1. Introduction
Macrophages are one of the important immune cells that play an essential role in the host defence system; however, excessive inflammation caused by over reactive macrophages and leads to inflammatory diseases or autoimmune disorders [
1]. Unlike traditional proinflammatory cytokines, interleukin (IL)-1β and IL-18 production are regulated by post-translational modification catalysed by protease caspase-1. The caspase-1 activity is regulated by a group of cytoplasmic multiprotein complexes called inflammasomes [
2]. The NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome is the best-characterized inflammasome composed of NLRP3, apoptosis-associated speck-like protein (ASC) and caspase-1. The NLRP3 inflammasome recognizes and responses to diverse pathogen associated molecular patterns from infectious microorganisms or sterile danger signals generated by host cells [
2]. IL-1β and IL-18 produced by inflammasomes not only activate innate immunity but also influence T cell adaptive immunity [
3].
The NLRP3 inflammasome activation is dependent on the priming and activation signals. The priming signal is triggered mainly by the Toll-like receptors and induces the gene and protein expressions of NLRP3 and IL-1β precursor (proIL-1β) through reactive oxygen species (ROS)-, mitogen-activated protein kinase (MAPK)- and nuclear factor kappa B (NF-κB)-dependent pathways [
4]. Only priming signal, however, is insufficient to activate the NLRP3 inflammasome unless the cells are activated by the activation signal, such as gout-causing monosodium urate (MSU) crystals, diabetes-causing saturated fatty acids, Alzheimer’s disease-causing amyloid-β and bacterial toxin nigericin [
5]. The activation signals trigger the downstream signalling events, such as lysosomal destabilization and mitochondrial dysfunction that induce NLRP3 inflammasome assembly and eventually caspase-1 activation [
2]. As the NLRP3 inflammasome recognizes and responses to the board range medicinally relevant stimuli, dysregulated NLRP3 inflammasome activation participates many human inflammatory diseases, including gout, type II diabetes, atherosclerosis and neurodegenerative disorders [
5]. Therefore, targeting the NLRP3 inflammasome has therapeutic significance in the management of dysregulated NLRP3 inflammasome complications.
The current therapeutic strategies for NLRP3-associated complications are based on the non-steroidal anti-inflammatory drugs, colchicines or glucocorticoids, which are not satisfactory and cause significant side effects [
6]. Therefore, the development of a novel NLRP3 inflammasome inhibitor is a therapeutic option to counteract dysregulated NLRP3-associated diseases. Conjugated polyenes are an interesting class of widely occurring natural products. Previous reports on conjugated polyenes indicate that they have various biological properties, such as their antibacterial, antifungal, and antitumour activities [
7]. Presently, some conjugated polyenes, including rapamycin and fumagillin, are commercially available. Clark et al. previously reported the isolation and structure elucidation of several polyenylfurans and polyenylpyrroles from the soil microbe
Gymnoascus reessii [
8]. We have also shown that the related polyenes auxarconjugatins A and B, which contain a chloropyrrole group, possess cytotoxic properties [
9], whereas furan-containing gymnoconjugatins possess no significant activity [
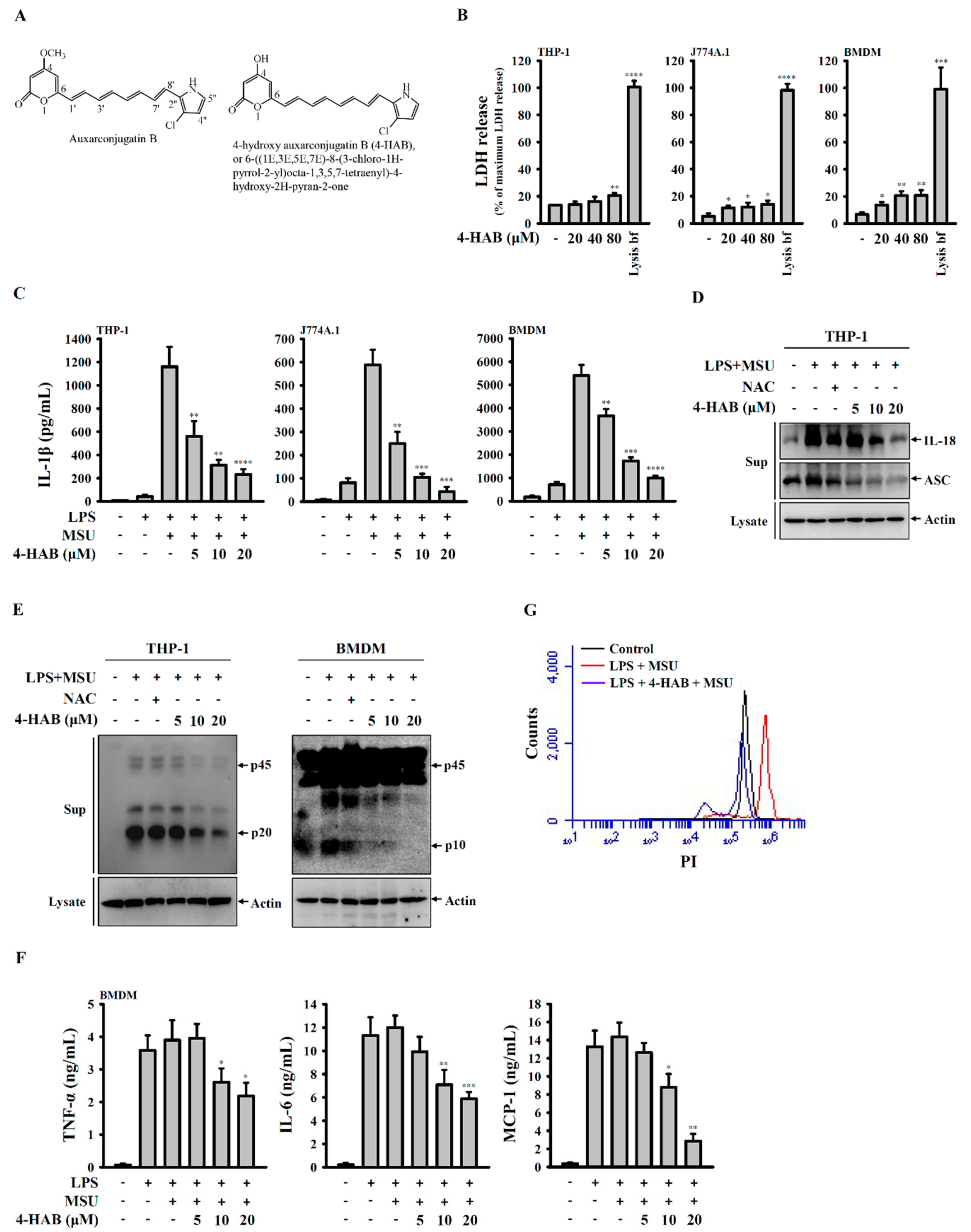
8]. The auxarconjugatin B derivative 4-hydroxy auxarconjugatin B, or 6-((1E,3E,5E,7E)-8-(3-chloro-1H-pyrrol-2-yl)octa-1,3,5,7-tetraenyl)-4-hydroxy-2H-pyran-2-one (4-HAB,
Figure 1A), is a novel, low-molecular-weight polyenylpyrrole agent [
9]. Our previous data showed that 4-HAB exerts strong anti-inflammatory effects by inhibiting lipopolysaccharide (LPS)-induced inflammation in macrophages and dendritic cells [
10]. However, little is known about the effects of 4-HAB on the NLRP3 inflammasome and the underlying molecular mechanism of these effects. As part of our efforts is to identify novel NLRP3 inflammasome inhibitors [
11,
12,
13,
14,
15] and based on the known anti-inflammatory effects of 4-HAB, we hypothesized that 4-HAB can inhibit the NLRP3 inflammasome.
2. Materials and Methods
2.1. Reagents and Chemicals
Escherichia coli 0111:B4 lipopolysaccharide (LPS), N-acetyl-L-cysteine (NAC), acridin orange (AO), monodansylcadaverine (MDC), 3-Methyladenine (3-MA), 6-Chloro-2,3,4,9-tetrahydro-1H-Carbazole-1-carboxamide (EX-527), phorbol myristate acetate (PMA) and propidium iodide (PI) and uric acid were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rapamycin and puromycin were purchased from InvivoGen (San Diego, CA, USA). GeneJammer® transfection reagent was purchased from Agilent Technologies (Santa Clara, CA, USA). Antibodies against human IL-1β, ASC, IL-18, Actin and horseradish peroxidase-labeled secondary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against human caspase-1 were obtained from Cell Signaling Technology (Beverly, MA, USA). Antibodies against NLRP3 and mouse caspase-1 were purchased from Adipogen International (San Diego, CA, USA). Antibody against mouse IL-1β was purchased from R&D systems (Minneapolis, MN, USA). Antibody against LC3B was purchased from Novus Biologicals (Littleton, CO, USA). Antibodies against Gr1 and CD45 were purchased from eBioscience (San Diego, CA, USA). JC-1 and Antibodies against Cathepsin B and Sirt1 were purchased from Millipore (Bedford, MA, USA). MitoTracker Deep Red, MitoTracker Green, MitoSOX and Pierce™ LAL Chromogenic Endotoxin Quantitation Kit were purchased from Thermo Scientific (Rockford, IL, USA). Magic Red Cathepsin B detection kit was purchased from ImmunoChemistry Technologies (Bloomington, MN, USA). The CytoScan LDH Cytotoxicity Assay kit was purchased from G-Bioscience (St. Louis, MO, USA).
2.2. Cell Lines and Culture
The murine J774A.1 macrophages and human THP-1 monocytes were purchased from the American Type Culture Collection (Rockville, MD, USA) and cultured in RPMI 1640 medium contained with 10% heat-inactivated fetal bovine serum at 37 °C in a 5% CO2 incubator. To induce monocytes differentiation into macrophages, THP-1 monocytes were treated with 50 nM PMA for 48 h. Non-adherent cells were removed by aspiration, and adherent macrophages were washed with RPMI 1640 medium before stabilizing for additional 48 h in cell culture medium. Bone marrow-derived macrophages (BMDM) were prepared from marrow collected from C57BL/6 mice femur and tibia incubated for seven days in culture medium containing M-CSF (Peprotech, London, UK). For generating gene knockout cells, cells were transfected with CRISPR/Cas9 knockout plasmids targeting LC3 (for J774A.1 macrophages: sc-426563 and sc-417828-HDR, Santa Cruz Biotechnology; for THP-1 monocytes: sc-4178288 and sc-417828-HDR, Santa Cruz Biotechnology) or Sirt1 (for J774A.1 macrophages: sc-430046 and sc-430046-HDR, Santa Cruz Biotechnology). The CRISPR/Cas9 knockout plasmids transfected cells were selected by puromycin and the expression levels of LC3 and Sirt1 were checked by Western blot.
2.3. General Procedure for the Synthesis of 4-hydroxy Auxarconjugatin B, or 6-((1’E,3’E,5’E,7’E)-8’-(3-chloro-1H-pyrrol-2-yl)octa-1,3,5,7-tetraenyl)-4-hydroxy-2H-pyran-2-one (4-HAB)
Compound 4-HAB was synthesized according to the experimental procedures which we have reported previously [
9]. Briefly, tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3) (2.7 mg, 3.0 μmol) and triphenylarsine (AsPh
3) (4.6 mg, 15 μmol) were added to dry THF (1 mL) followed by 2-(2-bromovinyl)-3-chloro-1-(methylsulfonyl)-1H-pyrrole (56 mg, 0.195 mmol) and 1.8 M aqueous KOH (0.167 mL, 0.300 mL). 4-Hydroxy-6-((1E,3E,5E)-6-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)hexa-1,3,5-trienyl)-2H-pyran-2-one (0.15 mmol) dissolved in THF (0.5 mL) was then added dropwise to the reaction mixture over 5 min with stirring. The reaction mixture was stirred for 20 min at room temperature and quenched with saturated NH
4Cl. The reaction mixture was extracted with ethyl acetate (EtOAc), the organic layer dried over MgSO
4 and concentrated under reduced pressure. The residue obtained was dissolved in THF (1 mL), and 1 M tetra-n-butylammonium fluoride (TBAF) in THF (0.300 mL, 0.300 mmol) was added. The mixture was stirred at room temperature for 30 min, and thereafter, EtOAc was added and the mixture was washed thrice with water followed by saturated NaCl solution. The organic layer was dried over MgSO
4, concentrated under reduced pressure, and purified by column chromatography. Compound 4-HAB was characterized by 1H and 13C NMR and mass spectral data, which were identical to the previously reported values [
9].
2.4. MSU Crystals Preparation
MSU crystals were prepared according to the method described previously [
16]. In brief, 250 mg uric acid was dissolved in 45 mL of boiling water containing 0.3 mL of 5 M NaOH. After the solution was passed through a 0.2-µM filter, 1 mL of 5 M NaCl was added to the solution, and keeps the solution at 26 °C for 7 days. The resulting crystals collected and washed with ethanol and acetone. The crystals evaporated and sterilized by heating at 180 °C for 2 h and stored in a sterile environment until use. All MSU crystals were determined to be endotoxin free (<0.01 EU/10 mg) by the LAL assay.
2.5. Cytokines and Proteins Measurements
The levels of cytokines in the culture medium and peritoneal lavage fluids were measured by Enzyme-Linked ImmunoSorbent Assay (ELISA) as described in our previous study [
10]. For detection of protein expression in the culture medium, the medium was concentrated by the following protocol before performing Western blot [
11]. Briefly, prepared a mixture containing 400 µL culture medium, 400 µL methanol and 166 µL chloroform. The mixture was vortexed and 400 µL double-distilled water was added. The mixture was thoroughly vortexed and incubated on ice for 10 min before centrifugation at 13,000 rpm at 4 °C for 10 min. The supernatant was removed and mixed the pellet with 500 µL methanol before centrifugation at 13,000 rpm at 4 °C for 10 min. The supernatant was removed and the pellet was dried at 55 °C and dissolved in Western blot loading buffer, followed by incubation in boiling water for 30 min. The samples were analysed by Western blot as described in our previous study [
10]. For detection of protein expression in the lysates, the cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed with 100 μL ice-cold lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM Na
3VO
4, 1 μg/mL leupeptin and 1 mM PMSF) on ice for 10 min. The samples were pelleted by centrifuging at 12,000×
g at 4 °C for 15 min, and the protein concentrations of supernatants were determined using Bio-Rad protein assay dye. 50 μg proteins form each sample was analysed by Western blot as described in our previous study [
10].
2.6. ASC Oligomerization and Speck Formation
ASC oligomerization was analyzed by Western blot-based assay. In brief, insoluble ASC complexes were isolated from cell lysates by centrifugation and subsequent crosslinking with disuccinimidylsuberate as previously described [
17]. The samples were resolved on 15% sodium dodecyl sulfate polyacrylamide gel electrophoresis and processed for Western blot. For ASC speck formation, the cells were fixed in pre-warmed 4% paraformaldehyde (PFA) in PBS for 30 min, and then permeabilized by 0.2% Triton X-100. After blocking with 1% bovine serum albumin, ASC speck formation was measured by incubating the cells with ASC antibody and fluorescent-conjugated secondary antibody. The cells were visualized by an Olympus BX-41 microscope and the images were analyzed by Image J.
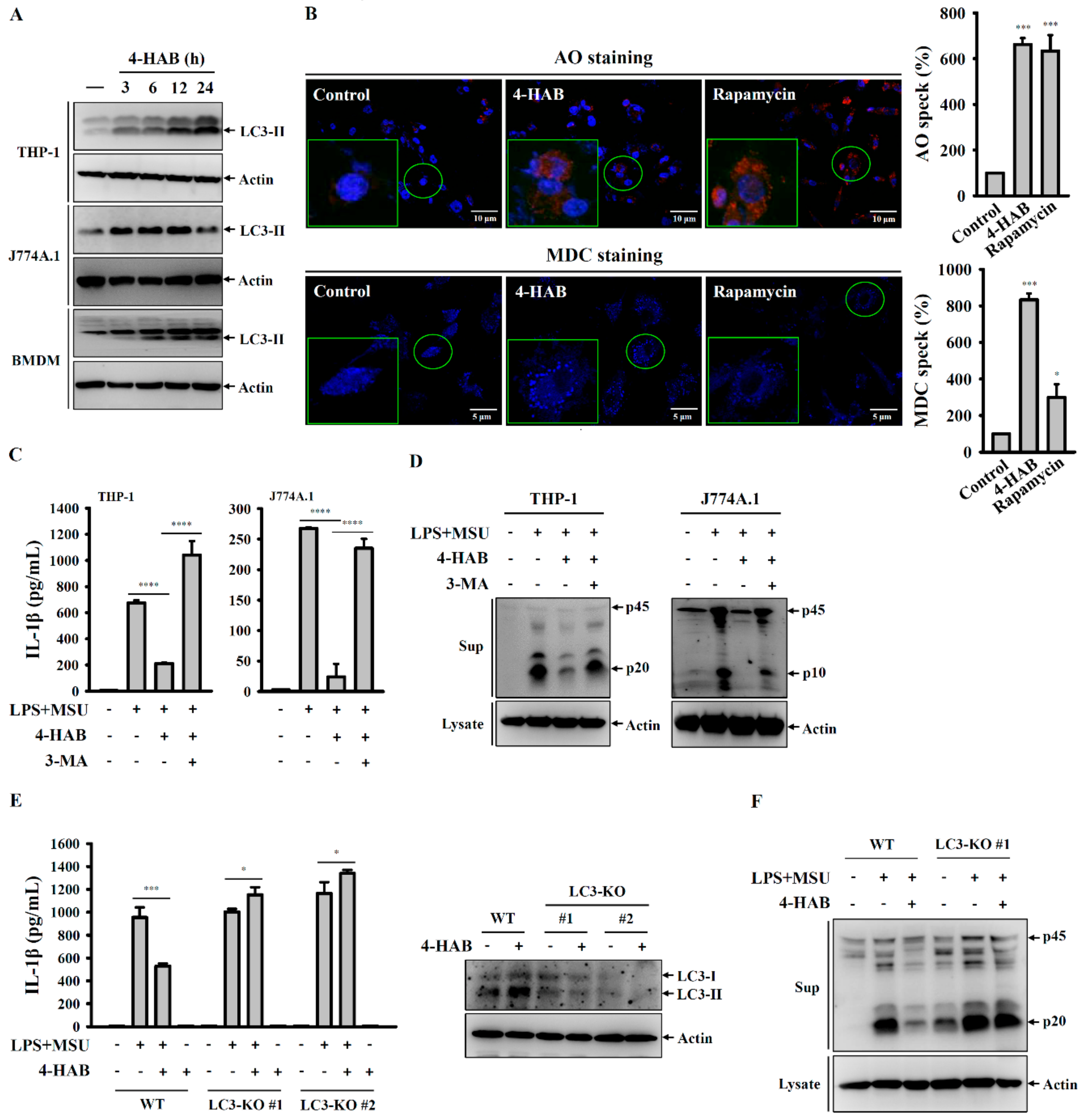
2.7. Autophagy Measurement by AO and MDC Staining
THP-1 macrophages were incubated for 24 h with 20 µM 4-HAB or 100 nM rapamycin. The cells were stained with 1 µg/mL AO or 50 µM MDC at 37 °C for 10 min followed by washing with PBS twice, and then fixed in pre-warmed 4% PFA in PBS for 30 min. In the AO stained cells, the nucleus was stained by 4′,6-diamidino-2-phenylindole (DAPI). The cells were visualized by Olympus FV 1000-IX81 confocal microscope.
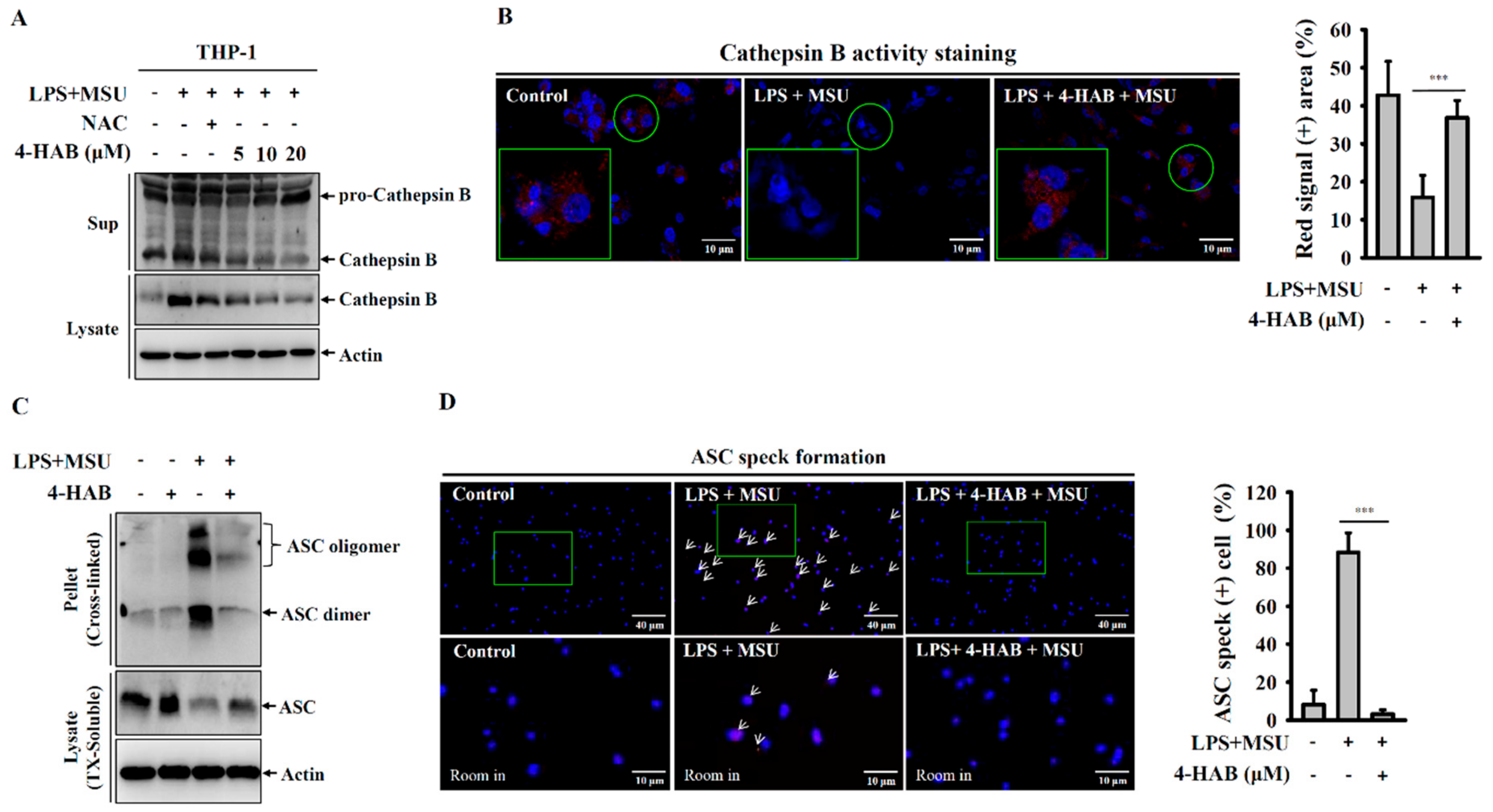
2.8. Cathepsin B Activity Assay
Cathepsin B activity were measured according to the product instruction manual of Magic Red Cathepsin B detection kit. In brief, THP-1 macrophages were incubated for 5 h with or without 1 μg/mL LPS followed by incubated with or without 20 µM 4-HAB for 30 min, then for 24 h with or without 100 μg/mL MSU crystals. The cells were stained for 10 min with Magic Red Cathepsin B detection kit at 37 °C, and then fixed for 30 min in pre-warmed 4% PFA in PBS. The cells were visualized by an Olympus FV 1000-IX81 confocal microscope.
2.9. Mitochondrial Function
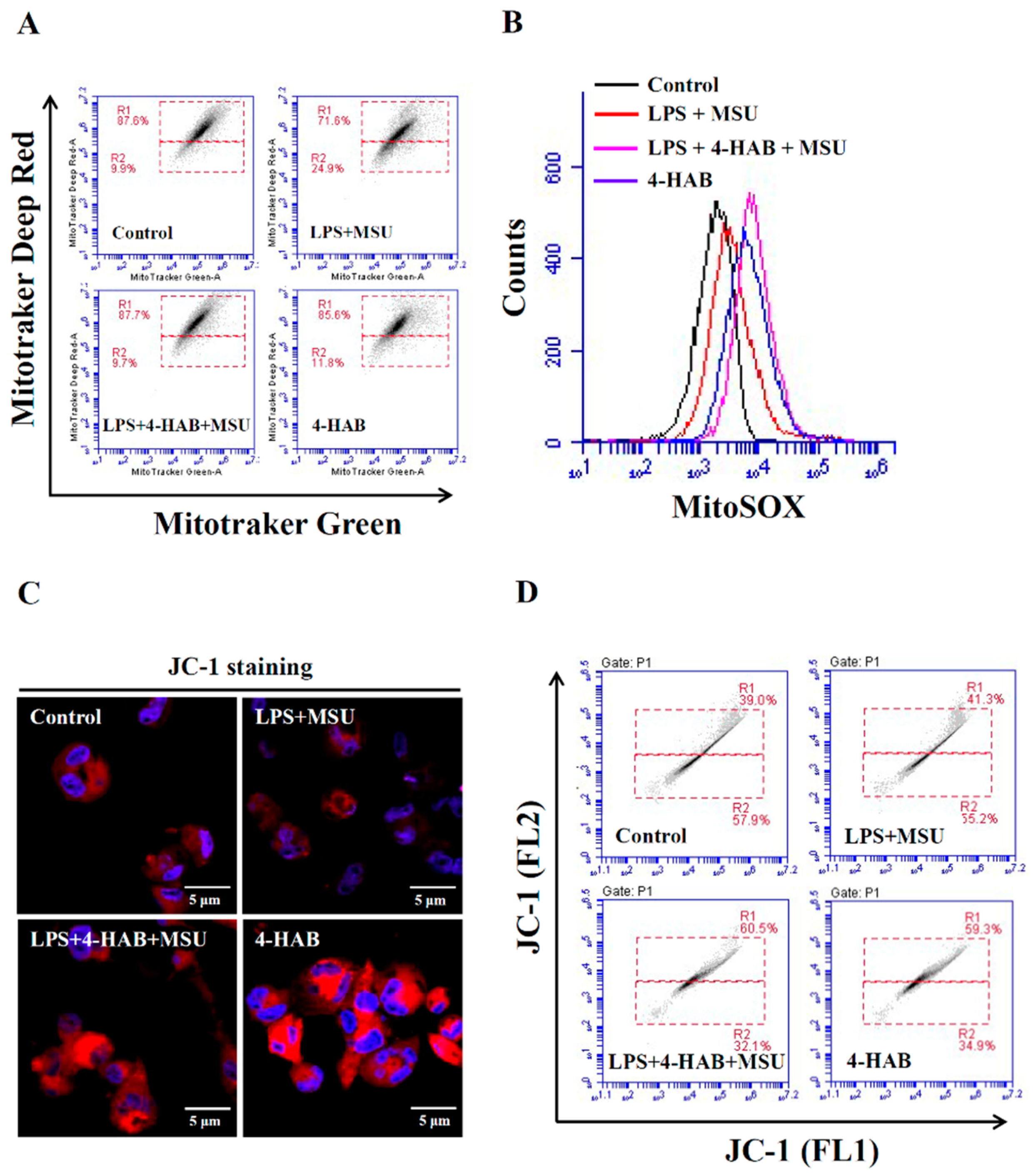
THP-1 macrophages were incubated with or without 1 μg/mL LPS for 5 h, followed by incubated with or without 20 µM 4-HAB for 30 min, then with or without 100 μg/mL MSU crystals for 24 h. To measure the inner transmembrane potential and mitochondrial mass, cells were stained with 25 nM MitoTracker Deep Red and 25 nM MitoTracker Green for 15 min. To measure the mitochondrial ROS, cells were stained with 5 μM MitoSOX for 15 min. The degree of mitochondrial membrane polarization in THP-1 macrophages was measured by staining with 2 μM JC-1. The signals of MitoTracker Deep Red, MitoTracker Green and MitoSOX were detected by flow cytometry, and signals of JC-1 were detected by an Olympus FV 1000-IX81 confocal microscope.
2.10. Cell Membrane Integrity Assay
THP-1 macrophages were incubated for 5 h with or without 1 μg/mL LPS followed by incubated with or without 20 µM 4-HAB for 30 min, then for 24 h with or without 100 μg/mL MSU crystals. To measure the cell membrane integrity, cells were fixed by 70% ethanol and stained with 2 μg/mL PI for 15 min. The signals of PI were detected by flow cytometry.
2.11. LDH Release Assay
Cells were incubated with 4-HAB, 10% H2O (spontaneous LDH release) or lysis buffer (maximum LDH release) for 24 h. To determine LDH release, culture medium was evaluated for the presence of the LDH using the CytoScan LDH Cytotoxicity Assay kit according to the manufacturer’s instructions. Briefly, prepared a mixture containing 50 µL culture medium and 50 µL LDH substrate. The mixture was incubated in dark for 30 min before 50 µL stop buffer was added. The LDH release was assessed by measuring the optical density (OD) at 490/680 nm using a microplate absorbance reader. The cytotoxicity % was calculated as 100× (sample OD—spontaneous OD)/(maximum OD—spontaneous OD).
2.12. In Vivo Mice Model of MSU Crystals-Induced Peritonitis
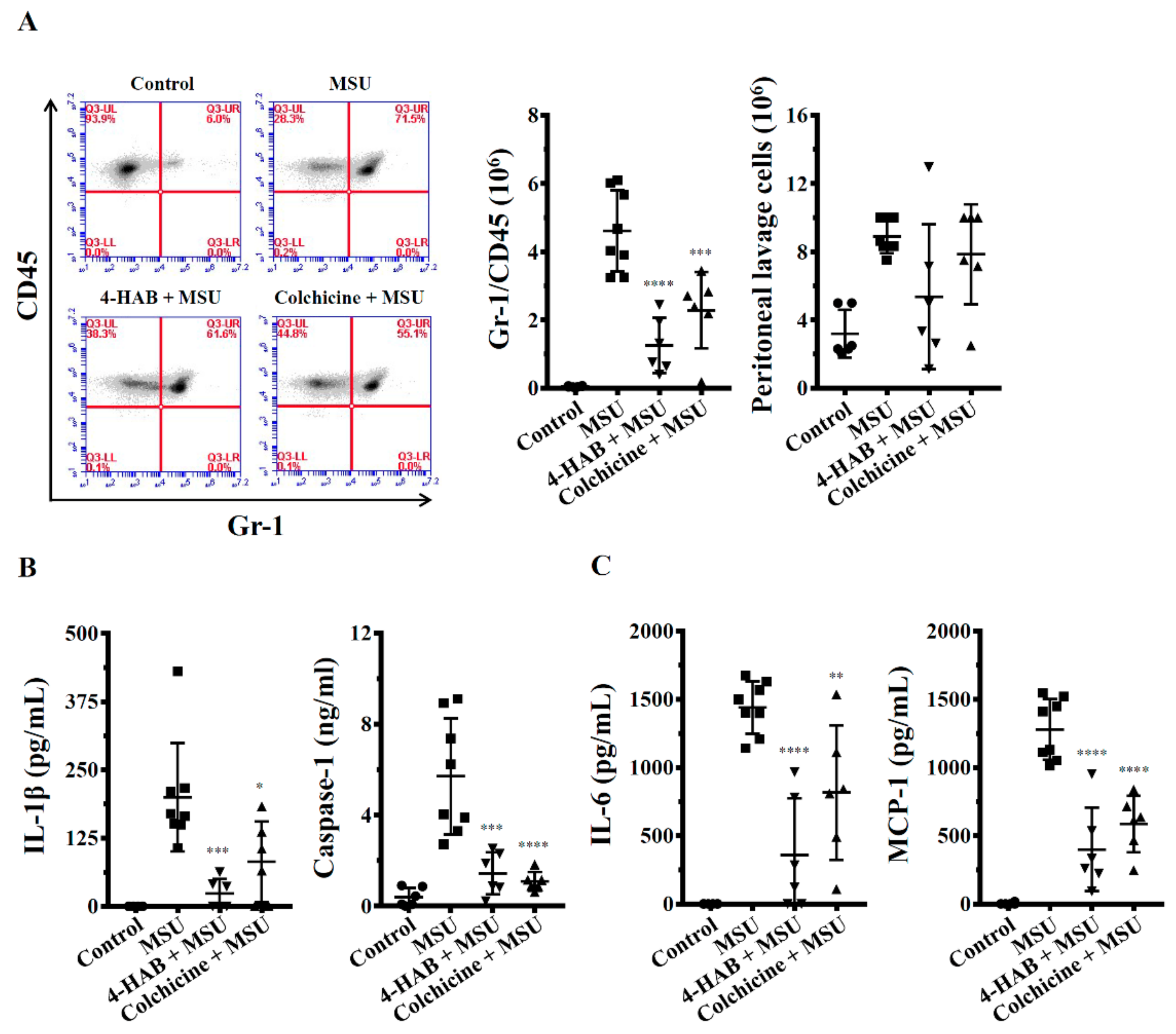
Male C57BL/6JNal mice aged eight weeks old were purchased from The National Laboratory Animal Center (Taipei, Taiwan). The mice housed in a room controlled for temperature (23 ± 3 °C) and relative humidity (40%–60%). Mice were acclimated in the animal facility for at least a week before the experiments. Animal experiments were performed with the approval of the Institutional Animal Care and Use Committee of the National Ilan University (approval number: No. 102-40), according to the NIH Guide for the Care and Use of Laboratory Animals. The mice were randomized into four groups: Group I: control, i.p. injection of 0.5% DMSO in sterile PBS (200 μL) at 0, 24 and 48 h; i.p. injection of sterile PBS (0.5 mL) at 1 and 49 h, n = 6. Group II: MSU crystals treatment, i.p. injection of 0.5% DMSO in sterile PBS (200 μL) at 0, 24 and 48 h; i.p. injection of sterile MSU crystals (3 mg in 0.5 mL PBS) at 1 and 49 h, n = 8. Group III: 4-HAB+MSU crystals treatment, i.p. injection of 4-HAB (20 mg/kg body weight) at 0, 24 and 48 h; i.p. injection of sterile MSU crystals (3 mg in 0.5 mL PBS) at 1 and 49 h, n = 6. Group IV: Colchicine+MSU crystals treatment, i.p. injection of colchicine (1 mg/kg body weight) at 48 h; i.p. injection of sterile MSU crystals (3 mg in 0.5 mL PBS) at 1 and 49 h, n = 6. Mice were euthanized at 53 h and the peritonea were lavaged with 3 mL ice-cold PBS. The absolute number of cells obtained counted in a hemocytometer before staining them with Gr1 and CD45 antibodies and analyzed by flow cytometry. All analysis was performed using BD CSampler Software (version 227). The expression levels of cytokines in peritoneal lavage fluids were measured by ELISA.
2.13. Statistical Analysis
GraphPad Prism 7.0 software was used for data analysis. Data are shown as mean ± SEM. Statistical significance was determined by t tests (two-tailed) for two groups or ANOVA (with Dunnett’s multiple comparisons test) for three or more groups. p values less than 0.05 were considered to be statistically significant.
4. Discussion
Gouty inflammation is characterized by intense pain caused by the deposition of MSU crystals into the articular joint and surrounding tissues [
25]. Uptake of MSU crystals by macrophages induces proinflammatory cytokine and chemokine productions, which leads to the further recruitment of immune cells into the joint and escalates the inflammation [
25,
26]. The current therapeutic approaches against gouty inflammation are given mainly to reduce hyperuricaemia and inflammatory status [
25]. For instance, allopurinol reduces the serum uric acid levels by inhibiting xanthine oxidase, but it is unable to reduce the inflammation during acute phase and may cause fever, skin rashes, allergic reactions, hepatitis and nephropathy [
5,
25]. Thus, non-steroidal anti-inflammatory (NSAID) drugs (e.g., indomethacin) and alkaloid drugs (e.g., colchicine) are frequently used as first-line therapies for the treatment of acute gouty inflammation [
25]. In addition, it has been demonstrated that the NLRP3 inflammasome plays important roles in MSU crystals-induced gouty inflammation [
27,
28]. Targeting NLRP3 inflammasome produced IL-1β and IL-18 by recombinant IL-1 receptor antagonist (anakinra), neutralizing IL-1β antibody (canakinumab), soluble decoy IL-1 receptor (rilonacept), IL-18-binding protein and anti-IL-18 receptor antibody are also useful in the treatment of gouty inflammation [
5]. However, the high cost of such treatments limits their wide clinical use. Novel therapeutic agents for gouty inflammation are therefore needed. As part of our study programme to identify novel therapeutic agents for the NLRP3-asscoiated complications, herein we investigated the effects of 4-HAB on NLRP3 inflammasome and the finding of this study clearly show that 4-HAB inhibited MSU crystals-induced NLRP3 inflammasome activation in intro and in vivo.
NLRP3 inflammasome activation results in not only IL-1β and IL-18 secretion but also pyroptosis, a caspase-1-dependent cell death characterized by the loss of membrane integrity [
18]. Notably, the inflammasome component ASC is released from the membrane damage macrophages upon NLRP3 inflammasome activation and amplify the inflammatory response by stimulating the surrounding cells [
29]. We found that 4-HAB reduced the PI uptake in MSU crystals-activated macrophages, indicating the reduced membrane integrity loss (
Figure 1F). In addition, 4-HAB also inhibited MSU crystals-induced ASC release (
Figure 1C), suggesting that 4-HAB can prevent the inflammatory response amplified by the release ASC [
29]. These data suggest that 4-HAB inhibits pyroptosis in MSU crystals-activated macrophages.
Mitochondrial damage induced by activation signal is one of the vital events for the NLPR3 inflammasome activation [
19,
20]. The uptake of MSU crystals by macrophages induces mitochondrial ROS generation and increases the oxidative status of mitochondrial DNA. The oxidized mitochondrial DNA release into cytosol because of the mitochondrial membrane integrity loss. The oxidized mitochondrial DNA binds to NLRP3, promotes NLRP3 inflammasome assembly and activates the NLRP3 inflammasome [
25]. Although a low level of ROS promotes NLRP3 inflammasome activation, a recent study demonstrated that a high level of ROS generated by Streptococcus pneumonia infection inhibited the NLRP3 inflammasome through the oxidation of the inflammasome components ASC and caspases [
30]. We found that although 4-HAB increased mitochondrial ROS production (
Figure 2B), it significantly reduced mitochondrial integrity loss induced by MSU crystals (
Figure 2A). These results indicated that 4-HAB inhibits the NLRP3 inflammasome through reduced oxidized mitochondrial DNA release and that a high level of mitochondrial ROS induced by 4-HAB may induce oxidation of NLRP3 inflammasome components, which contributes to 4-HAB-mediated NLRP3 inflammasome inhibition. Phagocytosis of MSU crystals by macrophages results in lysosomal rupture, and leads to the release of cathepsin B, a known activator of the NLRP3 inflammasome [
27,
28]. We found that 4-HAB protected against lysosomal rupture and reduced cathepsin B release (
Figure 3A,B). Lysosomal rupture also activates the TAK1-JNK pathway, which is necessary for complete activation of the NLRP3 inflammasome through the oligomerization of ASC [
31]. We demonstrated that 4-HAB reduced the oligomerization of ASC (
Figure 3C,D); however, the effect of 4-HAB on the activation of TAK1-JNK needs further investigation. Notably, our previous study showed that 4-HAB inhibited proIL-1β in LPS-activated macrophages; however, 4-HAB did not inhibit NLRP3 expression [
10]. These results indicated that 4-HAB inhibited MSU-mediated NLRP3 inflammasome activation was mainly through reducing the activation signal, but not through inhibiting the LPS-mediated priming signal.
Autophagy is a self-protective mechanism of the cells that maintains the cellular homeostasis by removing or recycling intracellular dysfunctional components [
32]. The enzyme converts pro-LC3B to LC3B-I by cleaving its C-terminus, after which LC3B-I can be conjugated to phosphatidylethanolamine by cysteine protease, thereby forming LC3B-II. This characteristic conversion of endogenous LC3-I to LC3-II is an autophagy biomarker that is used to monitor autophagy activity [
32]. Autophagy negatively regulates the NLRP3 inflammasome through removing damaged mitochondria to prevent the release of mitochondrial ROS and mitochondrial DNA into the cytoplasm, ultimately limiting NLRP3 inflammasome assembly [
32,
33]. NLRP3 inflammasome inhibition and autophagy induction is cooperative and of great relevance for the development of novel therapeutic strategies against NLRP3-asscoiated complications. We demonstrated that 4-HAB is a novel autophagy inducer that attenuates gouty inflammation by inhibiting the NLRP3 inflammasome. In addition, Sirt1, a class III histone deacetylase, is generally known as a vital regulator of inflammatory processes [
34]. Sirt1 inhibits the NLRP3 inflammasome activation in mesenchymal stem cells and vascular endothelial cells [
35,
36]. Sirt1 activator treatment significantly suppressed caspase-1 activation in mouse macrophages, linking Sirt1 to regulation of the NLRP3 inflammasome [
37]. Furthermore, Sirt1 inhibited TNF-α-induced IL-1β production through the autophagy-dependent degradation of NLRP3 in vascular adventitial fibroblasts [
23]. Consistent with these observations, our results showed that 4-HAB increased Sirt1 expression (
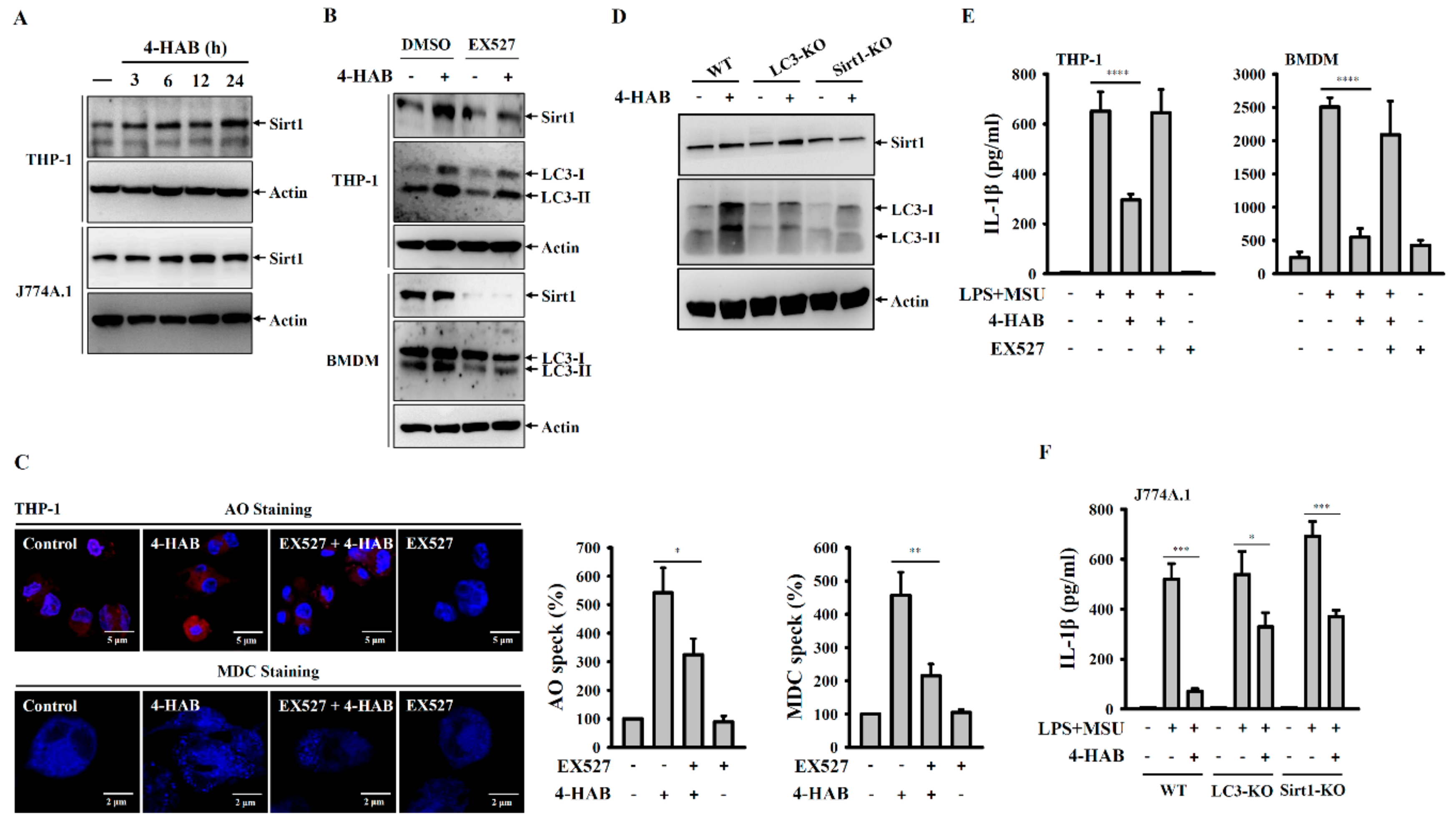
Figure 5A), and the Sirt1 inhibitor EX527 and Sirt1 knockout diminished autophagy induction and the inhibitory effects of 4-HAB on the NLRP3 inflammasome (
Figure 5E,F). Although we have gained promising data concerning the protective effect of Sirt1 on 4-HAB-mediated NLRP3 inflammasome inhibition, further studies are needed to address the molecular mechanism underlying 4-HAB-mediated regulation of Sirt1 expression.
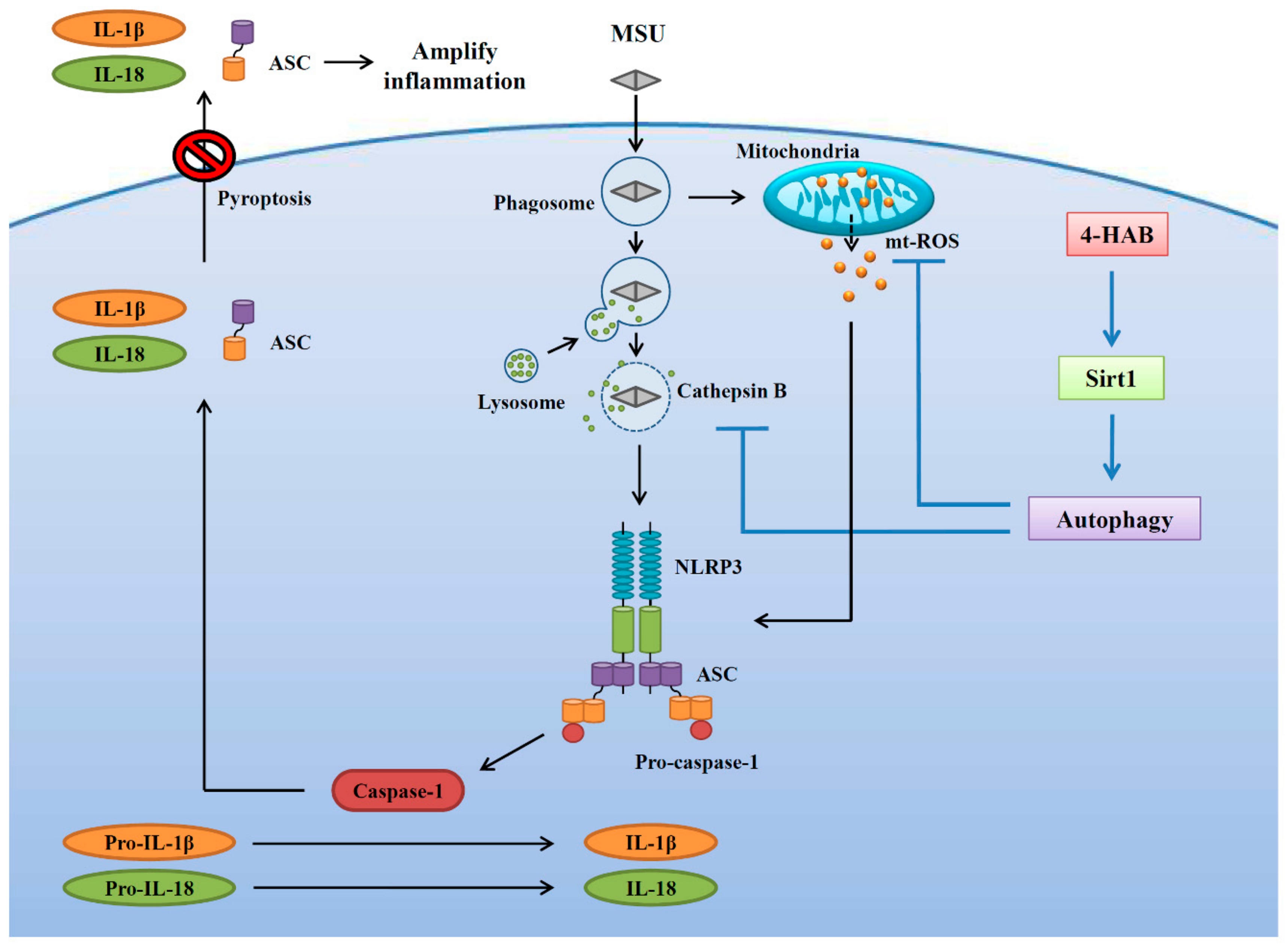
In conclusion, this study identified 4-HAB as an anti-NLRP3 inflammasome agent. Specifically, 4-HAB attenuated MSU crystals-mediated NLRP3 inflammasome activation by blocking mitochondrial damage, lysosomal rupture-mediated cathepsin B release and ASC oligomerization. Furthermore, 4-HAB inhibited the NLRP3 inflammasome by increasing the Sirt1/autophagy axis. The inhibitory effects of 4-HAB on the NLRP3 inflammasome were further confirmed in a mouse model of MSU crystals-induced peritonitis. A schematic representation of the present study is shown in
Figure 7. Thus, 4-HAB is a potential pharmacological agent for the management of gouty inflammation. However, the limitation of this study is the lack of toxicity test in a mice model. Although we did not observe any toxic response in our current mice study, a long term 4-HAB treatment or a high dosage 4-HAB treatment in mice should be conducted to verify the in vivo safety. Another imitation of this study is only using the mouse model of MSU crystals-induced peritonitis to test the in vivo activity of 4-HAB, it is better to confirm the anti-gouty inflammation activity of 4-HAB using a subcutaneous air pouch model in mouse.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






