSCC). Ann. Oncol. 2018, 29, viii729. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Sambandam, V.; Frederick, M.J.; Shen, L.; Tong, P.; Rao, X.; Peng, S.; Singh, R.; Mazumdar, T.; Huang, C.; Li, Q.; et al. PDK1 Mediates NOTCH1-Mutated Head and Neck Squamous Carcinoma Vulnerability to Therapeutic PI3K/mTOR Inhibition. Clin. Cancer Res. 2019, 25, 3329–3340. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Xia, R.; Li, J.; Long, Z.; Ren, H.; Chen, W.; Mao, L. Common and complex Notch1 mutations in Chinese oral squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Gaykalova, D.A.; Ochs, M.F.; Mambo, E.; Arnaoutakis, D.; Liu, Y.; Loyo, M.; Agrawal, N.; Howard, J.; Li, R.; et al. Activation of the NOTCH pathway in head and neck cancer. Cancer Res. 2014, 74, 1091–1104. [Google Scholar] [CrossRef] [Green Version]
- Artavanis-Tsakonas, S. The molecular biology of the Notch locus and the fine tuning of differentiation in Drosophila. Trends Genet. 1988, 4, 95–100. [Google Scholar] [CrossRef]
- Leong, K.G.; Karsan, A. Recent insights into the role of Notch signaling in tumorigenesis. Blood 2006, 107, 2223–2233. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.D.; Yau, C.; Bowlby, R.; Liu, Y.; Brennan, K.; Fan, H.; Taylor, A.M.; Wang, C.; Walter, V.; Akbani, R.; et al. Genomic, Pathway Network, and Immunologic Features Distinguishing Squamous Carcinomas. Cell Rep. 2018, 23, 194–212.e196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, X.; Wood, L.D.; Lindman, M.; Jones, S.; Buckhaults, P.; Polyak, K.; Sukumar, S.; Carter, H.; Kim, D.; Karchin, R.; et al. Somatic mutations in the Notch, NF-KB, PIK3CA, and Hedgehog pathways in human breast cancers. Genes Chromosomes Cancer 2012, 51, 480–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. Tan-1, the Human Homolog of the Drosophila Notch Gene, Is Broken by Chromosomal Translocations in T-Lymphoblastic Neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.T.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordonez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Bea, S.; Gonzalez-Diaz, M.; et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Lobry, C.; Oh, P.; Mansour, M.R.; Look, A.T.; Aifantis, I. Notch signaling: Switching an oncogene to a tumor suppressor. Blood 2014, 123, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Chillakuri, C.R.; Sheppard, D.; Lea, S.M.; Handford, P.A. Notch receptor-ligand binding and activation: Insights from molecular studies. Semin. Cell Dev. Biol. 2012, 23, 421–428. [Google Scholar] [CrossRef] [Green Version]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israel, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Karlstrom, H.; Lundkvist, J.; Mizutani, T.; Otaka, A.; Vestling, M.; Bernstein, A.; Donoviel, D.; Lendahl, U.; Honjo, T. Notch receptor cleavage depends on but is not directly executed by presenilins. Proc. Natl. Acad. Sci. USA 2002, 99, 4014–4019. [Google Scholar] [CrossRef] [Green Version]
- Doyen, C.M.; Depierre, D.; Yatim, A.; Heurteau, A.; Lelievre, J.D.; Levy, Y.; Cuvier, O.; Benkirane, M. NOTCH assembles a transcriptional repressive complex containing NuRD and PRC1 to repress genes involved in cell proliferation and differentiation. bioRxiv 2019, 513549. [Google Scholar] [CrossRef] [Green Version]
- Fattizzo, B.; Rosa, J.; Giannotta, J.A.; Baldini, L.; Fracchiolla, N.S. The Physiopathology of T- Cell Acute Lymphoblastic Leukemia: Focus on Molecular Aspects. Front. Oncol. 2020, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.F.; Chiang, S.L.; Lin, C.Y.; Chang, J.G.; Chung, C.M.; Ko, A.M.; Lin, Y.Z.; Lee, C.H.; Lee, K.W.; Chen, M.K.; et al. Somatic Mutations and Genetic Variants of NOTCH1 in Head and Neck Squamous Cell Carcinoma Occurrence and Development. Sci. Rep. 2016, 6, 24014. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, K.; Ota, Y.; Kajiwara, K.; Hirayama, N.; Kimura, M. Frequent mutations in NOTCH1 ligand-binding regions in Japanese oral squamous cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 452, 980–985. [Google Scholar] [CrossRef]
- IPT of the International; Maitra, A.; Biswas, N.K.; Amin, K.; Kowtal, P.; Kumar, S.; Das, S.; Sarin, R.; Majumder, P.P.; Bagchi, I.; et al. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat. Commun. 2013, 4, 2873. [Google Scholar] [CrossRef]
- Perdomo, S.; Anantharaman, D.; Foll, M.; Abedi-Ardekani, B.; Durand, G.; Reis Rosa, L.A.; Holmila, R.; Le Calvez-Kelm, F.; Tajara, E.H.; Wunsch-Filho, V.; et al. Genomic analysis of head and neck cancer cases from two high incidence regions. PLoS ONE 2018, 13, e0191701. [Google Scholar] [CrossRef] [Green Version]
- Seiwert, T.Y.; Zuo, Z.; Keck, M.K.; Khattri, A.; Pedamallu, C.S.; Stricker, T.; Brown, C.; Pugh, T.J.; Stojanov, P.; Cho, J.; et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin. Cancer Res. 2015, 21, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Morris, L.G.T.; Chandramohan, R.; West, L.; Zehir, A.; Chakravarty, D.; Pfister, D.G.; Wong, R.J.; Lee, N.Y.; Sherman, E.J.; Baxi, S.S.; et al. The Molecular Landscape of Recurrent and Metastatic Head and Neck Cancers: Insights from a Precision Oncology Sequencing Platform. JAMA Oncol. 2017, 3, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Izumchenko, E.; Sun, K.; Jones, S.; Brait, M.; Agrawal, N.; Koch, W.; McCord, C.L.; Riley, D.R.; Angiuoli, S.V.; Velculescu, V.E.; et al. Notch1 mutations are drivers of oral tumorigenesis. Cancer Prev. Res. 2015, 8, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, R.; Bao, R.; Faber, P.W.; Bindokas, V.P.; Bechill, J.; Lingen, M.W.; Spiotto, M.T. Notch1 Activation or Loss Promotes HPV-Induced Oral Tumorigenesis. Cancer Res. 2015, 75, 3958–3969. [Google Scholar] [CrossRef] [Green Version]
- Aster, J.C.; Xu, L.; Karnell, F.G.; Patriub, V.; Pui, J.C.; Pear, W.S. Essential roles for ankyrin repeat and transactivation domains in induction of T-cell leukemia by notch1. Mol. Cell. Biol. 2000, 20, 7505–7515. [Google Scholar] [CrossRef] [Green Version]
- Ferrando, A.A. The role of NOTCH1 signaling in T-ALL. Hematology 2009, 353–361. [Google Scholar] [CrossRef] [Green Version]
- Luca, V.C.; Kim, B.C.; Ge, C.; Kakuda, S.; Wu, D.; Roein-Peikar, M.; Haltiwanger, R.S.; Zhu, C.; Ha, T.; Garcia, K.C. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 2017, 355, 1320–1324. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S. Making sense out of missense mutations: Mechanistic dissection of Notch receptors through structure-function studies in Drosophila. Dev. Growth Differ. 2020, 62, 15–34. [Google Scholar] [CrossRef] [Green Version]
- De Celis, J.F.; Bray, S.J. The Abruptex domain of Notch regulates negative interactions between Notch, its ligands and Fringe. Development 2000, 127, 1291–1302. [Google Scholar]
- Zheng, Y.; Wang, Z.; Ding, X.; Zhang, W.; Li, G.; Liu, L.; Wu, H.; Gu, W.; Wu, Y.; Song, X. A novel Notch1 missense mutation (C1133Y) in the Abruptex domain exhibits enhanced proliferation and invasion in oral squamous cell carcinoma. Cancer Cell Int. 2018, 18, 6. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalu, N.N.; Mazumdar, T.; Peng, S.; Tong, P.; Shen, L.; Wang, J.; Banerjee, U.; Myers, J.N.; Pickering, C.R.; Brunell, D.; et al. Comprehensive pharmacogenomic profiling of human papillomavirus-positive and -negative squamous cell carcinoma identifies sensitivity to aurora kinase inhibition in KMT2D mutants. Cancer Lett. 2018, 431, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Sano, D.; Pickering, C.R.; Jasser, S.A.; Henderson, Y.C.; Clayman, G.L.; Sturgis, E.M.; Ow, T.J.; Lotan, R.; Carey, T.E.; et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin. Cancer Res. 2011, 17, 7248–7264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Yang, X.; Si, H.; Saleh, A.D.; Xiao, W.; Coupar, J.; Gollin, S.M.; Ferris, R.L.; Issaeva, N.; Yarbrough, W.G.; et al. Genomic and Transcriptomic Characterization Links Cell Lines with Aggressive Head and Neck Cancers. Cell Rep. 2018, 25, 1332–1345.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Moorthy, S.; Li, Q.; Saade, R.; Wang, J.; Rao, X.; Tanaka, N.; Zhang, J.; Tang, L.; Pickering, C.R.; et al. Abstract 5506: NOTCH1 activation in head and neck squamous cell carcinoma leads to growth inhibition, changes in gene expression associated with early differentiation, and acquisition of stem cell-like properties. Cancer Res. 2018, 78, 5506. [Google Scholar] [CrossRef]
- Huang, C.; Moorthy, S.; Li, Q.; Saade, R.; Wang, J.; Rao, X.; Tanaka, N.; Zhang, J.; Tang, L.; Pickering, C.R.; et al. Abstract. NOTCH1 activation inhibits head and neck squamous cell carcinoma growth. In Proceedings of the Cprit Innovations in Cancer Prevention and Research Conference, Austin, TX, USA, 13–14 November 2017. [Google Scholar]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef]
- Tian, J.; Liu, X.; Liu, X.; Jing, P.; Sa, N.; Wang, H.; Xu, W. Notch1 serves as a prognostic factor and regulates metastasis via regulating EGFR expression in hypopharyngeal squamous cell carcinoma. Onco Targets Ther. 2018, 11, 7395–7405. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.L.; Zhang, L.; Huang, C.F.; Ma, S.R.; Bu, L.L.; Liu, J.F.; Yu, G.T.; Liu, B.; Gutkind, J.S.; Kulkarni, A.B.; et al. NOTCH1 inhibition enhances the efficacy of conventional chemotherapeutic agents by targeting head neck cancer stem cell. Sci. Rep. 2016, 6, 24704. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.T.; Chen, M.K.; Yeh, K.T.; Chang, C.S.; Chang, T.H.; Lin, C.Y.; Wu, Y.C.; Su, B.W.; Lee, K.D.; Chang, P.J. Association of high levels of Jagged-1 and Notch-1 expression with poor prognosis in head and neck cancer. Ann. Surg. Oncol. 2010, 17, 2976–2983. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Stein, A.P.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Pearson, H.E.; Bahrar, H.; Fowler, T.L.; Bednarz, B.P.; et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2015, 21, 2601–2612. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Yang, X.F.; Huang, K.Q.; Ren, L.; Zhao, S.; Gou, W.F.; Shen, D.F.; Sun, H.Z.; Takano, Y.; Zheng, H.C. The upregulated alpha-catulin expression was involved in head-neck squamous cell carcinogenesis by promoting proliferation, migration, invasion, and epithelial to mesenchymal transition. Tumour Biol. 2016, 37, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Chen, Y.; Masood, R.; Sinha, U.K.; Kobielak, A. alpha-Catulin marks the invasion front of squamous cell carcinoma and is important for tumor cell metastasis. Mol. Cancer Res. MCR 2012, 10, 892–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rettig, E.M.; Chung, C.H.; Bishop, J.A.; Howard, J.D.; Sharma, R.; Li, R.J.; Douville, C.; Karchin, R.; Izumchenko, E.; Sidransky, D.; et al. Cleaved NOTCH1 Expression Pattern in Head and Neck Squamous Cell Carcinoma Is Associated with NOTCH1 Mutation, HPV Status, and High-Risk Features. Cancer Prev. Res. 2015, 8, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, P.; Nair, S.; Kaur, E.; Aich, J.; Dani, P.; Sethunath, V.; Gardi, N.; Chandrani, P.; Godbole, M.; Sonawane, K.; et al. Notch pathway activation is essential for maintenance of stem-like cells in early tongue cancer. Oncotarget 2016, 7, 50437–50449. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Do, S.I.; Lee, H.J.; Kang, H.J.; Koo, B.S.; Lim, Y.C. Notch1 signaling contributes to stemness in head and neck squamous cell carcinoma. Lab. Investig. 2016, 96, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Hong, H.S.; Liu, Z.X.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. TNFalpha enhances cancer stem cell-like phenotype via Notch-Hes1 activation in oral squamous cell carcinoma cells. Biochem. Biophys. Res. Commun. 2012, 424, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, S.; Steele, R.; Sowadski, M.; Crawford, S.E.; Varvares, M.; Ray, R.B. Identification of molecular signature of head and neck cancer stem-like cells. Sci. Rep. 2015, 5, 7819. [Google Scholar] [CrossRef] [Green Version]
- Kato, H.; Izumi, K.; Saito, T.; Ohnuki, H.; Terada, M.; Kawano, Y.; Nozawa-Inoue, K.; Saito, C.; Maeda, T. Distinct expression patterns and roles of aldehyde dehydrogenases in normal oral mucosa keratinocytes: Differential inhibitory effects of a pharmacological inhibitor and RNAi-mediated knockdown on cellular phenotype and epithelial morphology. Histochem. Cell Biol. 2013, 139, 847–862. [Google Scholar] [CrossRef]
- Baumeister, P.; Hollmann, A.; Kitz, J.; Afthonidou, A.; Simon, F.; Shakhtour, J.; Mack, B.; Kranz, G.; Libl, D.; Leu, M.; et al. High Expression of EpCAM and Sox2 is a Positive Prognosticator of Clinical Outcome for Head and Neck Carcinoma. Sci. Rep. 2018, 8, 14582. [Google Scholar] [CrossRef] [Green Version]
- Bayo, P.; Jou, A.; Stenzinger, A.; Shao, C.; Gross, M.; Jensen, A.; Grabe, N.; Mende, C.H.; Rados, P.V.; Debus, J.; et al. Loss of SOX2 expression induces cell motility via vimentin up-regulation and is an unfavorable risk factor for survival of head and neck squamous cell carcinoma. Mol. Oncol. 2015, 9, 1704–1719. [Google Scholar] [CrossRef]
- Panelos, J.; Massi, D. Emerging role of Notch signaling in epidermal differentiation and skin cancer. Cancer Biol. Ther. 2009, 8, 1986–1993. [Google Scholar] [CrossRef] [PubMed]
- Lefort, K.; Dotto, G.P. Notch signaling in the integrated control of keratinocyte growth/differentiation and tumor suppression. Semin. Cancer Biol. 2004, 14, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcolea, M.P.; Jones, P.H. Cell competition: Winning out by losing notch. Cell Cycle 2015, 14, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.D.; Lynch, C.N.S.; Craythorne, E.; Liakath-Ali, K.; Mallipeddi, R.; Barker, J.N.; Watt, F.M. Spatial constraints govern competition of mutant clones in human epidermis. Nat. Commun. 2017, 8, 1119. [Google Scholar] [CrossRef] [Green Version]
- Fukusumi, T.; Califano, J.A. The NOTCH Pathway in Head and Neck Squamous Cell Carcinoma. J. Dent. Res. 2018, 97, 645–653. [Google Scholar] [CrossRef]
- Del Alamo, D.; Rouault, H.; Schweisguth, F. Mechanism and significance of cis-inhibition in Notch signalling. Curr. Biol. 2011, 21, R40–R47. [Google Scholar] [CrossRef] [Green Version]
- Rettig, E.M.; Bishop, J.A.; Agrawal, N.; Chung, C.H.; Sharma, R.; Zamuner, F.; Li, R.J.; Koch, W.M.; Califano, J.A.; Guo, T.; et al. HEY1 is expressed independent of NOTCH1 and is associated with poor prognosis in head and neck squamous cell carcinoma. Oral Oncol. 2018, 82, 168–175. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Y.; Kong, D.; Sarkar, F.H. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 2010, 11, 745–751. [Google Scholar] [CrossRef]
- Jing, P.; Zhou, S.; Xu, P.; Cui, P.; Liu, X.; Liu, X.; Liu, X.; Wang, H.; Xu, W. PDK1 promotes metastasis by inducing epithelial-mesenchymal transition in hypopharyngeal carcinoma via the Notch1 signaling pathway. Exp. Cell Res. 2020, 386, 111746. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, G.; Zhou, L.; Li, P.; Yun, M.; Shi, Q.; Wang, T.; Wu, X. Notch signalling induces epithelialmesenchymal transition to promote metastasis in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 42, 2276–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Y.; Huang, W.X.; Zhou, X.; Chen, J.; Li, Z. Numb inhibits epithelial-mesenchymal transition via RBP-Jkappa-dependent Notch1/PTEN/FAK signaling pathway in tongue cancer. BMC Cancer 2019, 19, 391. [Google Scholar] [CrossRef] [Green Version]
- Inamura, N.; Kimura, T.; Wang, L.; Yanagi, H.; Tsuda, M.; Tanino, M.; Nishihara, H.; Fukuda, S.; Tanaka, S. Notch1 regulates invasion and metastasis of head and neck squamous cell carcinoma by inducing EMT through c-Myc. Auris Nasus Larynx 2017, 44, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Que, J.; et al. Interplay between Notch1 and Notch3 promotes EMT and tumor initiation in squamous cell carcinoma. Nat. Commun. 2017, 8, 1758. [Google Scholar] [CrossRef] [Green Version]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Jiao, Y.; Bettegowda, C.; Hutfless, S.M.; Wang, Y.; David, S.; Cheng, Y.; Twaddell, W.S.; Latt, N.L.; Shin, E.J.; et al. Comparative genomic analysis of esophageal adenocarcinoma and squamous cell carcinoma. Cancer Discov. 2012, 2, 899–905. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic Landscape of Esophageal Squamous Cell Carcinoma in a Japanese Population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- Vettore, A.L.; Ramnarayanan, K.; Poore, G.; Lim, K.; Ong, C.K.; Huang, K.K.; Leong, H.S.; Chong, F.T.; Lim, T.K.; Lim, W.K.; et al. Mutational landscapes of tongue carcinoma reveal recurrent mutations in genes of therapeutic and prognostic relevance. Genome Med. 2015, 7, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinhofer, I.; Stenzinger, A.; Eder, T.; Konschak, R.; Niehr, F.; Endris, V.; Distel, L.; Hautmann, M.G.; Mandic, R.; Stromberger, C.; et al. Targeted next-generation sequencing identifies molecular subgroups in squamous cell carcinoma of the head and neck with distinct outcome after concurrent chemoradiation. Ann. Oncol. 2016, 27, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Kaka, A.S.; Nowacki, N.B.; Kumar, B.; Zhao, S.; Old, M.O.; Agrawal, A.; Ozer, E.; Carrau, R.L.; Schuller, D.E.; Kumar, P.; et al. Notch1 Overexpression Correlates to Improved Survival in Cancer of the Oropharynx. Otolaryngol. Head Neck Surg. 2017, 156, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, Z.; Zhang, M.; Gross, N.; Gong, L.; Zhang, S.; Lei, D.; Zeng, Q.; Luo, X.; Li, G.; et al. High Notch1 expression affects chemosensitivity of head and neck squamous cell carcinoma to paclitaxel and cisplatin treatment. Biomed. Pharm. 2019, 118, 109306. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.N.; Burch, M.B.; Cooper, T.S.; Della Manna, D.L.; Wei, S.; Ojesina, A.I.; Rosenthal, E.L.; Yang, E.S. Notch Signaling Activation Is Associated with Patient Mortality and Increased FGF1-Mediated Invasion in Squamous Cell Carcinoma of the Oral Cavity. Mol. Cancer Res. 2016, 14, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, M.; Doescher, J.; Jira, D.; Meier, M.A.; Piontek, G.; Reiter, R.; Schlegel, J.; Pickhard, A. HES1 mRNA expression is associated with survival in sinonasal squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2016, 122, 491–499. [Google Scholar] [CrossRef]
- Wirth, M.; Jira, D.; Ott, A.; Piontek, G.; Pickhard, A. High NOTCH1 mRNA Expression Is Associated with Better Survival in HNSCC. Int. J. Mol. Sci. 2018, 19, 830. [Google Scholar] [CrossRef] [Green Version]
- Krikelis, D.; Kotoula, V.; Bobos, M.; Fountzilas, E.; Markou, K.; Karasmanis, I.; Angouridakis, N.; Vlachtsis, K.; Kalogeras, K.T.; Nikolaou, A.; et al. Protein and mRNA expression of notch pathway components in operable tumors of patients with laryngeal cancer. Anticancer Res. 2014, 34, 6495–6503. [Google Scholar]
- Moore, G.; Annett, S.; McClements, L.; Robson, T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells 2020, 9, 1503. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Furman, R.R.; Sharman, J.P.; Coutre, S.E.; Cheson, B.D.; Pagel, J.M.; Hillmen, P.; Barrientos, J.C.; Zelenetz, A.D.; Kipps, T.J.; Flinn, I.; et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2014, 370, 997–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, A.K.; Kahl, B.S.; de Vos, S.; Wagner-Johnston, N.D.; Schuster, S.J.; Jurczak, W.J.; Flinn, I.W.; Flowers, C.R.; Martin, P.; Viardot, A.; et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N. Engl. J. Med. 2014, 370, 1008–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquard, F.E.; Jucker, M. PI3K/AKT/mTOR signaling as a molecular target in head and neck cancer. Biochem. Pharmacol. 2020, 172, 113729. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Byers, L.A.; Ng, P.K.; Mills, G.B.; Peng, S.; Diao, L.; Fan, Y.H.; Stemke-Hale, K.; Heymach, J.V.; Myers, J.N.; et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol. Cancer Ther. 2014, 13, 2738–2750. [Google Scholar] [CrossRef] [Green Version]
- Keysar, S.B.; Astling, D.P.; Anderson, R.T.; Vogler, B.W.; Bowles, D.W.; Morton, J.J.; Paylor, J.J.; Glogowska, M.J.; Le, P.N.; Eagles-Soukup, J.R.; et al. A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins. Mol. Oncol. 2013, 7, 776–790. [Google Scholar] [CrossRef]
- Muellner, M.K.; Uras, I.Z.; Gapp, B.V.; Kerzendorfer, C.; Smida, M.; Lechtermann, H.; Craig-Mueller, N.; Colinge, J.; Duernberger, G.; Nijman, S.M. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat. Chem. Biol. 2011, 7, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Johnson, F.M.; Opyrchal, M.; Dowlati, A.; Hierro, C.; Forester, M.; Blagden, S.P.; Wicki, A.; Schmitz, D.; Adjei, A.A. Abstract B109: Oral dual PI3K/mTOR inhibitor bimiralisib demonstrates tolerability and a signal of activity in head and neck squamous cell cancer with NOTCH1 loss-of-function mutation. Mol. Cancer Ther. 2019, 18, B109. [Google Scholar] [CrossRef]
- Nishimura, K.; Tsuchiya, Y.; Okamoto, H.; Ijichi, K.; Gosho, M.; Fukayama, M.; Yoshikawa, K.; Ueda, H.; Bradford, C.R.; Carey, T.E.; et al. Identification of chemoresistant factors by protein expression analysis with iTRAQ for head and neck carcinoma. Br. J. Cancer 2014, 111, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.P.; Sun, Y.L.; Fu, L.; Gu, F.; Zhang, L.; Hao, X.S. Correlation of Notch1 expression and activation to cisplatin-sensitivity of head and neck squamous cell carcinoma. Ai Zheng 2009, 28, 100–103. [Google Scholar]
- Gu, F.; Ma, Y.; Zhang, Z.; Zhao, J.; Kobayashi, H.; Zhang, L.; Fu, L. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncol. Rep. 2010, 23, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Hanna, G.J.; Lizotte, P.; Cavanaugh, M.; Kuo, F.C.; Shivdasani, P.; Frieden, A.; Chau, N.G.; Schoenfeld, J.D.; Lorch, J.H.; Uppaluri, R.; et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI Insight 2018, 3, e98811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Hong, X.; Song, Z.; Xu, Y.; Li, C.; Wang, G.; Zhang, Y.; Zhao, X.; Zhao, Z.; Zhao, J.; et al. Identification of deleterious NOTCH mutation as novel predictor to efficacious immunotherapy in NSCLC. Clin. Cancer Res. 2020, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]


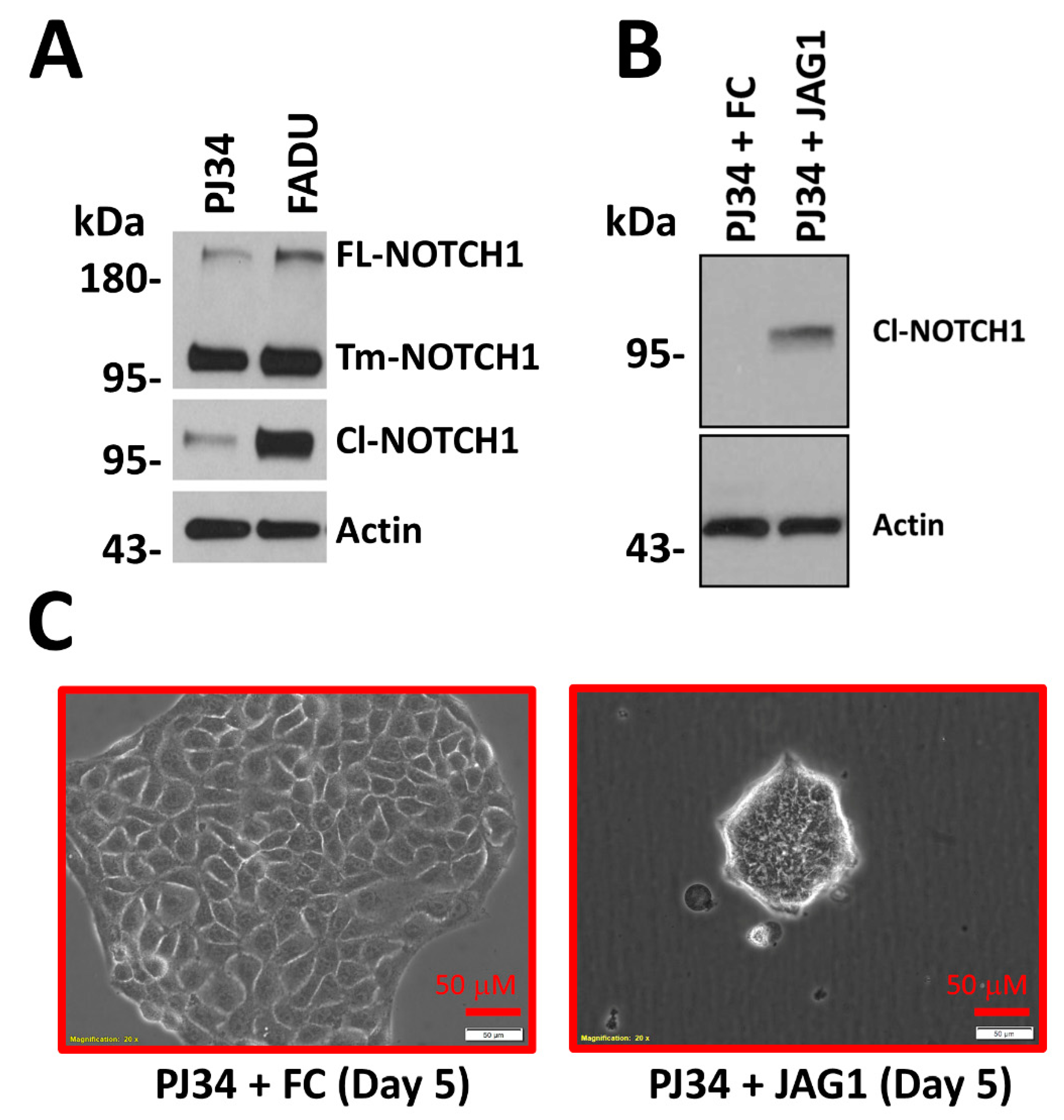
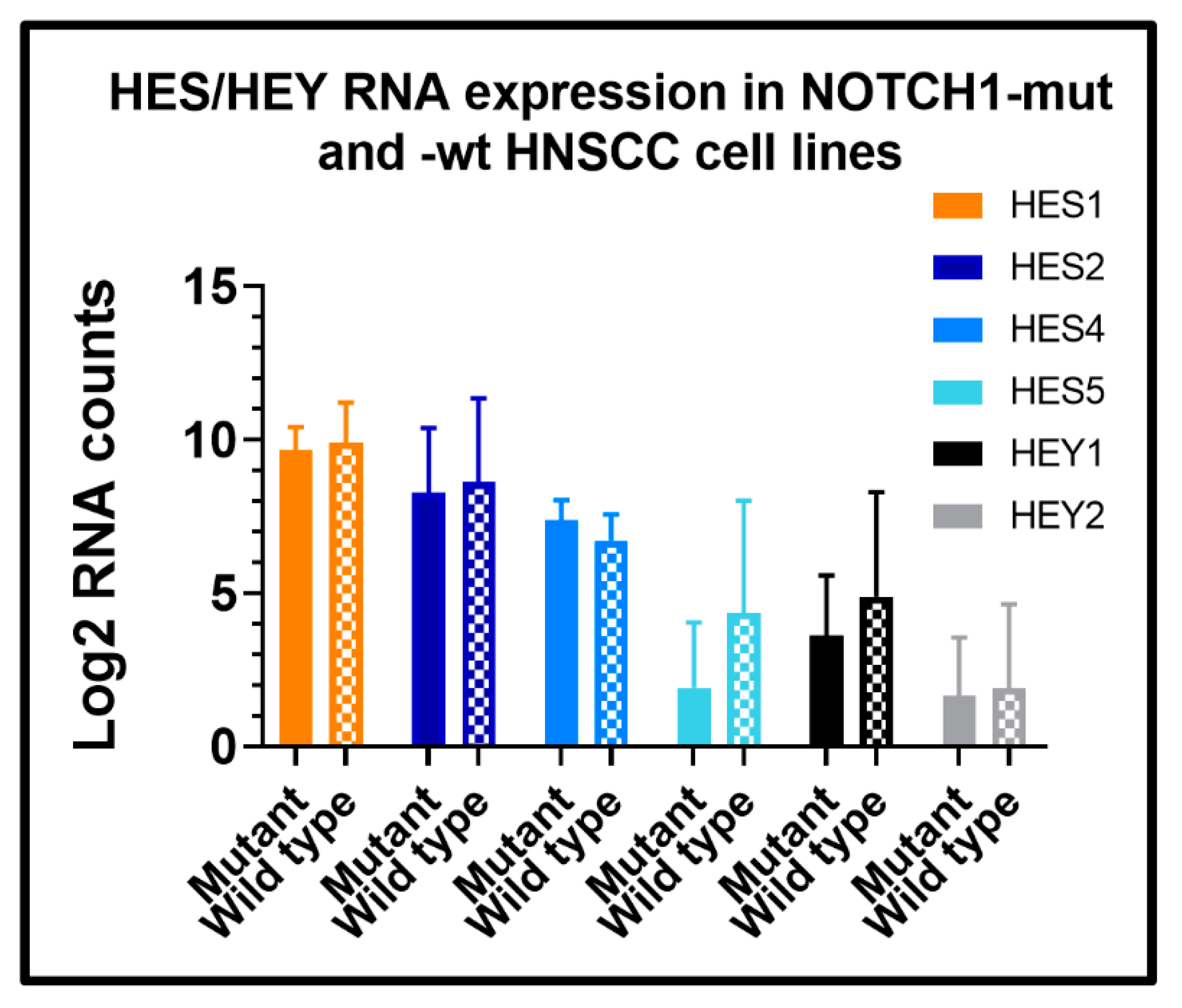

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV-Negative HNSCC | OPSCC/HPV-Positive HNSCC | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Year | Study | Subsite | Cohort Characteristics | #Patients | %Truncating | %Missense | %Total | #Patients | %Truncating | %Missense | %Total | Clinical Associations with NOTCH1 Mutations |
| 2011 | [10] | Mixed | USA | 90 | 10% | 9% | 19% | 30 | 1% | 13% | 14% | N.D. |
| 2011 | [11] | Mixed | USA | 60 | 7% | 7% | 14% | 14 | 0% | 7% | 7% | N.D. |
| 2013 | [38] | Gingivo-buccal | South Asian (mixed HPV neg and pos) | 50 | 8% | 8% | 16% | N.D. | N.D. | N.D. | N.D. | N.D. |
| 2014 | [16] | OCSCC | Chinese | 51 | 10% | 37% | 43% | N.D. | N.D. | N.D. | N.D. | NOTCH1 mutations significantly associated with worse OS and DFS, and LN mets |
| 2014 | [37] | OCSCC | Japanese OCSCC; limited exons sequenced | 84 | 0% | 10% | 10% | N.D. | N.D. | N.D. | N.D. | NOTCH1 mutations associated with better DFS, but no difference in OS |
| 2015 | [40] | Mixed | USA | 69 | 4% | 12% | 16% | 51% | 4% | 4% | 8% | N.D. |
| 2015 | [42] | Mixed | Chinese | 50 | 6% | 48% | 54% | N.D. | N.D. | N.D. | N.D. | N.D. |
| 2015 | [13] | Mixed | USA | 246 | 9% | 12% | 20% | 20 | 0% | 9% | 9% | No association found |
| 2016 | [36] | Mixed | Chinese | 128 | 5% | 12% | 22% | N.D. | N.D. | N.D. | N.D. | NOTCH1 mutations significantly associated with reduced OS and increased recurrence |
| 2017 | [41] | Mixed | Recurrent HNSCC; USA | 30 | 10% | 10% | 20% | 21 | 0% | 10% | 10% | N.D. for HNSCC |
| 2018 | [39] | Mixed | European and Latino | 165 | 7% | 16% | 23% | 15 | 0 | 2 | 13% | N.D. |
| 2018 | [43] | Mixed | USA | 445 | 8% | 10% | 18% | 51 | 0% | 6% | 6% | N.D. |
| HNSCC Cell Line | HPV Status | Protein Change [52,54] | Zygosity | Variant Type | Variant Class | Domain | PROVEAN Score | SIFTA Score | Consensus | Domain | Predicted Function | Experimental Evidence [12,55,56] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MDA1686 | Neg | H2018FS | Heterozygous | Frame shift del | Truncating | ANK4 | N/A | N/A | N/A | ANK4 | Inactivating | WB: NOTCH1 null |
| UMSCC25 | Neg | V489FS | Homozygous | Frame shift del | Truncating | Ca binding EGF_12 | N/A | N/A | N/A | Ca binding EGF_12 | Inactivating | |
| HN4 | Neg | C344FS | Homozygous | Frame shift ins | Truncating | Ca binding EGF_9 | N/A | N/A | N/A | Ca binding EGF_9 | Inactivating | WB: NOTCH1 null |
| PCI15A | Neg | Q1957 * | Heterozygous | Nonsense mutation | Truncating | ANK2 | N/A | N/A | N/A | ANK2 | Inactivating | WB: very weak NOTCH1 |
| PCI15B | Neg | Q1957 * | Homozygous | Nonsense mutation | Truncating | ANK2 | N/A | N/A | N/A | ANK2 | Inactivating | WB: NOTCH1 null |
| MDA1686 | Neg | E2008 * | Heterozygous | Nonsense mutation | Truncating | ANK4 | N/A | N/A | N/A | ANK4 | Inactivating | WB: NOTCH1 null |
| UMSCC85 | Neg | E694 * | Homozygous | Nonsense mutation | Truncating | Ca binding EGF_18 | N/A | N/A | N/A | Ca binding EGF_18 | Inactivating | |
| UMSCC47 | HPV16 | G192 * | Homozygous | Nonsense mutation | Truncating | Ca binding EGF_5 | N/A | N/A | N/A | Ca binding EGF_5 | Inactivating | WB: NOTCH1 null |
| UMSCC22A | Neg | E1679 * | Homozygous | Nonsense mutation | Truncating | HD | N/A | N/A | N/A | HD | Inactivating | WB: NOTCH1 null |
| UMSCC22B | Neg | E1679 * | Homozygous | Nonsense mutation | Truncating | HD | N/A | N/A | N/A | HD | Inactivating | |
| UMSCC25 | Neg | E488A | Homozygous | Missense mutation | SNV | Ca binding EGF_12 | Deleterious/ −4.65 | Damaging/0.004 | Impactful | Ca binding EGF_12 | Inactivating | |
| HN30 | Neg | C478F | Homozygous | Missense mutation | SNV | Ca binding EGF_12 | Deleterious/ −9.71 | Damaging/0.0 | Impactful | Ca binding EGF_12 | Inactivating | |
| HN31 | Neg | C478F | Homozygous | Missense mutation | SNV | Ca binding EGF_12 | Deleterious/ −9.71 | Damaging/0.0 | Impactful | Ca binding EGF_12 | Inactivating | ICN1 absent +/− ligand exp |
| SCC45 | HPV33 | G72R | Homozygous | Missense mutation | SNV | EGF_2 | Deleterious/ −6.81 | Damaging/0.0 | Impactful | EGF_2 | Inactivating | |
| MSK922 | Neg | C1536Y | Homozygous | Missense mutation | SNV | LNR3 | Deleterious/ −10.65 | Damaging/0.0 | Impactful | LNR3 | Weakly activating | |
| PCI13 | Neg | G1753W | Homozygous | Missense mutation | SNV | TM | Deleterious/ −5.83 | Damaging/0.01 | Impactful | TM | Inactivating | WB: very weak NOTCH1 |
| TR146 | Neg | A1524V | Heterozygous | Missense mutation | SNV | LNR3 | Neutral/ −0.55 | Tolerated/ 0.319 | Not Impactful | LNR3 | Impact unlikely | |
| JHU029 | Neg | L418del | Homozygous | In frame del | INDEL | Ca binding EGF_11 | N/A | N/A | N/A | Ca binding EGF_11 | Inactivating | |
| 1483 | Neg | F357del | Homozygous | In frame del | INDEL | Ca binding EGF_9 | N/A | N/A | N/A | Ca binding EGF_9 | Inactivating |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shah, P.A.; Huang, C.; Li, Q.; Kazi, S.A.; Byers, L.A.; Wang, J.; Johnson, F.M.; Frederick, M.J. NOTCH1 Signaling in Head and Neck Squamous Cell Carcinoma. Cells 2020, 9, 2677. https://doi.org/10.3390/cells9122677
Shah PA, Huang C, Li Q, Kazi SA, Byers LA, Wang J, Johnson FM, Frederick MJ. NOTCH1 Signaling in Head and Neck Squamous Cell Carcinoma. Cells. 2020; 9(12):2677. https://doi.org/10.3390/cells9122677
Chicago/Turabian StyleShah, Pooja A., Chenfei Huang, Qiuli Li, Sawad A. Kazi, Lauren A. Byers, Jing Wang, Faye M. Johnson, and Mitchell J. Frederick. 2020. "NOTCH1 Signaling in Head and Neck Squamous Cell Carcinoma" Cells 9, no. 12: 2677. https://doi.org/10.3390/cells9122677