Abstract
The threatening notoriety of pancreatic cancer mainly arises from its negligible early diagnosis, highly aggressive progression, failure of conventional therapeutic options and consequent very poor prognosis. The most important driver genes of pancreatic cancer are the oncogene KRAS and the tumor suppressors TP53, CDKN2A, and SMAD4. Although the presence of few drivers, several signaling pathways are involved in the oncogenesis of this cancer type, some of them with promising targets for precision oncology. Pancreatic cancer is recognized as one of immunosuppressive phenotype cancer: it is characterized by a fibrotic-desmoplastic stroma, in which there is an intensive cross-talk between several cellular (e.g., fibroblasts, myeloid cells, lymphocytes, endothelial, and myeloid cells) and acellular (collagen, fibronectin, and soluble factors) components. In this review; we aim to describe the current knowledge of the genetic/biological landscape of pancreatic cancer and the composition of its tumor microenvironment; in order to better direct in the intrinsic labyrinth of this complex tumor type. Indeed; disentangling the genetic and molecular characteristics of cancer cells and the environment in which they evolve may represent the crucial step towards more effective therapeutic strategies
1. Introduction
Pancreatic cancer (PC) is perhaps the most lethal among solid tumors worldwide; although incidence varies between different countries, PC is the seventh leading cause of global cancer-related deaths in industrialized countries, the fourth in Italy, and the third in the United States of America [1,2,3]. Long-term survival is poor, with a five-year survival rate ranging from 5% to 8% [3]. The threatening notoriety of PC primarily arises from the lack of sensitive and specific biomarker(s) for detection and its negligible early diagnosis: indeed, the vast majority of PC patients are not eligible for surgical resection at the time of the diagnosis and present with a locally advanced or metastatic stage [4]. Moreover, even in patients undergoing potentially curative surgical resection the disease usually recurs within 2–3 years in the majority of cases.
As highlighted above, such PC unique aggressiveness is due to both specific genetic and molecular traits (acquired through either a stepwise or a chromothripsis-mediated simultaneous loss of multiple tumor suppressors and gain of multiple oncogenes), but to complex interactions with a generally obstructive and immunosuppressive tumor microenvironment (TME), which confer a highly malignant phenotype, as well (Figure 1) [5,6].
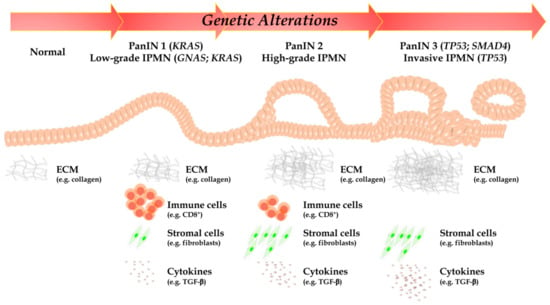
Figure 1.
According to PanIN and IPMN stage, a time-manner dependent accumulation of point mutations in oncogenes and tumor suppressor genes, epigenetic alterations and chromosomal structural variants are represented. Furthermore, PC is mainly represented by desmoplastic stroma and immunosuppressive TME: indeed, cell populations, collagen organization, and cytokines are profoundly different between normal pancreatic tissue and advanced PC. DC, dendritic cells; GNAS, guanine nucleotide binding protein, alpha stimulating; IPMN, intraductal papillary mucinous neoplasm; KRAS, Kirsten rat sarcoma; MDSC, myeloid-derived suppressor cells; PanIN, pancreatic intraepithelial lesion; PC, pancreatic cancer; PDAC, pancreatic ductal adenocarcinoma; PSC, pancreatic stellate cells; TME, tumor microenvironment; TP53, tumor suppressor protein 53.
Although combination chemotherapy has to some extent improved cure rates, survival, and quality of life in both early and advanced PC, anti-angiogenic drugs, molecularly targeted agents and immune checkpoint inhibitors have proven ineffective, so far, in PC [7,8]. Even the combination of therapies simultaneously targeting multiple crosstalking pathways has failed to confer clinical benefit in the absence of predictive biomarkers able to identify patients who will most likely respond to specific therapeutic combinations. This is well exemplified by the recent failure of a theoretically rational combination of MEK and AKT inhibitors [9]. In that respect, our group has recently identified PTEN-loss status as a predictive biomarker of synergistic pharmacologic interactions between mitogen-activated protein kinase (MAPK)- and phosphoinositide-3-kinase (PI3K)-inhibitors, in several preclinical models in vitro; when applied to PC the combination of data obtained in vitro and the relative rarity of complete loss of PTEN function in pancreatic ductal adenocarcinoma (PDAC) could have predicted the clinical failure of the MEK/AKT combination in unselected PC patients [9,10].
In recent years the genetic pathway to PC development (encompassing a relatively restricted set of recurrently altered genes, such as KRAS (Kirsten rat sarcoma), TP53 (tumor suppressor protein 53), CDKN2A (cyclin dependent kinase inhibitor 2A), and SMAD4 (SMAD family member 4), and molecular pathways) has been elucidated to an unprecedented extent; on the other hand, it is now clear that an intense crosstalk between stroma and cancer cells, mediated by soluble factors, leads to extensive fibrosis and deposition of extracellular matrix (ECM) proteins, resulting in poor perfusion and hypoxic conditions which, in turn, favor the evolution of aggressive clones (Figure 1). The molecular mechanisms of such interactions are only beginning to be elucidated and the Ariadne’s string linking specific tumor genetic/molecular background(s), to the production of specific sets of soluble factors and to the formation of an obstructive and immune suppressive TME will need to be precisely identified to develop new and effective therapeutic strategies to defeat such an aggressive and therapy resistant disease.
2. Driver Genes Alterations and Molecular Pathways to PC Development
2.1. Precursor Lesions
The high-aggressive PC represents a late event in a time-manner dependent sequence of genetic and molecular events, such as pancreatic intraductal neoplasia (PanIN) and intraductal papillary mucinous neoplasm (IPMN). PanIN is the most common PC precursor. It is a microscopic (<0.5 cm) intraductal lesion that can be found >80% of pancreas with invasive carcinoma [11,12]. PanIN is composed by cuboid to columnar mucinous cells; the new World Health Organization classification distinguishes low- from high-grade dysplasia to classify possible varying degrees of dysplasia [12]. Seminal papers on this topic showed molecular evidences of the progression from PanIN to PC, with early lesions (low-grade PanINs) displaying KRAS somatic mutations [13,14,15]. In PanIN carcinogenetic cascade, the TP53 and SMAD4 inactivations appear as very late events, often exclusive of an already existing invasion [16]. Another important PC precursor in certainly represented by IPMN. IPMN is a grossly-visible lesion (>1 cm by definition), with intraductal growth and papillary architecture, composed of mucinous cells. IPMN dysplasia also should be classified in low- and high-grade [12]. Based on the involvement in pancreatic ductal tree, IPMN could be categorized in: (1) main-duct IPMN (involvement of only Wirsung’s duct), (2) branch-duct IPMN (involvement of only secondary ducts), (3) mixed IPMN (contemporary involvement of the main and the branch ducts). This classification displays very important implications in clinical practice, indeed the main-duct IPMN shows higher risks towards evolution in PC, as compared to the others two [12,17]. From a histological point of view, IPMN can be classified into four subgroups: gastric, pancreatobiliary, intestinal, and oncocytic [12]. Even this classification shows a clinical impact, due to the association of the pancreatobiliary subtype with PC development [18,19]. From a molecular point of view, the most frequently mutated genes in IPMN are GNAS (guanine nucleotide binding protein, alpha stimulating) and KRAS, which are altered in up to 60% and to 80% of cases, respectively [12,20,21]. Notably, recent researches highlighted that the IPMN oncogenetic process is regulated by two different pathways: the first, linked to GNAS mutations, involves intestinal IPMN progressing to colloid adenocarcinomas (a PC variant reach in extracellular mucin), and the second, driven by KRAS mutations, is typical of pancreatobiliary IPMN and leads to conventional PC [20,21].
2.2. Driver Genes Alterations
Our knowledge of the molecular bases of PC has recently greatly improved, owing to advances in technology (next-generation sequencing—NGS) and consortia-based approaches, the latter enabling the collection of large cohorts of carefully annotated specimens. From a genetic point of view, PC appears as a complex disease, with a number of genes being altered through different mechanisms including point mutations, chromosomal aberrations, and epigenetic mechanisms, resulting in an “intermediate” tumor mutational burden [22]. Four genes, also called “PC genetic mountains”, are most commonly mutated: the KRAS oncogene, the TP53, the CDKN2A, and the SMAD4 tumor suppressor gene (Figure 1). Other genes altered at a lower but not-negligible prevalence are also called “PC genetic hills” [23,24]. Notably, alterations affecting the most important genetic drivers of PC can be demonstrated on tissue samples as well as by liquid biopsy, with reliable sensitivity and specificity [25].
2.2.1. KRAS
The KRAS is an oncogene located on chromosome 12, and is the most frequently mutated gene in PC (>90% of cases); the vast majority of activating mutations occurs at codons 12, 13, or 61 [23,24,25,26,27]. This oncogene encodes a small GTPase, that is switched on and off by cycling between the GTP-bound (active) and GDP-bound (inactive) forms. It acts as a transducer-moderator, interacting with cell surface receptors (receptor tyrosine kinases); once triggered, it stimulates several intracellular effector pathways, which drive very important modifications of cancer cells, such as increased proliferation, metabolism and migration, immune system evasion, and apoptosis blockade [28]. These downstream pathways include, among others, MAPK and PI3K. Notably, attempts at inhibiting the activity of the mutant KRAS oncoprotein have been unsuccessful, thus rendering such target undruggable so far and, perhaps, one of the most important reasons for PC’s high-mortality rate [29]. Very recently, new data on KRAS G12C-selective inhibitors and other new-generation KRAS-targeted approaches have been reported, raising renewed hope that we could one day target the main genetic PC driver directly or through repurposed drugs [30]. Mutations affecting KRAS represent an early event in PC carcinogenesis, as they are found also in precursor lesions with low-grade dysplasia, such as PanIN and IPMN with low-grade dysplasia [31].
2.2.2. TP53
The tumor suppressor gene TP53 is located on chromosome 17 and is mutated in a significant percentage of PC cases, ranging from 50–60% to 80% [32,33,34,35,36]. This so-called “master gene” encodes for P53, a crucial protein that controls cell growth, metabolism, senescence, DNA repair, and apoptosis in response to different types of cellular and extracellular processes, such as hypoxia and DNA damage [37]. The most common alterations affecting TP53 are represented by missense mutations and base substitutions in the DNA-binding domain, coupled with loss of the wild-type allele. Similar to KRAS mutations, TP53 alterations are currently considered “undruggable” from a therapeutic perspective, thus representing another reasonable explanation for PC’s high-mortality rate. Mutations affecting TP53 represent a late event in the PC carcinogenesis: indeed, they have been found in PanIN and IPMN with high-grade dysplasia or in the infiltrating component of PC only [31].
2.2.3. CDKN2A
CDKN2A is a tumor suppressor gene located on chromosome 9. It is altered in a several different types of tumor, and its inactivation is reported in up to 95% of PC variants. The inactivation of this tumor suppressor can occur following three different processes: (1) intragenic mutations associated with loss of the second allele (40%); (2) homozygous deletion (40%); (3) promoter hypermethylation associated with the loss of the second allele (in remaining cases) [38,39]. P16, the protein encoded by this gene, acts as a mediator of the “retinoblastoma” (RB) signaling pathway. By inhibiting the phosphorylation of RB, P16 stops the entry of cells into the S phase of the cell cycle. Alterations of this signaling pathway thus promotes an uncontrolled cell growth and neoplastic progression [40]. Of importance, fluorescence in situ hybridization is a common method for evaluating homozygous deletion of the CDKN2A locus [41]. Alterations affecting CDKN2A represent a late event in the oncogenesis of PC [31].
2.2.4. SMAD4
Alterations of the tumor suppressor gene SMAD4 (also called DPC4), located on chromosome 18, are reported in a variable percentage (30% to 60%) [34,35,36,42,43,44]. This gene is inactivated by homozygous deletion or intragenic mutations matched with loss of the second allele. The protein encoded by SMAD4 is an effector of the transforming growth factor (TGF)-β signaling pathway. In normal tissues, this pathway is important in preserving physiological homeostasis; indeed, it plays a crucial function in the control of cell proliferation. SMAD4 mutations and consequent perturbations of this pathway leads to a proliferative effect and by the activation of the process of the epithelial-to-mesenchymal transition (EMT) [45,46]. In this process, cancer cells lose their epithelial features and acquire a “mesenchymal” phenotype, which is essential for vascular invasion and overall metastatization. Recent studies have demonstrated that SMAD4 mutations increase the fitness of PC cells to colonize distant organs, thus characterizing PC cases with widespread metastatization, and are usually absent in cases in which the disease remains locally aggressive for a relatively long time during its course [47]. Mutations affecting SMAD4 represent a late event in the oncogenesis of PC: they have been found rarely in PanIN and IPMN with high-grade dysplasia, most commonly characterizing only the infiltrating component [31].
2.2.5. Other Mutations
Among less commonly mutated genes, some, such as AT-rich interaction domain 1A (ARID1A) and TGF-β receptor (R) I, have also been reported in different studies and are defined as “genetic hills” in the PC landscape. The most important “hill” is represented by the ARID1A gene, located on chromosome 3. It is a tumor suppressor gene and its mutations are usually associated with poor prognosis in several types of solid malignancies [34,47]. It encodes for a component of the of SWI/SNF (BAF) complex, a multi-protein complex with important functions in chromatin remodeling. This is an epigenetic modulator, which can regulate the gene expression without modifying the DNA sequence [48]. Mutations affecting ARID1A can result in a profound dysregulation of the chromatin remodeling mechanism, with important advantages for the proliferation of cancer cells [48,49].
PC molecular landscape is also characterized by complex chromosomal alterations. A recent whole-genome sequencing based study, conducted by the International Cancer Genome Consortium, has found 11,868 somatic structural variants at an average of 119 variants per PC case [36]. In this study, PC has been classified into 4 different subtypes based on structural variations: (1) stable; (2) locally rearranged; (3) scattered; (4) unstable. Particularly, in the “unstable” subgroup of PC (characterized by marked genomic instability), tumors showed a significantly increased proportion of breast cancer gene 1 (BRCA1), BRCA2, and partner and localizer of BRCA2 (PALB2) mutations. As an immediate consequence, patients who received platinum-based therapy experienced very significant clinical benefit. At the same time, this study first highlighted the therapeutic potential of PARP-inhibiting agents, also in PC: this class of drugs, indeed, has recently emerged as a potential targeted therapy in selected, genetically characterized, cases [49]. In terms of targeted therapy, the BRAF (v-raf murine sarcoma viral oncogene homolog B1) oncogene may also be mutated in PC, but its prevalence is less than 2% of all cases [50]. A recent study has shown that PC in older patients (>50 years) were significantly more likely to harbor genomic alterations in KRAS and SMAD4, whereas BRCA1 and BRCA2 alterations were more commonly detected in PC from younger patients (<50 years). A higher frequency of genomic alterations in KRAS, SMAD4 and CDKN2A were found in samples from female patients, whereas PC in male patients more often exhibited alterations in GNAS [50].
Some variants of PC also exhibit peculiar genetic alterations, not commonly observed in conventional PDAC. For example, the medullary PC variant often shows microsatellite instability (MSI), which can be targeted with immunotherapy [50,51,52,53,54]. The colloid PC variant, usually arising from IPMN, also has an increased rate of MSI and mutations in GNAS, located on chromosome 20, are also common in this variant [21,55,56]. Lastly, the PC variant with osteoclast-like giant cells is enriched for mutations in the SERPINA3 oncogene, although its peculiar morphological aspect has been mainly linked to its specific inflammatory background rather than its genetic profile, which is quite overlapping to conventional PC [57].
2.3. Dysregulation of Core Signaling Pathways
Thirteen main individual signaling pathways are currently reported altered in PC (Table 1) [34,50,58].

Table 1.
The most important genetic pathways altered in PC.
The first NGS study conducted by Jones and colleagues analyzed a cohort of 24 PC, finding an average of 63 genetic alterations per case [34]. This study showed that the most common mutations were base substitution, mainly involving the four PC driver gene KRAS, TP53, CDKN2A and SMAD4. A significant intra-tumor heterogeneity was also described. Of note, the most important result of this pioneering investigation was represented by the finding that the vast majority of mutated genes belonged to 12 so-called “core-signaling pathways”. Such pathways, integrating also the results of more recent publications, can be summarized as follows: (1) the MAPK signaling pathway, which is regulated by KRAS GTPase and still remains the most difficult target for tailored treatments; (2) the DNA damage control, which includes TP53 and represents a crucial event for the acquisition of the invasive capacities by PC, being a late event during PC carcinogenesis; (3) the regulatory pathway of G1/S cell phase transition, essential for the preparation of DNA synthesis, with the participation of P16; (4) the TGF-β signaling pathway, which is mainly regulated by the SMAD family of genes; (5) the pathway of apoptosis, in which the caspase proteins play a very important role and are of importance for the proliferation of cancer cells; (6) the hedgehog pathway, recently emerged as a potential target for precision oncology, which is mainly regulated by GLI3 gene products in PC; (7) the pathway of homophilic cell adhesion, in which the class of CDH and FAT genes are the most important effectors; (8) the pathway of integrin signaling, in which the encoded proteins regulate cell-to-cell adhesion and drive the process of local invasion of cancer cells; (9) the c-Jun N-terminal kinase (JNK) signaling, which is based on the energy transfer of phosphoryl groups and mainly involves MAP4K3 and tumor necrosis factor (TNF) as most important genes; (10) the pathway belonging to chromatin remodeler-complexes (SWI/SNF), in which ARID1A is the most commonly mutated gene; (11) the signaling pathway dependent on small GTPases (KRAS excluded); (12) the Wnt/Notch signaling pathway, fundamental also in other types of cancer and represented by the MYC and GATA6 genes as the main regulators; (13) to these pathways, another study has suggested to add a thirteenth, represented by the path of the embryonic driving regulators of the axon, which sees as the main signaling pathway the “so-called” SLIT/ROBO pathway [24,59]. Starting from the manuscript of Jones and colleagues, all the findings regarding the most important molecular pathway in PC have reinforced the vision of PC as a very complex disease with several different interactions and with a remarkable intra-tumor heterogeneity. Tailored therapeutic strategies in PC, based on a single gene only, have also for these reasons limited success for this patient setting [24]. Very recently, some clinical trials have initiated new approaches against PC, specifically using signaling pathway inhibitors (Table 2) [58].
Table 2.
Ongoing clinical trials investigating signaling pathway inhibitors in patients with PC.
Although the majority of these trials are of phase I, they represent one of the most important promising strategies of precision medicine for PC.
2.4. Molecularly-Defined PC Subtypes
Of interest is also reporting a recent attempt of classification of PC based on the frequency and distribution of structural rearrangements. Using data from whole-genome sequencing, Waddell and colleagues identified four different PC subtypes [36]. Subtype 1 was classified as “stable” and represented about 1/5 of cases. “Stable” was used to indicate that such neoplasms contained ≤50 structural variation events. Furthermore, this PC class often presented widespread aneuploidy suggesting defects in cell cycle/mitosis. Mutations of major PC driver genes were similar to the rest of the cohort. Subtype 2 was classified as “locally rearranged” and represented about 1/3 of all samples. This subtype presented a significant focal event on one or two chromosomes. About 1/3 of locally rearranged genomes contained regions of copy number gain that harbored known oncogenes, including focal amplifications with therapeutic targets (e.g., ERBB2). Another important molecular process discovered in this PC class was represented by chromothripsis. This process has been more recently described also in pancreatic neuroendocrine tumors, and causes fragmentation of a chromosome in many segments with consequent inaccurate reparations [60]. Subtype 3 was called as “scattered” and comprises, similarly to the “locally rearranged” class, about 1/3 of all cases. This PC subgroup presented a moderate range of non-random chromosomal damage and less than 200 structural variation events. At last, subtype 4 was called “unstable” and included about 15% of all samples. PC of this class harbored a large number of structural variation events (>200). This significant genomic instability suggested defects in DNA maintenance, with potential sensitivity to DNA-damaging agent. Of note, few patients within this group and with mutations affecting BRCA genes demonstrated clinical benefits from platinum-based therapy.
Several classifications have been also proposed based on mRNA expression analyses of bulk or microdissected tissues. Overall, when considering the characteristics of neoplastic cells all three classifications agree on the existence of two major lineages (classical/pancreatic progenitor and quasimesenchymal/basal-like/squamous) specified by different transcriptional programs and showing different prognosis and drug sensitivity [61,62,63]. In particular, the squamous/basal-like/quasi-mesenchymal lineage is characterized by loss of endodermal origin and activation of p63 and Dnp63 transcriptional programs. The analysis of bulk tissues also evidenced the existence of additional subtypes (Immunogenic and ADEX) which can be considered as classical/pp tumors but heavily infiltrated by non-neoplastic components, particularly leukocytes. Several studies have shown that a better understanding of the cell autonomous and non-cell autonomous determinants of molecular subtypes might lead to the design of better therapeutic intervention [64,65]. Along the same line, transcriptional rather than genetic signatures are being proven predictive of response to chemotherapy regimen or targeted therapy approaches [66].
3. TME
PC has long been described by pathologists as “a hypertrophic scar with sparse neoplastic cells”and stromal components may represent up to 85% of the tumor mass in PC, probably the most “desmoplastic” malignancy, among all solid tumors. PC desmoplastic stroma is a complex environment composed by both cellular and acellular components, such as cancer associated fibroblasts (CAF), immune cells (macrophages, neutrophils, myeloid-derived suppressor cells (MDSC), lymphocytes), and endothelial cells, on one hand, and collagen, fibronectin, and soluble factors (cytokines and chemokines), on the other (Figure 1). Such distinctive desmoplastic reaction around the tumor tissue is due to an intense autocrine and paracrine cross-talk between cancer and non-cancer components. Whether (or, more precisely, under what circumstances) TME represents a friend or a foe in terms of biological and clinical aggressiveness and response to therapy in PC remains a hotly debated topic. Indeed, TME elements with putative protumor properties are summarized in Table 3. Given the central role of TME in PC, understanding the biology behind tumor-stroma interactions (TSI) will be crucial to develop new therapeutic strategies for PC patients.
Table 3.
Contribution of protumor TME components in PC.
3.1. Cellular Components
3.1.1. Stromal Cells
PSC and CAF
Quiescent Pancreatic Stellate Cells (qPSC) are involved in maintenance of pancreatic tissue architecture and lipid and vitamin A storage; in response to external stimuli (known PC risk factors (e.g., ethanol and smoking), environmental stress (e.g., hypoperfusion and hypoxia), soluble cellular factors (e.g., interleukin (IL)-6 and TGF-β) and activation of molecular signaling pathways (e.g., Wnt/β-catenin and PI3K)), they skew towards activated PSC (aPSC), acquiring alpha-smooth muscle actin (α-SMA) expression and enhancing ECM deposition and the release of soluble factors involved in angiogenesis and EMT. In PC, aPSC represent the most important source of CAF: they represent the predominant fibroblast cell type in PC stroma and share similar features, including increased expression of the most important myofibroblastic marker α-SMA and of the CXCL12 cytokine involved in lymphocyte recruitment [67,70,71,72,109]. For these reasons, it is quite accepted that aPSC and CAF represent the same cellular population, even though their classification is still debated and some groups maintain a distinction between the two cell populations [110,111].
Whether CAF and aPSC exert pro- or anti-tumor activity in PC remains a matter of debate, owing to opposite lines of experimental evidence. In general, both CAF and aPSC are involved in tumor proliferation and growth, metastasis and immunity disruption, thereby causing chemoresistance (Figure 1) [73,112]. For example, Wei and colleagues demonstrated that CAF release CXCL12 which in turn upregulates the expression of the nuclear matrix attachment region-binding protein SATB-1 in PC cells; through a reciprocal feedback loop between fibroblasts and tumor cells, SATB-1 is involved in maintenance of CAF features (i.e., α-SMA, fibroblast activation protein and CXCL12) and gemcitabine resistance [113]. Chemoresistance is also induced by hypoxic conditions, as revealed by multiple studies in which the authors demonstrated that stroma depletion upregulates intratumor perfusion and gemcitabine accumulation at the tumor site, thus promoting cytotoxicity and prolonging survival [114]. Another mechanism involved in gemcitabine-resistance is linked to the decrease of aPSC-mediated apoptosis, as demonstrated by Liu and coworkers. In the same paper, the authors also demonstrated the key role of another soluble factor, TGF-β1, in TSI: indeed, the TGF-β1 inhibitor SB525334 partially blocks aPSC and tumor cells viability and aPSC-dependent invasion [115]. A further signaling pathway involved in affecting the interactions between PSC and pancreatic tumor cells is the MAPK cascade, as very recently demonstrated by Amrutkar and collaborators. Indeed, activation of ERK1/2 in PC cells, mediated by fibronectin aPSC-released, is involved in gemcitabine resistance: consistently, inhibition of aPSC-released fibronectin or ERK signaling may counteract chemoresistance and deserves further investigation as a potential therapeutic strategy [69]. In clinical PC series, CAF activation appears to correlate with reduced OS: indeed, produced by CAF promote tumor cell invasion and a high proportion of CAF in primary tumors correlates with nodal and distant metastases and a high lymph node ratio [116].
On the other hand, in other reports low levels of α-SMA+ myofibroblasts correlated with poor survival of PC patients, thereby leading to the hypothesis that fibrosis constitutes a “protective” TME, rather than a tumor promoter. Alpha-SMA+ myofibroblasts knock-out mice display multiple adverse outcomes, such as reduced immunosurveillance and gemcitabine resistance; chemoresistance could be reverted by a combination of gemcitabine plus anti-CTLA-4 treatment, which resulted in increased animal survival [117].
Endothelial Cells
Another important element in PC stroma is represented by endothelial cells and the mutual influence between endothelial cells and PC microenvironment is now well established. Indeed, the mere presence of conditioned medium derived from hypoxic PSC increases angiogenesis by inducing endothelial cells proliferation and migration in vitro and in vivo [118]. Conversely, conditioned medium derived from PC cells reduces endothelial cells proliferation [119]. However, the reciprocal influence between these cells on their growth is not yet defined: indeed, Craven and her collaborators showed that PC cells promote endothelial cells expansion. This group has recently demonstrated that the concomitant inhibition of TGF-β1R and janus kinase (JAK) 2 suppresses the mitogenic effects exerted by both endothelial and PC cells [120]. The endothelial cells surface marker, CD31, is significantly associated with better prognosis and improved overall survival (OS) in PC patients. Indeed, tumors, which express high levels of CD31, are associated with higher infiltration of anti-cancer immune cells (e.g., CD8+ and CD4+ T cells) [74]. Moreover, CD31 correlates with SMAD4 expression. Therefore, it is not surprising that SMAD4, frequently mutated in PC tissue, is associated with the number of endothelial cells in PC [120].
3.1.2. Immune Cells
In PC, cellular stromal components are also characterized by different immune cells which influence each other with endothelial cells to define an immunosuppressive TME.
Macrophages
Macrophages infiltrate PC attracted by vascular endothelial growth factor (VEGF)/VEGFR2 interactions [121] and affect its biological properties and clinical behavior. When polarized towards an M1 phenotype (HLA-DR+ and CD11c+), they exhibit pro-inflammatory properties; conversely, M2-polarized macrophages express CD163 and CD204 and exert tumor-promoting functions [108,122]. In PC TME hypoxic conditions and aPSC-derived cytokines, such as IL-10 and IL-13, polarize macrophages towards a M2, pro-tumor phenotype [123,124]. In addition, the expression of the toll-like receptor 4 on M2 macrophages promotes a vicious circle, stimulating further IL-10 release by tumor cells and promoting EMT through E-cadherin inhibition and Snail and vimentin upregulation [75]. In line with preclinical evidence, clinical data also confirm a dominant role of these cell populations within the PC immune landscape: indeed, macrophages are more represented in cancer tissue as compared to healthy pancreas, and their accumulation starts at the level of early stage lesions (PanIN) and increases during invasive cancer transformation [125]. In particular, macrophages exert an active role in conferring an aggressive behavior to primary PC through the regulation of the TGF-β [126]. Consistently, M2 populations represent a negative prognostic factor [76].
Neutrophils
Similar to macrophages, neutrophils represent another group of myeloid-derived circulating cells belonging to innate immunity. Despite their role in TME is still debate, the increasing knowledge of this cell type led to differentiate tumor associated neutrophils in N1 (with anti-tumor properties) and N2 (with pro-tumor properties), according with the type of cytotoxicity exerted and the ability to activate different immune cells [127]. Particular interest is actually attributed to neutrophil-to-lymphocyte ratio (NLR) as a prognostic biomarker in PC. In a recent metanalysis, Zhou and collaborators reported that patients with low NLR had significant smaller tumor size, longer OS and disease-free survival (DFS) as compared to high NLR patients [128]. Furthermore, the involvement of neutrophils in chemoresistance was recently investigated. Indeed, the combined blockade of CXCR2+ neutrophils and CCR2+ macrophages results in an increased antitumor immune response and response to the chemotherapy treatment with the FOLFIRINOX regimen, due to the reduction of infiltrating myeloid cells in the tumor [77].
Lymphocytes
Among immune cells, T lymphocytes are the most represented population and significantly influences the clinical outcome of PC patients [129]. According to the expression of specific surface markers, CD3+ T cells are classified in T helper CD4+, Treg CD4+/CD25+/Forkhead box P3 (Foxp3, and cytotoxic CD8+ [130]. CD4+ T cells mainly activate innate and acquired immunity, thus it is not surprising that their levels are lower in tumor tissue and peripheral blood of PC patients, as compared to healthy subjects; consistently, their infiltration is correlated with better OS [131,132]. On the other hand, in PC pro-tumor Th2 CD4+ (GATA3+) sub-populations are more represented than the tumor-killing Th1 (T-bet+) and their levels correlate with poor prognosis [133,134]. Tassi and her group indeed showed a potential benefit of immune reprogramming strategies aimed at reverting the predominant Th2 into a Th1 T cell phenotype preclinically, although clinical evidence of potential effectiveness is currently lacking [135]. Another subset of CD4+ cells characterized by the production of IL-17 (Th17) has been shown to play a pro-tumor role in PC, where both tumor-infiltrating Th17 CD4+ cells and circulating levels of Il-17 and IL-23 correlated with advanced stage and worse OS [78]. The well characterized Treg sub-population of immune cells contribute to an immunosuppressive TME by promoting CTLA-4 expression and TGF-β release [79,136]. In PC stroma the Treg population profoundly suppresses the activity of antitumor immune cells (i.e., CD4+, CD8+, macrophages and dendritic cells—DC) [79]. According to their tumor-promoting role, high levels of Treg cells are detected at the PC tumor site and circulating in the bloodstream, as compared to control subjects [137,138]. However, recent new data suggest that through their interactions with fibroblasts in PC TME, Tregs may actually counteract tumor development: indeed, it has been demonstrated that Treg depletion promotes pancreatic carcinogenesis, as a result of CAF activation and immunosuppressive myeloid infiltration [139].
CD8+ are cytotoxic T cells which may curtail the proliferation of tumor cells through two mechanisms: macrophages and interferon (IFN)-γ, activation [140]. Consistent with the ability of PC to evade tumor immunoediting, CD8+ T cells are rarely detectable in PC and their presence is associated with better prognosis (Figure 1) [141]. During tumor progression, PC evades CD8+ cytotoxicity, by causing both direct CD8+ aggregation in the desmoplastic tissue and indirect CD8+ inactivation through TGF-β secretion [142]. TGF-β release by PC cells in turn inhibits the transcription of CTL cytotoxic genes: perforin, granzyme A, granzyme B, Fas ligand, and IFN-γ, in vitro; these data support the hypothesis that TGF-β targeting could restore cytotoxic response involved in in vivo tumor clearance [143].
MDSC
MDSC are a heterogeneous group of immature cells classified in polymorphonuclear granulocytic MDSC and mononuclear monocytic MDSC, although they lack specific surface markers [144]. MDSC are represented in both human peripheral blood and TME and are involved in tumor growth, invasion and angiogenesis, through their activity on immune cells: for example, they promote the switch from M1 to M2 macrophage phenotype, promote Treg expansion and suppress natural killer (NK), CD4+, and CD8+ cells [81,82]. Indeed, in PC the levels of intratumor MDSC correlate with circulating Treg, but not with infiltrating CD8+ [145,146]. Moreover, circulating MDSC levels in PC patients are higher than in control subjects and increase upon disease progression [80]. Due to their contribution in promoting an immunosuppressive TME, blockade of the interactions between cancer cells and MDSC could improve clinical outcome by reactivating CD8+ cytotoxic activities. Baine and coworkers indeed demonstrated that the inhibition of tumor-derived granulocyte-macrophage colony-stimulating factor (GM-CSF, a cytokine highly expressed in PC and involved in disrupting antigen-specific T cells) prevents MDSC recruitment and tumor development [147].
DC
Antigen-presenting DC cells exert their effects in antitumor response mainly through MHC-mediated activation of CD4+ and CD8+ T cells [148]. Due to DC role in patrolling tissues to respond to danger stimuli, cancer cells develop strategies to evade immunosurveillance by blocking DC maturation and function; indeed, DC are rarely detected in PC TME [149,150]. Levels of both circulating and infiltrating DC correlates with better prognosis in both resectable and unresectable PC; furthermore, Fukunaka and coworkers reported a positive correlation between DC and CD4+/CD8+ tumor infiltration and an improved patient prognosis [132,151,152]. DC-based vaccination has been shown to gemcitabine’s therapeutic efficacy in the Panc02 poorly immunogenic murine tumor model [153]. Although preliminary clinical results with DC-based vaccination have shown tolerability and immunogenicity, clinical efficacy remains to be demonstrated [154,155].
3.2. Acellular Components
3.2.1. ECM
PC is histologically characterized by the abundance of ECM which, in addition to mechanical support, influences both the cellular and molecular composition of pancreatic stroma [156]. This three-dimensional acellular portion is composed by fibrous proteins (e.g., collagen, fibronectin, laminin, proteoglycans (e.g., decorin, biglycan, and lumican)) and water, and represents a dynamic structure in continuous remodeling, especially during tumor progression [157]. In normal pancreatic tissue, PSC regulate ECM turnover through the synthesis and metalloproteinase (MMP)-2/MMP-9-dependent degradation of ECM proteins [158,159]. Since PSC activation is an early event during the neoplastic transformation, it is not surprising that PSC and PC cells bidirectionally influence ECM dynamics to sustain cancer progression: indeed, aPSC enhance ECM proteins production and remodeling, hence providing a more favorable microenvironment for tumor cell growth [160]. The excessive PSC-dependent ECM proteins deposition also contributes to increase tissue hypoxia, which could explain the biological mechanism behind the failure of antiangiogenic therapies in PC [119].
Collagens
Collagens are one the best characterized and most abundant components of PC’s ECM and 28 types with at least a triple-helical domain are currently identified [161]. In TME, PC cells intensively interact with interstitial collagens and these connections significantly foster the pro-tumorigenic features, such as EMT and invasion, through both integrin-dependent and -independent signaling [162]. Indeed, collagens can either bind integrin receptors or the tyrosine kinase discoidin receptor 1 (DDR1) expressed by PC cells. The combination of different α and β subunit heterodimers allows integrin receptors to bind different types of collagens, in order to activate several intracellular signaling pathways. The canonical integrin-dependent biological mechanisms comprise focal adhesion kinase-mediated cell migration and EMT through downregulation of E-cadherin [83,84]. On the other hand, binding of collagen to DDR1 promotes invadopodia formation and MMP-2/MMP-9 expression [163,164]. Recent evidence has demonstrated that different collagen types can play contrasting roles in tumor. Collagens I, IV and V are involved in tumor progression by affecting adhesion, migration and cell viability and their levels increase and correlate with cancer stage and decreased patients’ survival. Ohlund and collaborators demonstrated that collagen IV colocalizes with integrin receptors on cancer cell surface and their interaction maintains cell proliferation and avoids apoptosis [85]. Similar effects are exerted by collagen V, which has been identified as a tumor promoter in PC: indeed, Berchtold and coworkers showed that PSC release collagen type V, which promotes invasion and proliferation of PC cells [86]. On the other hand, collagen XV decreases during PC progression, consistent with its role in hindering protumor features [165]. These controversial roles played by different types of collagens reflect the plasticity of ECM: indeed, the collagens architecture is continuously modified during cancer progression in order to favor the acquisition of a more aggressive behavior (Figure 1). In that respect, collagens mainly contribute to provide mechanical cues and ECM stiffness, which increase resistance to conventional treatments [87].
Proteoglycans
Many of the ECM-characterizing proteins are glycoproteins, a large group of molecules divided in four families, according to cellular and subcellular localization, genetic homology and protein activity [166]. Proteoglycans are heavily glycosylated proteins and their pattern of N- or O- glycosylation fluctuates during neoplastic transformation, in order to promote cell proliferation, angiogenesis and immune response [167]. Among proteoglycans, decorin, biglycan, and lumican are all small leucine-rich proteoglycan and the most important in PC [167]. Decorin acts as a monomer Zn2+ metalloprotein and is the first proteoglycan shown to be involved in the regulation of cell growth, by inhibiting TGF-β, ERBB2 dimerization and receptor tyrosine kinase signaling [168,169,170,171,172]. Since it mainly displays tumor suppressive properties, it is not surprising that low levels of its expression are detected in the stroma of many solid tumors, including PC, and its production is suppressed at a transcriptional level by TNF-α [166,173]. Köninger and coworker demonstrated that decorin is mostly released by PSC rather than by tumor cells in PC; however, cancer cells are sensitive to the decorin-dependent anti-tumor activity of chemotherapeutic drug carboplatin [174]. Biglycan shares > 65% of homology with decorin, and consistently also biglycan is able to bind and inactivate TGF-β [175]. More specifically, Weber and his collaborators showed that biglycan is expressed by fibroblasts in PC and overexpressed by cancer cells in response to TGF-β. Moreover, as a compensative host defense mechanism, biglycan affects cell growth in a TGF-β-independent manner, by inducing the cyclin-dependent kinase inhibitor p27 and a consequent cell cycle arrest [175]. Yamamoto and coworkers investigated the role of lumican in PC progression and demonstrated that lumican-transfected PANC-1 cells display a hyperactivated ERK pathway, which stimulates cell growth and lowers invasive ability [88]. Li and colleagues correlated the expression of lumican with prognosis in 131 untreated PC patients undergoing surgery and observed that patients with higher levels of this proteoglycan had significantly increased OS after tumor resection, as compared to those with low lumican expression levels (p = 0.0006). In the same paper, they also demonstrated that lumican increases apoptotic signals via hypoxia-inducible factor (HIF)1-α blockade in PC cell lines in vitro [176]. Hence, the role of lumican in PC is not yet well clarified, probably due to differential glycosylation pattern of lumican between the experimental conditions of different analyses [88].
Fibronectin
Fibronectin is the most abundant acellular component within PC tumor stroma and is mainly produced by aPSC [159,177,178]. Fibronectin is recognized by integrins family members which represent the best characterized fibronectin receptors; through this binding, different processes such as proliferation, migration, adhesion and survival, are modulated [178]. An anti-β1 integrin antibody profoundly modifies pancreatic TME, hence counteracting fibronectin polymerization and fibrillar collagen I formation in PANC 1/3 T3 spheroids [179]. Moreover, Cortes and his colleagues demonstrated that tamoxifen treatment reduces the hypoxic response of PSC, leading not only to a reduction of fibronectin alignment and thickness, which are associated with lower cellular invasion, but also to an increase of fibronectin protein expression in in vitro experiments [180]. In PC, fibronectin production is related to the activation of the NF-κB pathway: Jagadeeshan and coworkers demonstrated that p21-activated kinase 1, a serine/threonine kinase significantly upregulated in PC as compared to normal tissue, stimulates fibronectin production by promoting the direct NF-κB binding to fibronectin promoter [181]. Fibronectin upregulation in PC is associated with advanced clinicopathological stage and tumor aggressiveness [177]. Moreover, fibronectin-induced EMT correlates with poor OS in surgically resected PC patients [89]. Currently, the use of multi-gene expression profile datasets and meta-analyses has led to the identification of fibronectin as one of ten hubs that may be involved in PC progression and pathogenesis [182]. Along these lines, focal adhesion kinase (FAK) activation downstream of integrin binding to fibronectin and collagens correlates with high levels of fibrosis and poor CD+ cytotoxic T cell infiltration in PC; FAK inhibition limited tumor progression, markedly reduced tumor fibrosis, decreased the numbers of tumor-infiltrating immunosuppressive cells, and rendered the previously unresponsive KPC mouse model responsive to T cell immunotherapy and programmed cell death protein (PD)-1 antagonists. [183].
3.2.2. Cytokines
Cytokines are a family of soluble factors, classified into growth factors, chemokines, angiogenic factors and interferons and involved in tumor immune-landscape, by exerting pro- (IL-1β, IL-6, IL-8, and macrophage migration inhibitory factor—MIF) and anti-inflammatory (IL-10 and TGF-β) functions [184]. In TME, several cell types (e.g., immune, stromal, and tumor cells) release cytokines, involved in pleiotropic biological mechanisms through the binding to their specific cognate receptors in an autocrine and paracrine fashion. More in detail, cytokines affect the dynamic and complex immune network in TME, promoting PC immune-escape, PC progression, and drug response [184]. Evidence demonstrates that circulating pro-inflammatory cytokines are elevated in PC patients, as compared to healthy subjects, induce the PC cachexia clinical syndrome, and associate with clinical outcome, hence rendering these soluble factors new potential prognostic biomarkers and therapeutic targets (Figure 1) [185,186]. However, the role played by cytokines is often controversial, as the same factor (i.e., TNF-α) can be both a tumor promoter or a suppressor; thus, a recent systematic review suggested that considering a set of cytokines, rather than a single one, could be a more exhaustive and comprehensive approach [187]. Here, we summarize the biological implications of the most investigated cytokines potentially involved in PC disease course and prognosis.
IL-1β
IL-1β is a pro-inflammatory cytokine synthetized by monocytes, tissue macrophages, and PC cells; through the binding to the type 1 IL-1R, shared with IL-1α, the cleaved form of IL-1β activates NF-κB in PC cells [188]. Etoposide-resistant PC cell lines display elevated IL-1β production and NF-κB activity; blocking IL-1β signaling with an anti-IL-1R(I) antibody results in decreased NF-κB activity and restored chemosensitivity. Consistently, IL-1β produced by resistant A818-4 and PancTu-1 cell lines confers chemoresistance to the otherwise chemosensitive PT45-P1 cells, by enhancing the NF-κB [189]. IL-1β is also involved in promoting tumor aggressiveness by directly modulating the migration ability of both inflammatory and tumor cells through the regulation of the integrin superfamily. Indeed, IL-1β reduced integrin signaling in PC cell lines via JNK activation: JNK silencing counteracts IL-1β mediated vinculin and α5-integrin blockade [190]. Moreover, Schmid and his colleagues demonstrated that the recruitment and adhesion of myeloid cells is increased by IL-1β-induced integrin α4β1 activity [90]. In PC patients, IL-1β levels correlated with poor progression free survival (PFS; p = 0.056), especially when combined with high IL-6 levels (p < 0.001; see also below) [91].
IL-6
IL-6 is another proinflammatory cytokine, which activates JAK 2 and signal transducers and activators of transcription (STAT)1 and STAT3, through the IL-6 presentation to the signal-transducer gp130 by IL-6 receptor [191]. Once activated, JAK stimulates signaling through downstream pathways (MAPK, PI3K and STAT) involved in the promotion of cell cycle progression and proliferation. Furthermore, IL-6 is involved in PSC-mediated EMT in PC cells, as demonstrated by the reversion of STAT3-dependent EMT after either anti-IL-6 antibody treatment or STAT3 inhibition [92]. IL-6 may additionally contribute to the regulation of angiogenesis and metastatization, through the modulation of multiple soluble players. Indeed, IL-6 promotes the expression of angiogenesis-related genes such as VEGF, neuropilin (NRP)-1 and IL-10 [93]. Moreover, IL-6, IL-10, TGF-β, and VEGF together orchestrate the inhibition of dendropoiesis, thus shaping an immunosuppressive TME [94]. In PC patients, IL-6 levels are higher in metastatic disease than in locally advanced tumors (p = 0.0001) [94] and IL-6 released by myeloid cells in TME activates STAT3 signaling in PC cells, hence affecting poor patient’s outcome [192].
IL-8
Initially identified as a neutrophil-activating peptide, IL-8 is now a well-established proinflammatory chemokine, which signals through the binding to the CXCR1/2 receptors, thereby activating several G-protein-mediated signaling cascades involved in conferring several pro-tumoral properties to cancer cells [193]. Indeed, due to the presence of their receptors in several TME cell types (e.g., neutrophils, macrophages and endothelial cells), IL-8 regulates a plethora of mechanisms, such as cell proliferation and migration, angiogenesis, EMT and immune-escape [95]. PC cell lines release higher levels of IL-8 in culture medium, as compared to non-transformed HPDE cells; moreover, exogenous IL-8 increases the expression of the pro-angiogenic factor VEGF and its receptor NRP-2 and the levels of ERK phosphorylation, directly promoting tumor angiogenesis and cell proliferation, respectively [96]. Another group described a model in which stromal and tumor cells influence each other to enhance invasion and angiogenesis: in co-culture conditions, cancer cells stimulate fibroblasts to release higher levels of CXCL12 which, in turn, upregulates PC cell-derived IL-8 [194]. IL-8 is also involved in promoting ECM remodeling through MMP-2 and MMP-9 production [195]. The IL-8/CXCR1 axis is associated with the acquisition of CSC markers, such as CD44 and CD133, and functional abilities and correlates with lymph-node metastasis (LNM) (p = 0.012) and shorter OS in PC patients (p < 0.001) [97]. Furthermore, Chen and colleagues showed that PC patients express significantly higher IL-8 levels as compared to those with pancreatitis, in both tumor tissues and blood [196].
MIF
Another pleiotropic pro-inflammatory cytokine often overexpressed in PC is MIF. Although MIF was initially identified as a specific product of activated T lymphocytes, it is currently known that macrophages represent the main source of this soluble factor: TNF-α activation regulates MIF production and viceversa in an auto-amplification loop [197]. MIF interacts with the CD74 receptor, although complete activation requires the recruitment of different co-receptors, such as CD44 or CXCRs (e.g., CXCR2, CXCR4) [198]. MIF exerts its oncogenic role in PC through the downregulation of E-cadherin and the stimulation of vimentin and Zinc finger E-box-binding homeobox 1/2 production, hence causing EMT. Moreover, MIF-promoted PC aggressiveness is associated with increased resistance to gemcitabine treatment [98]. Recently, the detection of higher levels of MIF in PC tissue as compared to normal tissue increased the scientific interest in studying MIF as a new potential therapeutic target: MIF-overexpressing PC are more aggressive and result in poor OS [98,99]. Consistently, in vitro experiments demonstrated that the MIF post-transcriptional silencing could lead to PC regression through the induction of apoptosis and cell cycle arrest [199].
IL-10
Monocytes, macrophages, Th2 CD4+ T cells and activated B cells are the main source of IL-10 production in TME. The IL-10R1 is involved in the activation of specific IL-10 functions and the heterodimerization of IL-10R1 and IL-10R2 chains is required for outside-in cell signaling [200]. As an inhibitor of inflammatory responses, IL-10 counteracts the productions of proinflammatory cytokines (e.g., IFN-γ, IL-1β, and IL-6) from different cell types and DC maturation by blocking IL-12 expression [201]. IL-10 inhibits CD4+ T-cell response induced by mesothelin, an immunogen involved in promoting metastasis and tumor proliferation and a potential therapeutic target in PC [100]. Consistently, blocking IL-10 partially restores mesothelin-CAR T-cell activity [202]. IL-10 levels are increased in PC patients, as compared to healthy subjects, and correlate with poor survival of PC patients [101,102].
TGF-β
TGF-β plays an essential, but also complex and controversial, role in PC. TGF-β maintains homeostasis in normal tissue; however, being genetically unstable entities, cancer cells have the capacity to corrupt this suppressive influence. As a consequence, through the binding to the TGF-βRI and TGF-βRII heterotetrameric complexes, pathological forms of TGF-β signaling promote tumor growth and metastasis by activating downstream canonical Smad-dependent, as well as Smad-independent, intracellular pathways [203,204,205]. Activated phospho-Smad translocates into the nucleus, where it binds Smad-binding elements and transactivates TGF-β-dependent genes; non-Smad dependent signaling, on the other hand, mainly promotes activation of the MAPK pathway through RAS [206]. Regardless of the activated signaling, TGF-β acts as an EMT inducer [103]. Ellenrieder and collaborators demonstrated that MEK inhibition reduces TGF-β-dependent EMT and PC cells migration in KRAS mutant contexts [207]. TGF-β is also involved in modulating immune response against cancer cells: while DC stimulate CD8+ differentiation in co-culture with PC cells, TGF-β blockade improves antigen specific response and decreases T cells apoptosis; as a consequence, DC vaccination in combination with TGF-β blockade results in increased DFS in murine tumor models [104]. In PC, TGF-β levels correlate with clinical outcome and different groups demonstrated that PC patients with high levels of soluble TGF-β have a worse prognosis, as compared to those with low levels [105,106].
TNF-α
Similar to TGF-β, TNF-α plays a controversial role in PC stage progression and therapeutic response. Indeed, TNF-α exerts its activity as both a pro-tumoral and an anti-tumoral soluble factor [208]. This dual role is mainly due to the presence of two different receptors, TNFR1 and TNFR2, associated with pro-inflammatory and anti-inflammatory signaling, respectively [209]. As a type II transmembrane protein, TNF-α can bind both TNFR1 and TNFR2, whereas as the soluble form derived from proteolytic cleavage is able to activate TNFR1 exclusively [209,210]. While the ubiquitously expressed TNFR1 is mainly involved in apoptotic signaling, the role of TNFR2, mainly expressed on immune cells, is poorly understood [211]. PC cells-derived TNF-α enhances the tumor cell growth and invasiveness both in vitro and in vivo [107]. M1 macrophages also induce matrix remodeling through EMT in a TNF-α-dependent manner, highlighting the importance of TNF-α as a mediator in pancreatic TME [108]. The levels of TNF-α are higher in serum derived from PC patients, as compared to chronic pancreatitis or healthy patients [212]. However, gemcitabine treatment upregulates TNF-α production, resulting in a potent antitumor activity [213]. TNF-α-dependent anti-tumoral activity is limited by NF-κB activation, which can be overcome by the combination between gene therapy through an adenoviral vector-expressing TNF-α and nafamostat mesilate, a NF-κB inhibitor which upregulates apoptosis both in in vitro and in vivo [214].
4. Current Treatment Standard of Care and Novel Therapeutic Strategies
The overall five-year survival rate for PC patients remains below 10% (all stages included) and although median OS in advanced disease has improved with modern chemotherapy it remains below one year [2]. Approximately 80% of PC patients presents with surgically unresectable or metastatic cancer at diagnosis and among resected patients most will recur with metastatic disease within 3 years from surgery. Standard treatment strategies usually become rapidly ineffective, due to the early development of drug resistance; the cornerstone of PC treatment is conventional cytotoxic chemotherapy, and no predictive biomarkers are currently available to select patients for targeted anti-cancer treatments or immunotherapy. Thus, novel therapeutic approaches are urgently needed to improve the dismal prognosis of PC patients.
So far, results from clinical trials testing agents targeting cell autonomous signaling aberrations and angiogenesis have been quite disappointing in PC. Clinical trials investigating inhibitors of the EGFR family of receptors, of the MAPK and of the Hedgehog signaling pathways have failed to demonstrate an improvement in OS in PC patients [215,216,217]. Similarly, targeting the VEGF/VEGFR axis alone or in combination with an anti-EGFR, have also met with little clinical success; nevertheless, a small randomized phase II trial did demonstrate potential advantages for the multikinase angiogenesis inhibitor sunitinib in the first-line maintenance setting [218,219].
So far, alterations in the DNA damage repair machinery appear to be the most promising target(s) in PC [220]. Five to 15% of PC are characterized by either somatic or germline BRCA mutations and several case studies reported responses to poly-ADP-ribose-polymerase (PARP) inhibitors [221,222]. While a phase II trial of veliparib failed to demonstrate increased response in PC patients previously treated with chemotherapy, maintenance therapy with olaparib after a platinum-containing 1st line regimen prolonged OS in patients with non-progressing PC patients with germline BRCA1/2 mutations in the phase III trial POLO; olaparib is thus the first PARP inhibitor being granted approval by United States Food and Drug Administration and European Medicines Agency for the treatment of advanced PC [49,223]. Additional trials are ongoing to evaluate combination therapy with PARP inhibitors and chemotherapy or other targeted therapy (NCT01489865, NCT0158805). Similarly, a variable proportion of PC patients, whose tumors are characterized by defective mismatch DNA repair machinery and microsatellite instability (MSI-H), appears to experience prolonged responses to immune checkpoint inhibition by anti-PD1 antibodies [224].
More recently, attempts at modulating TSI with agents targeting different TME components, have also failed: pegvorhyaluronidase alfa, an agent targeting tumor hyaluronan, napabucasin, an agent targeting cancer cell stemness, recombinant pegylated IL-10, and a combination of anti-CTLA-4 and anti-PD-L1 antibodies have all recently shown lack of efficacy in clinical trials in advanced PC patients.
The largest effort of integrated genomic analysis in understanding the molecular pathology of PC has recently confirmed the TGF-β as the most recurrently mutated signal transduction pathway in PC [63]. The therapeutic efficacy of the TGF-β canonical Smad-dependent pathway inhibition by using the small molecule selective inhibitor of the TGF-βRI LY2109761 (Lilly, Indianapolis, USA) was initially demonstrated in preclinical models of PC [225]. Moreover, the role of a unique Smad-independent TGF-β pathway represented by the TGF-β activated kinase 1 (TAK1) was dissected in PC [226]. This serine/threonine kinase has a critical role in sustaining resistance to chemo- and radiotherapy in PC, by integrating signals from various cytokines and controlling, in turn, the activation of different transcription factors, including NF-κB [227,228,229]. More recently, a fundamental role of TAK1 was demonstrated towards PC chemoresistance through the kinase-independent modulation of two transcriptional regulators emerging as central determinants of malignancy in this disease, namely the transcriptional regulator yes associated protein (YAP) and the transcriptional coactivator with a PDZ-binding domain (TAZ) [230]. In light of these observations, TGF-β signaling represents a key target for personalized treatment of PC. Different therapeutic strategies have been developed and demonstrated efficacy in several preclinical and clinical trials in PC. Among the approaches used to target TGF-β, it is worth to mention trabedersen (AP12009), a TGF-βII antisense RNA molecule, which inhibits proliferation and migration of human PC cells and reduces growth and metastasis in human PC orthotopic mouse models [231]. In addition, a trabedersen phase I dose-escalation trial is ongoing in advanced PC patients (NCT00844064). Soluble TGF-βII was also used as another therapeutic strategy to inhibit of TGF-β binding to cellular TGF-βR in human PC cells, thereby suppressing tumor growth, angiogenesis and migratory capability of PC [232,233]. In addition, a phase I/II trial is ongoing in advanced solid tumors, including PC, with the anti- TGF-β antibody NIS793, as single agent or in combination with anti-PD-L1 (NCT02947165). Among TGF-β inhibitors acting at the intracellular signaling level, inhibitors of TGF-βRI kinase (i.e., SD208, SD093 and LY580276) significantly reduced the aggressiveness of PC cells in preclinical models. In particular, LY580276 is an ATP-competitive inhibitor of the TGF-βRI/ALK5 shown to arrest EMT and migration in PC cells [234,235]. A multi-center, open-label, phase Ib clinical trial is ongoing to evaluate the safety, tolerability, and exploratory efficacy of vactosertib (TEW-7197)—a small molecule inhibitor of TGF-βRI kinase—in combination with FOLFOX second-line chemotherapy in patients with metastatic PC after first-line gemcitabine and nab-paclitaxel (MP-PDAC-01, NCT03666832 [236]. The inhibitor of TGF-βRI/II kinase LY2109761 demonstrated to reduce tumor growth, metastasis and survival in combination with chemotherapy in an orthotopic murine model of advanced PC [225]. Despite the great number of TGF-βR inhibitors developed, only a few of them progressed to clinical investigations mainly due to relevant cardiac adverse events in animals. Compared to others small molecule inhibitors, galunisertib (LY2157299 monohydrate), an oral small molecule inhibitor of the TGF-βRI kinase that specifically downregulates the phosphorylation of Smad2, has demonstrated a reduced cardiovascular toxicity profile in preclinical models and it was the first-in-class to start its clinical development in PC patients [237]. The phase Ib/randomized phase II, double-blind trial in patients with advanced or unresectable PC, H9H-MC-JBAJ, demonstrated that the combination of galunisertib and gemcitabine improved OS versus gemcitabine monotherapy with an expected manageable toxicity [238]. Based on these positive results, the phase I clinical trial I8X-MC-JECA has been designed and is ongoing in PC patients, to test the safety of the next generation small TGF-βRI inhibitor LY3200882, in combination with the most recent standard first-line chemotherapy, gemcitabine and nab-paclitaxel (NCT02937272). Translational studies have been conducted to measure plasma proteins and miRNA markers at baseline and during treatment in patients enrolled in the H9H-MC-JBAJ study [239]. Predictive markers, useful to select patients who may benefit more from TGF-βR inhibition, have been identified. In particular, macrophage inflammatory protein (MIP)-1-α (a proinflammatory cytokine involved in the recruitment of monocytes and macrophages) and INFγ-induced protein (IP)-10 (involved in the recruitment of Treg) were two of the four most significant predictive biomarkers for chemoresistance in the population receiving single agent gemcitabine (for MIP-1-α mOS 95% CI high vs. low, 3.6 (2.5–4.0) vs. 10.1 (7.2–17.0) months, HR 95% CI = 3.93 (2.13–7.22) p < 0.01; for IP-10 mOS 95% CI high vs. low, 3.6 (2.5–7.7) vs. 12.5 (7.1–17.0) months, HR 95% CI = 2.78 (1.55–4.99) p < 0.001) [240]. On the contrary, treatment with galunisertib demonstrated to dramatically revert this resistance as MIP-1-α and IP-10 were the most significant positive predictive biomarkers for the combination of this agent with chemotherapy (for MIP-1-α mOS 95%CI galunisertib + gemcitabine vs. gemcitabine, 9.2 (5.5–12.4) vs. 3.6 (2.5–4.0) months, HR 95% CI = 0.38 (0.22–0.66) p < 0.01; for IP-10, 10.9 (5.5–12.4) vs. 3.6 (2.5–4.0) months, HR 95% CI = 0.4 (0.23–0.67) p < 0.0015) [239]. These results suggested an involvement of the immune system in the response of PC to galunisertib. Unfortunately, results from clinical trials using cancer vaccines or immune-checkpoint inhibitors (anti-CTLA4 and anti-PD-L1) in PC have been disappointing so far, thus immunotherapy remains a therapeutic challenge in PC patients [241,242,243,244]. The challenge for these approaches resides in the fact that, unlike melanoma, PC is characterized by a highly immune-suppressive microenvironment with a very low number of effector T cells. For this reason, research is moving towards the use of microenvironment re-educators, such as TGF-β inhibitors, in combination with checkpoint inhibitors, as a strategy for “immune-excluded” tumors, aiming to converting a non-immunologic tumor into an immune-responsive disease. Preclinical data from PC mouse models demonstrated that silencing of TGF-βRI induced a recruitment of CD8+ lymphocytes around PC [245]. Based on all these results, it has been conducted a phase Ib dose-escalation and cohort-expansion clinical trial of galunisertib in combination with the anti-PD-L1 antibody durvalumab in refractory metastatic PC patients (H9H-MC-JBEG). In the 32 patients treated with the recommended phase II dose, one partial response and 7 stable diseases were observed (disease control rate 25%); PFS was 1.9 months (95% CI: 1.5, 2.2) and mOS was longer than 12 months (95% CI: 3.6, NR). This combination has demonstrated to be active and well-tolerated [246]. Although these results represent the first signal of activity for an immune checkpoint-inhibitor combination strategy in an “immune-excluded” tumor as PC, its efficacy would need to be confirmed in larger randomized trials in advanced PC patients. Nonetheless, having demonstrated that TGF-β signaling inhibition is an active strategy in combination with classic chemotherapy and immune checkpoint-inhibitors in unselected advanced PC patient populations, the identification of biomarkers that could predict which patients would benefit more from these combination strategies remains of unique importance for the future development of more personalized treatments.
5. Conclusions
PC remains arguably the deadliest of solid tumors. Advances in the understanding of the genomic and transcriptomic complexity and heterogeneity of this disease, as well as elucidation of basic biological mechanisms underlying PC transformation and progression to a highly malignant phenotype, are of paramount importance to drive further clinical research and improve survival. However, clinical experiments clearly show that, with few exceptions, targeting individual players within the extremely complex and multifaceted crosstalk between the cancer cell genetic background, soluble factors, and cellular and acellular components of a usually highly desmoplastic stroma represents an oversimplification endowed with little potential for clinical efficacy.
Research efforts should thus concentrate on dissecting the currently elusive Ariadne’s string that links early occurring genetic aberrations of transformed epithelial cells, with their ability to secrete specific soluble factors (growth factors, cytokines, chemokines) which, in turn, will shape the molecular and cellular composition of the TME; TME, on the other hand, will respond to tumor growth (and presumably to the selective pressure applied by treatments) by activating different sets of soluble factors which, in turn, will influence tumor cell behavior and reshape the TME. In depth understanding examples of how the tumor genetic background can influence TME composition and response to treatments include, for example, evidence that loss of the PTEN tumor suppressor results in immunotherapy resistance, by directly inducing the T-cell inhibitor PD-L1 in several malignancies, including in mouse pancreatic cancer models [247,248,249]. Another mechanism by which tumor genotype can directly affect TME composition is the expression of specific cytokines, often involved in characterizing an immunosuppressive environment [250]. In that respect, it has been demonstrated that the oncogenic KRAS mutations are associated to an increased production of the GM-CSF, which causes the expansion of immunosuppressive Gr1+CD11b+ myeloid cells in mouse PC models; GM-CSF inhibition, in turn, could prevent MDSC recruitment and tumor development [251].
TME, on the other hand, may activate tumor cell signaling pathways which, in turn, cause profound TME remodeling: for example, FAK by PC cell’s integrin binding to fibronectin and collagens induces fibrosis and poor CD8+ cytotoxic T cell infiltration; inhibition of intracellular FAK signaling reduces tumor fibrosis, decreases the numbers of tumor-infiltrating immunosuppressive cells, and restores PC sensitivity T cell immunotherapy and PD-1 antagonists [183].
Cytokines secreted by either PC cells or stromal elements in the TME have pleiotropic, and sometimes unpredictable, effects: for example, both CAF and aPSC act in an autocrine and paracrine fashion in order to modulate the activity of the other cellular types in TME, such as endothelial and immune cells [252]. Cancer cells release proangiogenic soluble factors to induce neovascular formation in hypoxic condition of growth; on the other hand, hypoxia is one of the external stimuli of PSC activation and induces aPSC to secrete proangiogenic elements, which not only promote endothelial cells proliferation, but also sustain the maintenance of aPSC features [253].
Finally, TME components (both soluble factors and cellular and structural components, often display a double-edged function: for example, Tregs usually create an immunosuppressive, therapy resistant environment, but their depletion in PC may actually promotes pancreatic carcinogenesis, as a result of CAF activation and immunosuppressive myeloid infiltration. As another example, IL-10 function as an anti-inflammatory mediator, counteracting the production and activity of pro-inflammatory cytokines such as IL-1β and IL-6, which have been shown to favor PC progression and aggressiveness, has led to its development as a therapeutic agent; however, its double-edged activity as a suppressor of CD4 responses and DC maturation may possibly explain its therapeutic failure in recent clinical trials in PC.
It affords from the above that finding such elusive Ariadne’s string will be of paramount importance in moving PC therapeutic scenario forward, avoiding a dreadful waste of time and patients’ lives within dead-end paths. In addition to better understanding biology, we will have to improve our clinical methodology, by designing smart and effective clinical trials to: (1) provide clinical proof of concept supporting the proposed biological mechanism; (2) identify relevant biomarkers; (3) lay solid foundations for success in phase III clinical trials.
Author Contributions
Conceptualization, C.B. and F.C. (Fabiana Conciatori); writing-original draft preparation, C.B., F.C. (Fabiana Conciatori), C.L., F.S. and M.M; writing-review and editing, C.B., F.C. (Fabiana Conciatori), C.L., V.C. and M.M.; visualization, R.S., V.V., I.F., G.F. and F.C. (Francesco Cognetti); supervision, D.M., A.S., L.C. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Italian Association for Cancer Research (AIRC) (grant number L.C. IG 18622, D.M. IG 23719 and 5 × 1000 12182) and by the Ricerca Finalizzata 2016 grant through the Italian Ministry of Health, and by the “Nastro Viola” and “Voglio il Massimo” associations of patients’ donations to D.M. Fabiana Conciatori and Chiara Bazzichetto were supported by an AIRC fellowship for Italy.
Acknowledgments
The authors wish to thank IRCCS Scientific Director office for supporting the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- AIOM. I Numeri del Cancro in Italia 2019, 9th ed.; Intermedia EDITORE: Rodengo Saiano (BS), Italy, 2019; pp. 1–388. [Google Scholar]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef] [PubMed]
- Uzunparmak, B.; Sahin, I.H. Pancreatic cancer microenvironment: A current dilemma. Clin. Transl. Med. 2019, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Reiter, J.G.; Iacobuzio-Donahue, C.A. Pancreatic cancer: Pancreatic carcinogenesis - several small steps or one giant leap? Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, N.E.; Beniata, O.V.; Vitsos, P.; Tsitsilonis, O.; Samara, P. Harnessing the immune system to improve cancer therapy. Ann. Transl. Med. 2016, 4, 261. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.; Fisher, G.; Hezel, A.; Chang, S.C.; Vlahovic, G.; et al. Durvalumab With or Without Tremelimumab for Patients With Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Ciuffreda, L.; Del Curatolo, A.; Falcone, I.; Conciatori, F.; Bazzichetto, C.; Cognetti, F.; Corbo, V.; Scarpa, A.; Milella, M. Lack of growth inhibitory synergism with combined MAPK/PI3K inhibition in preclinical models of pancreatic cancer. Ann. Oncol. 2017, 28, 2896–2898. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Matteoni, S.; Sacconi, A.; De Luca, T.; Bazzichetto, C.; Corbo, V.; Simbolo, M.; Sperduti, I.; et al. PTEN status is a crucial determinant of the functional outcome of combined MEK and mTOR inhibition in cancer. Sci. Rep. 2017, 7, 43013. [Google Scholar] [CrossRef]
- Andea, A.; Sarkar, F.; Adsay, V.N. Clinicopathological correlates of pancreatic intraepithelial neoplasia: A comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod. Pathol. 2003, 16, 996–1006. [Google Scholar] [CrossRef]
- WHO. Digestive System Tumours, 5th ed.; IARC: Lyon, France, 2019. [Google Scholar]
- Yokode, M.; Akita, M.; Fujikura, K.; Kim, M.J.; Morinaga, Y.; Yoshikawa, S.; Terada, T.; Matsukiyo, H.; Tajiri, T.; Abe-Suzuki, S.; et al. High-grade PanIN presenting with localised stricture of the main pancreatic duct: A clinicopathological and molecular study of 10 cases suggests a clue for the early detection of pancreatic cancer. Histopathology 2018, 73, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Klimstra, D.S.; Longnecker, D.S. K-ras mutations in pancreatic ductal proliferative lesions. Am. J. Pathol. 1994, 145, 1547–1550. [Google Scholar]
- Moskaluk, C.A.; Hruban, R.H.; Kern, S.E. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997, 57, 2140–2143. [Google Scholar] [PubMed]
- Hosoda, W.; Chianchiano, P.; Griffin, J.F.; Pittman, M.E.; Brosens, L.A.; Noe, M.; Yu, J.; Shindo, K.; Suenaga, M.; Rezaee, N.; et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J. Pathol. 2017, 242, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M. Intraductal Papillary Mucinous Neoplasm of the Pancreas as the Main Focus for Early Detection of Pancreatic Adenocarcinoma. Pancreas 2018, 47, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Hatori, T.; Fujita, I.; Yamamoto, M.; Kobayashi, M.; Ohike, N.; Morohoshi, T.; Egawa, S.; Unno, M.; Takao, S.; et al. Prognostic relevance of morphological types of intraductal papillary mucinous neoplasms of the pancreas. Gut 2011, 60, 509–516. [Google Scholar] [CrossRef]
- Distler, M.; Kersting, S.; Niedergethmann, M.; Aust, D.E.; Franz, M.; Ruckert, F.; Ehehalt, F.; Pilarsky, C.; Post, S.; Saeger, H.D.; et al. Pathohistological subtype predicts survival in patients with intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann. Surg. 2013, 258, 324–330. [Google Scholar] [CrossRef]
- Wu, J.; Matthaei, H.; Maitra, A.; Dal Molin, M.; Wood, L.D.; Eshleman, J.R.; Goggins, M.; Canto, M.I.; Schulick, R.D.; Edil, B.H.; et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci. Transl. Med. 2011, 3, 92ra66. [Google Scholar] [CrossRef]
- Tan, M.C.; Basturk, O.; Brannon, A.R.; Bhanot, U.; Scott, S.N.; Bouvier, N.; LaFemina, J.; Jarnagin, W.R.; Berger, M.F.; Klimstra, D.; et al. GNAS and KRAS Mutations Define Separate Progression Pathways in Intraductal Papillary Mucinous Neoplasm-Associated Carcinoma. J. Am. Coll. Surg. 2015, 220, 845–854. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Pea, A.; Cheng, L.; Luchini, C. Dissecting the molecular landscape of pancreatic cancer: Towards a precision medicine approach. Exp. Rev. Prec. Med. Drug Develop. 2019, 4, 113–119. [Google Scholar] [CrossRef]
- Luchini, C.; Veronese, N.; Nottegar, A.; Cappelletti, V.; Daidone, M.G.; Smith, L.; Parris, C.; Brosens, L.A.A.; Caruso, M.G.; Cheng, L.; et al. Liquid Biopsy as Surrogate for Tissue for Molecular Profiling in Pancreatic Cancer: A Meta-Analysis Towards Precision Medicine. Cancers 2019, 11, 1152. [Google Scholar] [CrossRef] [PubMed]
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988, 53, 549–554. [Google Scholar] [CrossRef]
- Smit, V.T.; Boot, A.J.; Smits, A.M.; Fleuren, G.J.; Cornelisse, C.J.; Bos, J.L. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988, 16, 7773–7782. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Riva, G.; Pea, A.; Pilati, C.; Fiadone, G.; Lawlor, R.T.; Scarpa, A.; Luchini, C. Histo-molecular oncogenesis of pancreatic cancer: From precancerous lesions to invasive ductal adenocarcinoma. World J. Gastrointest. Oncol. 2018, 10, 317–327. [Google Scholar] [CrossRef]
- Scarpa, A.; Capelli, P.; Mukai, K.; Zamboni, G.; Oda, T.; Iacono, C.; Hirohashi, S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am. J. Pathol. 1993, 142, 1534–1543. [Google Scholar]
- Pellegata, N.S.; Sessa, F.; Renault, B.; Bonato, M.; Leone, B.E.; Solcia, E.; Ranzani, G.N. K-ras and p53 gene mutations in pancreatic cancer: Ductal and nonductal tumors progress through different genetic lesions. Cancer Res. 1994, 54, 1556–1560. [Google Scholar] [PubMed]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tsutsumi, S.; Kawaguchi, T.; Nagasaki, K.; Tatsuno, K.; Yamamoto, S.; Sang, F.; Sonoda, K.; Sugawara, M.; Saiura, A.; et al. Whole-exome sequencing of human pancreatic cancers and characterization of genomic instability caused by MLH1 haploinsufficiency and complete deficiency. Genome Res. 2012, 22, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Caldas, C.; Hahn, S.A.; da Costa, L.T.; Redston, M.S.; Schutte, M.; Seymour, A.B.; Weinstein, C.L.; Hruban, R.H.; Yeo, C.J.; Kern, S.E. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat. Genet. 1994, 8, 27–32. [Google Scholar] [CrossRef]
- Ueki, T.; Toyota, M.; Sohn, T.; Yeo, C.J.; Issa, J.P.; Hruban, R.H.; Goggins, M. Hypermethylation of multiple genes in pancreatic adenocarcinoma. Cancer Res. 2000, 60, 1835–1839. [Google Scholar]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef]
- Hustinx, S.R.; Hruban, R.H.; Leoni, L.M.; Iacobuzio-Donahue, C.; Cameron, J.L.; Yeo, C.J.; Brown, P.N.; Argani, P.; Ashfaq, R.; Fukushima, N.; et al. Homozygous deletion of the MTAP gene in invasive adenocarcinoma of the pancreas and in periampullary cancer: A potential new target for therapy. Cancer Biol. Ther. 2005, 4, 83–86. [Google Scholar] [CrossRef]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef]
- Hahn, S.A.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Schutte, M.; Rozenblum, E.; Seymour, A.B.; Weinstein, C.L.; Yeo, C.J.; Hruban, R.H.; et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res. 1996, 56, 490–494. [Google Scholar] [PubMed]
- Iacobuzio-Donahue, C.A.; Song, J.; Parmiagiani, G.; Yeo, C.J.; Hruban, R.H.; Kern, S.E. Missense mutations of MADH4: Characterization of the mutational hot spot and functional consequences in human tumors. Clin. Cancer Res. 2004, 10, 1597–1604. [Google Scholar] [CrossRef]
- Lawlor, R.T.; Veronese, N.; Nottegar, A.; Malleo, G.; Smith, L.; Demurtas, J.; Cheng, L.; Wood, L.D.; Silvestris, N.; Salvia, R.; et al. Prognostic Role of High-Grade Tumor Budding in Pancreatic Ductal Adenocarcinoma: A Systematic Review and Meta-Analysis with a Focus on Epithelial to Mesenchymal Transition. Cancers 2019, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Massague, J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer 2003, 3, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef]
- Wilson, B.G.; Roberts, C.W. SWI/SNF nucleosome remodellers and cancer. Nat. Rev. Cancer 2011, 11, 481–492. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Singhi, A.D.; George, B.; Greenbowe, J.R.; Chung, J.; Suh, J.; Maitra, A.; Klempner, S.J.; Hendifar, A.; Milind, J.M.; Golan, T.; et al. Real-Time Targeted Genome Profile Analysis of Pancreatic Ductal Adenocarcinomas Identifies Genetic Alterations That Might Be Targeted With Existing Drugs or Used as Biomarkers. Gastroenterology 2019, 156, 2242–2253. [Google Scholar] [CrossRef]
- Goggins, M.; Offerhaus, G.J.; Hilgers, W.; Griffin, C.A.; Shekher, M.; Tang, D.; Sohn, T.A.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am. J. Pathol. 1998, 152, 1501–1507. [Google Scholar]
- Wilentz, R.E.; Goggins, M.; Redston, M.; Marcus, V.A.; Adsay, N.V.; Sohn, T.A.; Kadkol, S.S.; Yeo, C.J.; Choti, M.; Zahurak, M.; et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: A newly described and characterized entity. Am. J. Pathol. 2000, 156, 1641–1651. [Google Scholar] [CrossRef]
- Luchini, C.; Capelli, P.; Scarpa, A. Pancreatic Ductal Adenocarcinoma and Its Variants. Surg. Pathol. Clin. 2016, 9, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO recommendations on microsatellite instability testing for immunotherapy in cancer, and its relationship with PD-1/PD-L1 expression and tumour mutational burden: A systematic review-based approach. Ann. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Amato, E.; Molin, M.D.; Mafficini, A.; Yu, J.; Malleo, G.; Rusev, B.; Fassan, M.; Antonello, D.; Sadakari, Y.; Castelli, P.; et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J. Pathol. 2014, 233, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, W.; Sasaki, E.; Murakami, Y.; Yamao, K.; Shimizu, Y.; Yatabe, Y. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows Arch. 2015, 466, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Luchini, C.; Pea, A.; Lionheart, G.; Mafficini, A.; Nottegar, A.; Veronese, N.; Chianchiano, P.; Brosens, L.A.; Noe, M.; Offerhaus, G.J.A.; et al. Pancreatic undifferentiated carcinoma with osteoclast-like giant cells is genetically similar to, but clinically distinct from, conventional ductal adenocarcinoma. J. Pathol. 2017, 243, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Candido, J.B.; Morton, J.P.; Bailey, P.; Campbell, A.D.; Karim, S.A.; Jamieson, T.; Lapienyte, L.; Gopinathan, A.; Clark, W.; McGhee, E.J.; et al. CSF1R(+) Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 2018, 23, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Andricovich, J.; Perkail, S.; Kai, Y.; Casasanta, N.; Peng, W.; Tzatsos, A. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell 2018, 33, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef] [PubMed]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer-stromal interaction and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596. [Google Scholar] [CrossRef]
- Xue, R.; Jia, K.; Wang, J.; Yang, L.; Wang, Y.; Gao, L.; Hao, J. A Rising Star in Pancreatic Diseases: Pancreatic Stellate Cells. Front. Physiol. 2018, 9, 754. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, B.; Hu, Q.; Qin, Y.; Xu, W.; Liu, W.; Yu, X.; Xu, J. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics 2018, 8, 5072–5087. [Google Scholar] [CrossRef] [PubMed]
- Katsuta, E.; Qi, Q.; Peng, X.; Hochwald, S.N.; Yan, L.; Takabe, K. Pancreatic adenocarcinomas with mature blood vessels have better overall survival. Sci. Rep. 2019, 9, 1310. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Invest. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int. J. Mol. Sci. 2011, 12, 7424–7437. [Google Scholar] [CrossRef]
- Drake, C.G.; Jaffee, E.; Pardoll, D.M. Mechanisms of immune evasion by tumors. Adv. Immunol. 2006, 90, 51–81. [Google Scholar] [CrossRef]
- Markowitz, J.; Brooks, T.R.; Duggan, M.C.; Paul, B.K.; Pan, X.; Wei, L.; Abrams, Z.; Luedke, E.; Lesinski, G.B.; Mundy-Bosse, B.; et al. Patients with pancreatic adenocarcinoma exhibit elevated levels of myeloid-derived suppressor cells upon progression of disease. Cancer Immunol. Immunother. 2015, 64, 149–159. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef]
- Lu, J.; Zhou, S.; Siech, M.; Habisch, H.; Seufferlein, T.; Bachem, M.G. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. Br. J. Cancer 2014, 110, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Menke, A.; Philippi, C.; Vogelmann, R.; Seidel, B.; Lutz, M.P.; Adler, G.; Wedlich, D. Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer Res. 2001, 61, 3508–3517. [Google Scholar] [PubMed]
- Ohlund, D.; Franklin, O.; Lundberg, E.; Lundin, C.; Sund, M. Type IV collagen stimulates pancreatic cancer cell proliferation, migration, and inhibits apoptosis through an autocrine loop. BMC Cancer 2013, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, S.; Grunwald, B.; Kruger, A.; Reithmeier, A.; Hahl, T.; Cheng, T.; Feuchtinger, A.; Born, D.; Erkan, M.; Kleeff, J.; et al. Collagen type V promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Cancer Lett. 2015, 356, 721–732. [Google Scholar] [CrossRef]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef]
- Yamamoto, T.; Matsuda, Y.; Kawahara, K.; Ishiwata, T.; Naito, Z. Secreted 70kDa lumican stimulates growth and inhibits invasion of human pancreatic cancer. Cancer Lett. 2012, 320, 31–39. [Google Scholar] [CrossRef]
- Javle, M.M.; Gibbs, J.F.; Iwata, K.K.; Pak, Y.; Rutledge, P.; Yu, J.; Black, J.D.; Tan, D.; Khoury, T. Epithelial-mesenchymal transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann. Surg. Oncol. 2007, 14, 3527–3533. [Google Scholar] [CrossRef]
- Schmid, M.C.; Avraamides, C.J.; Foubert, P.; Shaked, Y.; Kang, S.W.; Kerbel, R.S.; Varner, J.A. Combined blockade of integrin-alpha4beta1 plus cytokines SDF-1alpha or IL-1beta potently inhibits tumor inflammation and growth. Cancer Res. 2011, 71, 6965–6975. [Google Scholar] [CrossRef]
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of IL-6 and IL-1beta can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069. [Google Scholar] [CrossRef]
- Hamada, S.; Masamune, A.; Yoshida, N.; Takikawa, T.; Shimosegawa, T. IL-6/STAT3 Plays a Regulatory Role in the Interaction Between Pancreatic Stellate Cells and Cancer Cells. Dig. Dis. Sci. 2016, 61, 1561–1571. [Google Scholar] [CrossRef]
- Feurino, L.W.; Zhang, Y.; Bharadwaj, U.; Zhang, R.; Li, F.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q.; Min, L. IL-6 stimulates Th2 type cytokine secretion and upregulates VEGF and NRP-1 expression in pancreatic cancer cells. Cancer Biol. Ther. 2007, 6, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Bellone, G.; Smirne, C.; Mauri, F.A.; Tonel, E.; Carbone, A.; Buffolino, A.; Dughera, L.; Robecchi, A.; Pirisi, M.; Emanuelli, G. Cytokine expression profile in human pancreatic carcinoma cells and in surgical specimens: Implications for survival. Cancer Immunol. Immunother. 2006, 55, 684–698. [Google Scholar] [CrossRef] [PubMed]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.; Feurino, L.W.; Wang, H.; Fisher, W.E.; Brunicardi, F.C.; Chen, C.; Yao, Q. Interleukin-8 increases vascular endothelial growth factor and neuropilin expression and stimulates ERK activation in human pancreatic cancer. Cancer Sci. 2008, 99, 733–737. [Google Scholar] [CrossRef]
- Chen, L.; Fan, J.; Chen, H.; Meng, Z.; Chen, Z.; Wang, P.; Liu, L. The IL-8/CXCR1 axis is associated with cancer stem cell-like properties and correlates with clinical prognosis in human pancreatic cancer cases. Sci. Rep. 2014, 4, 5911. [Google Scholar] [CrossRef]
- Funamizu, N.; Hu, C.; Lacy, C.; Schetter, A.; Zhang, G.; He, P.; Gaedcke, J.; Ghadimi, M.B.; Ried, T.; Yfantis, H.G.; et al. Macrophage migration inhibitory factor induces epithelial to mesenchymal transition, enhances tumor aggressiveness and predicts clinical outcome in resected pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 785–794. [Google Scholar] [CrossRef]
- Wang, D.; Wang, R.; Huang, A.; Fang, Z.; Wang, K.; He, M.; Xia, J.T.; Li, W. Upregulation of macrophage migration inhibitory factor promotes tumor metastasis and correlates with poor prognosis of pancreatic ductal adenocarcinoma. Oncol. Rep. 2018, 40, 2628–2636. [Google Scholar] [CrossRef]
- Chen, Y.; Ayaru, L.; Mathew, S.; Morris, E.; Pereira, S.P.; Behboudi, S. Expansion of anti-mesothelin specific CD4+ and CD8+ T cell responses in patients with pancreatic carcinoma. PLoS ONE 2014, 9, e88133. [Google Scholar] [CrossRef]
- Dima, S.O.; Tanase, C.; Albulescu, R.; Herlea, V.; Chivu-Economescu, M.; Purnichescu-Purtan, R.; Dumitrascu, T.; Duda, D.G.; Popescu, I. An exploratory study of inflammatory cytokines as prognostic biomarkers in patients with ductal pancreatic adenocarcinoma. Pancreas 2012, 41, 1001–1007. [Google Scholar] [CrossRef]
- Feng, L.; Qi, Q.; Wang, P.; Chen, H.; Chen, Z.; Meng, Z.; Liu, L. Serum levels of IL-6, IL-8, and IL-10 are indicators of prognosis in pancreatic cancer. J. Int. Med. Res. 2018, 46, 5228–5236. [Google Scholar] [CrossRef]
- Maier, H.J.; Wirth, T.; Beug, H. Epithelial-mesenchymal transition in pancreatic carcinoma. Cancers 2010, 2, 2058–2083. [Google Scholar] [CrossRef] [PubMed]
- Pu, N.; Zhao, G.; Gao, S.; Cui, Y.; Xu, Y.; Lv, Y.; Nuerxiati, A.; Wu, W. Neutralizing TGF-beta promotes anti-tumor immunity of dendritic cells against pancreatic cancer by regulating T lymphocytes. Cent. Eur. J. Immunol. 2018, 43, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Li, Y.; Tan, D.; Dong, X.; Chang, P.; Kar, S.; Li, D. Biomarkers of TGF-beta signaling pathway and prognosis of pancreatic cancer. PLoS ONE 2014, 9, e85942. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Bang, J.-H.; Nam, A.-R.; Park, J.E.; Kim, M.H.; Bang, Y.-J.; Oh, D.-Y. The role of soluble TGF-beta and its dynamics for predicting the prognosis in unresectable pancreatic cancer patients treated with chemotherapy. J. Clin. Oncol. 2018, 36, 288. [Google Scholar] [CrossRef]
- Egberts, J.H.; Cloosters, V.; Noack, A.; Schniewind, B.; Thon, L.; Klose, S.; Kettler, B.; von Forstner, C.; Kneitz, C.; Tepel, J.; et al. Anti-tumor necrosis factor therapy inhibits pancreatic tumor growth and metastasis. Cancer Res. 2008, 68, 1443–1450. [Google Scholar] [CrossRef]
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Kruger, U.; Becker, T.; Ebsen, M.; Rocken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 2014, 135, 843–861. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Qu, C.; Wang, Q.; Meng, Z.; Wang, P. Cancer-Associated Fibroblasts in Pancreatic Cancer: Should They Be Deleted or Reeducated? Integr. Cancer Ther. 2018, 17, 1016–1019. [Google Scholar] [CrossRef]
- Parente, P.; Parcesepe, P.; Covelli, C.; Olivieri, N.; Remo, A.; Pancione, M.; Latiano, T.P.; Graziano, P.; Maiello, E.; Giordano, G. Crosstalk between the Tumor Microenvironment and Immune System in Pancreatic Ductal Adenocarcinoma: Potential Targets for New Therapeutic Approaches. Gastroenterol. Res. Pract. 2018, 2018, 7530619. [Google Scholar] [CrossRef]
- Bynigeri, R.R.; Jakkampudi, A.; Jangala, R.; Subramanyam, C.; Sasikala, M.; Rao, G.V.; Reddy, D.N.; Talukdar, R. Pancreatic stellate cell: Pandora’s box for pancreatic disease biology. World J. Gastroenterol. 2017, 23, 382–405. [Google Scholar] [CrossRef]
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065. [Google Scholar] [CrossRef]
- McCarroll, J.A.; Naim, S.; Sharbeen, G.; Russia, N.; Lee, J.; Kavallaris, M.; Goldstein, D.; Phillips, P.A. Role of pancreatic stellate cells in chemoresistance in pancreatic cancer. Front. Physiol. 2014, 5, 141. [Google Scholar] [CrossRef]
- Liu, S.L.; Cao, S.G.; Li, Y.; Sun, B.; Chen, D.; Wang, D.S.; Zhou, Y.B. Pancreatic stellate cells facilitate pancreatic cancer cell viability and invasion. Oncol. Lett. 2019, 17, 2057–2062. [Google Scholar] [CrossRef]
- Bolm, L.; Cigolla, S.; Wittel, U.A.; Hopt, U.T.; Keck, T.; Rades, D.; Bronsert, P.; Wellner, U.F. The Role of Fibroblasts in Pancreatic Cancer: Extracellular Matrix Versus Paracrine Factors. Transl. Oncol. 2017, 10, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Hirota, M.; Shimosegawa, T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am. J. Physiol. Gastrointest Liver Physiol. 2008, 295, G709–G717. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar] [CrossRef]
- Craven, K.E.; Gore, J.; Wilson, J.L.; Korc, M. Angiogenic gene signature in human pancreatic cancer correlates with TGF-beta and inflammatory transcriptomes. Oncotarget 2016, 7, 323–341. [Google Scholar] [CrossRef]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar] [CrossRef]
- Barros, M.H.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage polarisation: An immunohistochemical approach for identifying M1 and M2 macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [PubMed]
- Looi, C.K.; Chung, F.F.; Leong, C.O.; Wong, S.F.; Rosli, R.; Mai, C.W. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 162. [Google Scholar] [CrossRef]
- Glazer, E.S.; Reed, L.F.; Freeburg, C.J.; Husain, S.M.; Deneve, J.L.; Dickson, P.V.; Shibata, D. Effect of macrophages on pancreatic cancer. J. Clin. Oncol. 2018, 36, 324. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wei, Q.; Fan, J.; Cheng, S.; Ding, W.; Hua, Z. Prognostic role of the neutrophil-to-lymphocyte ratio in pancreatic cancer: A meta-analysis containing 8252 patients. Clin. Chim. Acta 2018, 479, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A., 3rd; Yarchoan, M.; Lee, V.; Laheru, D.A.; Jaffee, E.M. Strategies for Increasing Pancreatic Tumor Immunogenicity. Clin. Cancer Res. 2017, 23, 1656–1669. [Google Scholar] [CrossRef]
- Chang, J.H.; Jiang, Y.; Pillarisetty, V.G. Role of immune cells in pancreatic cancer from bench to clinical application: An updated review. Medicine 2016, 95, e5541. [Google Scholar] [CrossRef]
- Bang, S.; Kim, H.S.; Choo, Y.S.; Park, S.W.; Chung, J.B.; Song, S.Y. Differences in immune cells engaged in cell-mediated immunity after chemotherapy for far advanced pancreatic cancer. Pancreas 2006, 32, 29–36. [Google Scholar] [CrossRef]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004, 28, E26–E31. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef]
- Tassi, E.; Braga, M.; Longhi, R.; Gavazzi, F.; Parmiani, G.; Di Carlo, V.; Protti, M.P. Non-redundant role for IL-12 and IL-27 in modulating Th2 polarization of carcinoembryonic antigen specific CD4 T cells from pancreatic cancer patients. PLoS ONE 2009, 4, e7234. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Vegran, F.; Hichami, A.; Ladoire, S.; Derangere, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xu, X.; Guo, S.; Zhang, C.; Tang, Y.; Tian, Y.; Ni, B.; Lu, B.; Wang, H. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS ONE 2014, 9, e91551. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Hirooka, S.; Yamaki, S.; Yui, R.; Yamao, J.; Kim, S.; Kwon, A.H. Circulating CD4+CD25+ regulatory T cells in patients with pancreatic cancer. Pancreas 2012, 41, 409–415. [Google Scholar] [CrossRef]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.; et al. Regulatory T cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Wormann, S.M.; Diakopoulos, K.N.; Lesina, M.; Algul, H. The immune network in pancreatic cancer development and progression. Oncogene 2014, 33, 2956–2967. [Google Scholar] [CrossRef]
- Hou, Y.C.; Chao, Y.J.; Hsieh, M.H.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Low CD8(+) T Cell Infiltration and High PD-L1 Expression Are Associated with Level of CD44(+)/CD133(+) Cancer Stem Cells and Predict an Unfavorable Prognosis in Pancreatic Cancer. Cancers 2019, 11, 541. [Google Scholar] [CrossRef]
- Ademmer, K.; Ebert, M.; Muller-Ostermeyer, F.; Friess, H.; Buchler, M.W.; Schubert, W.; Malfertheiner, P. Effector T lymphocyte subsets in human pancreatic cancer: Detection of CD8+CD18+ cells and CD8+CD103+ cells by multi-epitope imaging. Clin. Exp. Immunol. 1998, 112, 21–26. [Google Scholar] [CrossRef]
- Thomas, D.A.; Massague, J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Pergamo, M.; Miller, G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2017, 24, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Song, X.; Yu, H.; Zhang, M.; Li, F.; Cao, C.; Jiang, Q. Dendritic cell-based cancer immunotherapy for pancreatic cancer. Arab. J. Gastroenterol. 2018, 19, 1–6. [Google Scholar] [CrossRef]
- Dallal, R.M.; Christakos, P.; Lee, K.; Egawa, S.; Son, Y.I.; Lotze, M.T. Paucity of dendritic cells in pancreatic cancer. Surgery 2002, 131, 135–138. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yanagimoto, H.; Satoi, S.; Toyokawa, H.; Yamao, J.; Kim, S.; Terakawa, N.; Takahashi, K.; Kwon, A.H. Circulating myeloid dendritic cells as prognostic factors in patients with pancreatic cancer who have undergone surgical resection. J. Surg. Res. 2012, 173, 299–308. [Google Scholar] [CrossRef]
- Hirooka, S.; Yanagimoto, H.; Satoi, S.; Yamamoto, T.; Toyokawa, H.; Yamaki, S.; Yui, R.; Inoue, K.; Michiura, T.; Kwon, A.H. The role of circulating dendritic cells in patients with unresectable pancreatic cancer. Anticancer Res. 2011, 31, 3827–3834. [Google Scholar]
- Bauer, C.; Bauernfeind, F.; Sterzik, A.; Orban, M.; Schnurr, M.; Lehr, H.A.; Endres, S.; Eigler, A.; Dauer, M. Dendritic cell-based vaccination combined with gemcitabine increases survival in a murine pancreatic carcinoma model. Gut 2007, 56, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, S.; Kitago, M.; Sakurai, T.; Matsuda, T.; Fujita, T.; Higuchi, H.; Taguchi, J.; Takeuchi, H.; Itano, O.; Aiura, K.; et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015, 106, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, R.; Koizumi, T.; Koya, T.; Sano, K.; Koido, S.; Nagai, K.; Kobayashi, M.; Okamoto, M.; Sugiyama, H.; Shimodaira, S. WT1-pulsed Dendritic Cell Vaccine Combined with Chemotherapy for Resected Pancreatic Cancer in a Phase I Study. Anticancer Res. 2018, 38, 2217–2225. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, T.E.; Fosang, A.J. Proteoglycans: Many forms and many functions. FASEB J. 1992, 6, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Pirola, R.C.; Wilson, J.S. Pancreatic stellate cells: A starring role in normal and diseased pancreas. Front. Physiol. 2012, 3, 344. [Google Scholar] [CrossRef]
- Mekapogu, A.R.; Pothula, S.P.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Multifunctional role of pancreatic stellate cells in pancreatic cancer. Ann. Pancreat. Cancer 2019, 2, 10. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef]
- Linder, S.; Castanos-Velez, E.; von Rosen, A.; Biberfeld, P. Immunohistochemical expression of extracellular matrix proteins and adhesion molecules in pancreatic carcinoma. Hepatogastroenterology 2001, 48, 1321–1327. [Google Scholar]
- Yang, J.C.; Zhang, Y.; He, S.J.; Li, M.M.; Cai, X.L.; Wang, H.; Xu, L.M.; Cao, J. TM4SF1 Promotes Metastasis of Pancreatic Cancer via Regulating the Expression of DDR1. Sci. Rep. 2017, 7, 45895. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Yang, J.C.; Ramachandran, V.; Arumugam, T.; Deng, D.F.; Li, Z.S.; Xu, L.M.; Logsdon, C.D. TM4SF1 Regulates Pancreatic Cancer Migration and Invasion In Vitro and In Vivo. Cell Physiol. Biochem. 2016, 39, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Clementz, A.G.; Mutolo, M.J.; Leir, S.H.; Morris, K.J.; Kucybala, K.; Harris, H.; Harris, A. Collagen XV inhibits epithelial to mesenchymal transition in pancreatic adenocarcinoma cells. PLoS ONE 2013, 8, e72250. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015, 42, 11–55. [Google Scholar] [CrossRef]
- Pan, S.; Brentnall, T.A.; Chen, R. Glycoproteins and glycoproteomics in pancreatic cancer. World J. Gastroenterol. 2016, 22, 9288–9299. [Google Scholar] [CrossRef]
- Yang, V.W.; LaBrenz, S.R.; Rosenberg, L.C.; McQuillan, D.; Hook, M. Decorin is a Zn2+ metalloprotein. J. Biol. Chem. 1999, 274, 12454–12460. [Google Scholar] [CrossRef]
- Goldoni, S.; Owens, R.T.; McQuillan, D.J.; Shriver, Z.; Sasisekharan, R.; Birk, D.E.; Campbell, S.; Iozzo, R.V. Biologically active decorin is a monomer in solution. J. Biol. Chem. 2004, 279, 6606–6612. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Mann, D.M.; Ruoslahti, E. Negative regulation of transforming growth factor-beta by the proteoglycan decorin. Nature 1990, 346, 281–284. [Google Scholar] [CrossRef]
- Santra, M.; Eichstetter, I.; Iozzo, R.V. An anti-oncogenic role for decorin. Down-regulation of ErbB2 leads to growth suppression and cytodifferentiation of mammary carcinoma cells. J. Biol. Chem. 2000, 275, 35153–35161. [Google Scholar] [CrossRef]
- Csordas, G.; Santra, M.; Reed, C.C.; Eichstetter, I.; McQuillan, D.J.; Gross, D.; Nugent, M.A.; Hajnoczky, G.; Iozzo, R.V. Sustained down-regulation of the epidermal growth factor receptor by decorin. A mechanism for controlling tumor growth in vivo. J. Biol. Chem. 2000, 275, 32879–32887. [Google Scholar] [CrossRef]
- Mauviel, A.; Santra, M.; Chen, Y.Q.; Uitto, J.; Iozzo, R.V. Transcriptional regulation of decorin gene expression. Induction by quiescence and repression by tumor necrosis factor-alpha. J. Biol. Chem. 1995, 270, 11692–11700. [Google Scholar] [CrossRef] [PubMed]
- Koninger, J.; Giese, N.A.; di Mola, F.F.; Berberat, P.; Giese, T.; Esposito, I.; Bachem, M.G.; Buchler, M.W.; Friess, H. Overexpressed decorin in pancreatic cancer: Potential tumor growth inhibition and attenuation of chemotherapeutic action. Clin. Cancer Res. 2004, 10, 4776–4783. [Google Scholar] [CrossRef] [PubMed]
- Kolb, M.; Margetts, P.J.; Sime, P.J.; Gauldie, J. Proteoglycans decorin and biglycan differentially modulate TGF-beta-mediated fibrotic responses in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L1327–L1334. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Truty, M.A.; Kang, Y.; Chopin-Laly, X.; Zhang, R.; Roife, D.; Chatterjee, D.; Lin, E.; Thomas, R.M.; Wang, H.; et al. Extracellular lumican inhibits pancreatic cancer cell growth and is associated with prolonged survival after surgery. Clin. Cancer Res. 2014, 20, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Ansari, D.; Zhou, Q.; Sasor, A.; Said Hilmersson, K.; Andersson, R. Stromal fibronectin expression in patients with resected pancreatic ductal adenocarcinoma. World J. Surg. Oncol. 2019, 17, 29. [Google Scholar] [CrossRef] [PubMed]
- Topalovski, M.; Brekken, R.A. Matrix control of pancreatic cancer: New insights into fibronectin signaling. Cancer Lett. 2016, 381, 252–258. [Google Scholar] [CrossRef]
- Durymanov, M.; Kroll, C.; Permyakova, A.; O’Neill, E.; Sulaiman, R.; Person, M.; Reineke, J. Subcutaneous Inoculation of 3D Pancreatic Cancer Spheroids Results in Development of Reproducible Stroma-Rich Tumors. Transl. Oncol. 2019, 12, 180–189. [Google Scholar] [CrossRef]
- Cortes, E.; Lachowski, D.; Robinson, B.; Sarper, M.; Teppo, J.S.; Thorpe, S.D.; Lieberthal, T.J.; Iwamoto, K.; Lee, D.A.; Okada-Hatakeyama, M.; et al. Tamoxifen mechanically reprograms the tumor microenvironment via HIF-1A and reduces cancer cell survival. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Jagadeeshan, S.; Krishnamoorthy, Y.R.; Singhal, M.; Subramanian, A.; Mavuluri, J.; Lakshmi, A.; Roshini, A.; Baskar, G.; Ravi, M.; Joseph, L.D.; et al. Transcriptional regulation of fibronectin by p21-activated kinase-1 modulates pancreatic tumorigenesis. Oncogene 2015, 34, 455–464. [Google Scholar] [CrossRef]
- Ma, Y.P.; Peng, L.; Luo, X.; Xu, J.; Peng, Y.; Tang, X. Identification of potential hub genes associated with the pathogenesis and prognosis of pancreatic duct adenocarcinoma using bioinformatics meta-analysis of multi-platform datasets. Oncol. Lett. 2019, 18, 6741–6751. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Bazzichetto, C.; Conciatori, F.; Falcone, I.; Cognetti, F.; Milella, M.; Ciuffreda, L. Advances in Tumor-Stroma Interactions: Emerging Role of Cytokine Network in Colorectal and Pancreatic Cancer. J. Oncol. 2019, 2019, 5373580. [Google Scholar] [CrossRef] [PubMed]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Farajzadeh Valilou, S.; Keshavarz-Fathi, M.; Silvestris, N.; Argentiero, A.; Rezaei, N. The role of inflammatory cytokines and tumor associated macrophages (TAMs) in microenvironment of pancreatic cancer. Cytokine Growth Factor Rev. 2018, 39, 46–61. [Google Scholar] [CrossRef]
- Yako, Y.Y.; Kruger, D.; Smith, M.; Brand, M. Cytokines as Biomarkers of Pancreatic Ductal Adenocarcinoma: A Systematic Review. PLoS ONE 2016, 11, e0154016. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Vorndamm, J.; Muerkoster, S.; Yu, H.; Schmidt, W.E.; Folsch, U.R.; Schafer, H. Autocrine production of interleukin 1beta confers constitutive nuclear factor kappaB activity and chemoresistance in pancreatic carcinoma cell lines. Cancer Res. 2002, 62, 910–916. [Google Scholar]
- Verma, G.; Bhatia, H.; Datta, M. Gene expression profiling and pathway analysis identify the integrin signaling pathway to be altered by IL-1beta in human pancreatic cancer cells: Role of JNK. Cancer Lett. 2012, 320, 86–95. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Muller-Newen, G.; Schaper, F.; Graeve, L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J. 1998, 334, 297–314. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Kloppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Ha, H.; Debnath, B.; Neamati, N. Role of the CXCL8-CXCR1/2 Axis in Cancer and Inflammatory Diseases. Theranostics 2017, 7, 1543–1588. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Ochi, N.; Sawai, H.; Yasuda, A.; Takahashi, H.; Funahashi, H.; Takeyama, H.; Tong, Z.; Guha, S. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int. J. Cancer 2009, 124, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, Y.; Sasaki, T.; Morinaka, K.; Kitadai, Y.; Mukaida, N.; Chayama, K. Potential involvement of IL-8 and its receptors in the invasiveness of pancreatic cancer cells. Int. J. Oncol. 2003, 4, 765–771. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, M.; Yu, G.Z.; Qin, X.R.; Jin, G.; Chen, P.; Zhu, M.H. Interleukin-8, a promising predictor for prognosis of pancreatic cancer. World J. Gastroenterol. 2012, 18, 1123–1129. [Google Scholar] [CrossRef] [PubMed]
- Calandra, T.; Bernhagen, J.; Mitchell, R.A.; Bucala, R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. J. Exp. Med. 1994, 179, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Jankauskas, S.S.; Wong, D.W.L.; Bucala, R.; Djudjaj, S.; Boor, P. Evolving complexity of MIF signaling. Cell Signal. 2019, 57, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Denz, A.; Pilarsky, C.; Muth, D.; Ruckert, F.; Saeger, H.D.; Grutzmann, R. Inhibition of MIF leads to cell cycle arrest and apoptosis in pancreatic cancer cells. J. Surg. Res. 2010, 160, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.R. The molecular basis of IL-10 function: From receptor structure to the onset of signaling. Curr. Top. Microbiol. Immunol. 2014, 380, 191–212. [Google Scholar] [CrossRef]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Batchu, R.B.; Gruzdyn, O.V.; Mahmud, E.M.; Chukr, F.; Dachepalli, R.; Manmari, S.K.; Mostafa, G.; Weaver, D.W.; Gruber, S.A. Inhibition of Interleukin-10 in the tumor microenvironment can restore mesothelin chimeric antigen receptor T cell activity in pancreatic cancer in vitro. Surgery 2018, 163, 627–632. [Google Scholar] [CrossRef]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. TGF-beta in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFbeta in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-beta/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and Its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef]
- Ellenrieder, V.; Hendler, S.F.; Boeck, W.; Seufferlein, T.; Menke, A.; Ruhland, C.; Adler, G.; Gress, T.M. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001, 61, 4222–4228. [Google Scholar]
- Zhao, X.; Fan, W.; Xu, Z.; Chen, H.; He, Y.; Yang, G.; Yang, G.; Hu, H.; Tang, S.; Wang, P.; et al. Inhibiting tumor necrosis factor-alpha diminishes desmoplasia and inflammation to overcome chemoresistance in pancreatic ductal adenocarcinoma. Oncotarget 2016, 7, 81110–81122. [Google Scholar] [CrossRef]
- Chopra, M.; Lang, I.; Salzmann, S.; Pachel, C.; Kraus, S.; Bauerlein, C.A.; Brede, C.; Garrote, A.L.; Mattenheimer, K.; Ritz, M.; et al. Tumor necrosis factor induces tumor promoting and anti-tumoral effects on pancreatic cancer via TNFR1. PLoS ONE 2013, 8, e75737. [Google Scholar] [CrossRef]
- Shadhu, K.; Xi, C. Inflammation and pancreatic cancer: An updated review. Saudi. J. Gastroenterol. 2019, 25, 3–13. [Google Scholar] [CrossRef]
- Ham, B.; Fernandez, M.C.; D’Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27. [Google Scholar]
- Talar-Wojnarowska, R.; Gasiorowska, A.; Smolarz, B.; Romanowicz-Makowska, H.; Kulig, A.; Malecka-Panas, E. Tumor necrosis factor alpha and interferon gamma genes polymorphisms and serum levels in pancreatic adenocarcinoma. Neoplasma 2009, 56, 56–62. [Google Scholar] [CrossRef]
- Murugesan, S.R.; King, C.R.; Osborn, R.; Fairweather, W.R.; O’Reilly, E.M.; Thornton, M.O.; Wei, L.L. Combination of human tumor necrosis factor-alpha (hTNF-alpha) gene delivery with gemcitabine is effective in models of pancreatic cancer. Cancer Gene Ther. 2009, 16, 841–847. [Google Scholar] [CrossRef]
- Furukawa, K.; Ohashi, T.; Haruki, K.; Fujiwara, Y.; Iida, T.; Shiba, H.; Uwagawa, T.; Kobayashi, H.; Yanaga, K. Combination treatment using adenovirus vector-mediated tumor necrosis factor-alpha gene transfer and a NF-kappaB inhibitor for pancreatic cancer in mice. Cancer Lett. 2011, 306, 92–98. [Google Scholar] [CrossRef]
- Infante, J.R.; Somer, B.G.; Park, J.O.; Li, C.P.; Scheulen, M.E.; Kasubhai, S.M.; Oh, D.Y.; Liu, Y.; Redhu, S.; Steplewski, K.; et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur. J. Cancer 2014, 50, 2072–2081. [Google Scholar] [CrossRef]
- O’Neil, B.H.; Scott, A.J.; Ma, W.W.; Cohen, S.J.; Leichman, L.; Aisner, D.L.; Menter, A.R.; Tejani, M.A.; Cho, J.K.; Granfortuna, J.; et al. A phase II/III randomized study to compare the efficacy and safety of rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 2505. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized Phase Ib/II Study of Gemcitabine Plus Placebo or Vismodegib, a Hedgehog Pathway Inhibitor, in Patients With Metastatic Pancreatic Cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Sutherland, S.; Schrag, D.; Hurwitz, H.; Innocenti, F.; Mulcahy, M.F.; O’Reilly, E.; Wozniak, T.F.; et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: Phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J. Clin. Oncol. 2010, 28, 3617–3622. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Cereda, S.; Milella, M.; Novarino, A.; Passardi, A.; Mambrini, A.; Di Lucca, G.; Aprile, G.; Belli, C.; Danova, M.; et al. Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: A phase II randomised trial. Eur. J. Cancer 2013, 49, 3609–3615. [Google Scholar] [CrossRef]
- Sehdev, A.; Gbolahan, O.; Hancock, B.A.; Stanley, M.; Shahda, S.; Wan, J.; Wu, H.H.; Radovich, M.; O’Neil, B.H. Germline and Somatic DNA Damage Repair Gene Mutations and Overall Survival in Metastatic Pancreatic Adenocarcinoma Patients Treated with FOLFIRINOX. Clin. Cancer Res. 2018, 24, 6204–6211. [Google Scholar] [CrossRef]
- Fogelman, D.R.; Wolff, R.A.; Kopetz, S.; Javle, M.; Bradley, C.; Mok, I.; Cabanillas, F.; Abbruzzese, J.L. Evidence for the efficacy of Iniparib, a PARP-1 inhibitor, in BRCA2-associated pancreatic cancer. Anticancer Res. 2011, 31, 1417–1420. [Google Scholar]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Lu, M.; Lin, S.C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.C.; Myszka, D.; Han, J.; Wu, H. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol. Cell 2007, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients With Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results From the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Carbone, C.; Piro, G.; Chiao, P.J.; Melisi, D. TAK-ing aim at chemoresistance: The emerging role of MAP3K7 as a target for cancer therapy. Drug Resist. Updat. 2017, 33–35, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Carbone, C.; Melisi, D. NF-kappaB as a target for pancreatic cancer therapy. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S1–S10. [Google Scholar] [CrossRef]
- Melisi, D.; Chiao, P.J. NF-kappa B as a target for cancer therapy. Expert Opin. Ther. Targets 2007, 11, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Xia, Q.; Paradiso, G.; Ling, J.; Moccia, T.; Carbone, C.; Budillon, A.; Abbruzzese, J.L.; Chiao, P.J. Modulation of pancreatic cancer chemoresistance by inhibition of TAK1. J. Natl. Cancer Inst. 2011, 103, 1190–1204. [Google Scholar] [CrossRef]
- Santoro, R.; Zanotto, M.; Simionato, F.; Zecchetto, C.; Merz, V.; Cavallini, C.; Piro, G.; Sabbadini, F.; Boschi, F.; Scarpa, A.; et al. Modulating TAK1 expression inhibits YAP and TAZ oncogenic functions in pancreatic cancer. Mol. Cancer Ther. 2019. [Google Scholar] [CrossRef]
- Schlingensiepen, K.H.; Jaschinski, F.; Lang, S.A.; Moser, C.; Geissler, E.K.; Schlitt, H.J.; Kielmanowicz, M.; Schneider, A. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Sci. 2011, 102, 1193–1200. [Google Scholar] [CrossRef]
- Rowland-Goldsmith, M.A.; Maruyama, H.; Kusama, T.; Ralli, S.; Korc, M. Soluble type II transforming growth factor-beta (TGF-beta) receptor inhibits TGF-beta signaling in COLO-357 pancreatic cancer cells in vitro and attenuates tumor formation. Clin. Cancer Res. 2001, 7, 2931–2940. [Google Scholar]
- Rowland-Goldsmith, M.A.; Maruyama, H.; Matsuda, K.; Idezawa, T.; Ralli, M.; Ralli, S.; Korc, M. Soluble type II transforming growth factor-beta receptor attenuates expression of metastasis-associated genes and suppresses pancreatic cancer cell metastasis. Mol. Cancer Ther. 2002, 1, 161–167. [Google Scholar] [PubMed]
- Subramanian, G.; Schwarz, R.E.; Higgins, L.; McEnroe, G.; Chakravarty, S.; Dugar, S.; Reiss, M. Targeting endogenous transforming growth factor beta receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004, 64, 5200–5211. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.J.; Li, L.; Kapoun, A.M.; Medicherla, S.; Reddy, M.; Li, G.; O’Young, G.; Quon, D.; Henson, M.; Damm, D.L.; et al. Inhibition of transforming growth factor beta signaling reduces pancreatic adenocarcinoma growth and invasiveness. Mol. Pharmacol. 2007, 72, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-G.; Sergeeva, O.; Luo, G.; Choi, S.H.; Lee, Z.; Kim, S.-J.; Letterio, J.; Malek, E. Abstract 2647: TGF-β type I receptor inhibitor (TEW-7197) diminishes myeloma progression by multiple immunomodulatory mechanisms in combination with ixazomib. Cancer Res. 2017, 77, 2647. [Google Scholar] [CrossRef]
- Kovacs, R.J.; Maldonado, G.; Azaro, A.; Fernandez, M.S.; Romero, F.L.; Sepulveda-Sanchez, J.M.; Corretti, M.; Carducci, M.; Dolan, M.; Gueorguieva, I.; et al. Cardiac Safety of TGF-beta Receptor I Kinase Inhibitor LY2157299 Monohydrate in Cancer Patients in a First-in-Human Dose Study. Cardiovasc. Toxicol. 2015, 15, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Oettle, H.; Kozloff, M.; Cleverly, A.; et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br. J. Cancer 2018, 119, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Garcia-Carbonero, R.; Macarulla, T.; Pezet, D.; Deplanque, G.; Fuchs, M.; Trojan, J.; Kozloff, M.; Simionato, F.; Cleverly, A.; et al. TGFbeta receptor inhibitor galunisertib is linked to inflammation- and remodeling-related proteins in patients with pancreatic cancer. Cancer Chemother. Pharmacol. 2019, 83, 975–991. [Google Scholar] [CrossRef]
- Piro, G.; Simionato, F.; Carbone, C.; Frizziero, M.; Malleo, G.; Zanini, S.; Casolino, R.; Santoro, R.; Mina, M.M.; Zecchetto, C.; et al. A circulating TH2 cytokines profile predicts survival in patients with resectable pancreatic adenocarcinoma. Oncoimmunology 2017, 6, e1322242. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- O’Reilly, E.M.; Oh, D.Y.; Dhani, N.; Renouf, D.J.; Lee, M.A.; Sun, W.J.; Fisher, G.A.; Hezel, A.F.; Chang, S.C.; Vlahovic, G.; et al. A randomized phase 2 study of durvalumab monotherapy and in combination with tremelimumab in patients with metastatic pancreatic ductal adenocarcinoma (mPDAC): ALPS study. J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef]
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840. [Google Scholar] [CrossRef]
- Principe, D.R.; DeCant, B.; Mascarinas, E.; Wayne, E.A.; Diaz, A.M.; Akagi, N.; Hwang, R.; Pasche, B.; Dawson, D.W.; Fang, D.; et al. TGFbeta Signaling in the Pancreatic Tumor Microenvironment Promotes Fibrosis and Immune Evasion to Facilitate Tumorigenesis. Cancer Res. 2016, 76, 2525–2539. [Google Scholar] [CrossRef]
- Melisi, D.; Hollebecque, A.; Oh, D.-Y.; Calvo, E.; Varghese, A.M.; Borazanci, E.H.; Mercade, T.M.; Simionato, F.; Park, J.O.; Bendell, J.C.; et al. A phase Ib dose-escalation and cohort-expansion study of safety and activity of the transforming growth factor (TGF) β receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J. Clin. Oncol. 2019, 37, 4124. [Google Scholar] [CrossRef]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef]
- Song, M.; Chen, D.; Lu, B.; Wang, C.; Zhang, J.; Huang, L.; Wang, X.; Timmons, C.L.; Hu, J.; Liu, B.; et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE 2013, 8, e65821. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, C.; Liu, F.; Zhao, Y.; Liu, J.; Hua, Y.; Liu, J.; Huang, J.; Ge, C. A blockade of PD-L1 produced antitumor and antimetastatic effects in an orthotopic mouse pancreatic cancer model via the PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2017, 10, 2115–2126. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Schnittert, J.; Bansal, R.; Prakash, J. Targeting Pancreatic Stellate Cells in Cancer. Trends Cancer 2019, 5, 128–142. [Google Scholar] [CrossRef]
- Tang, D.; Wang, D.; Yuan, Z.; Xue, X.; Zhang, Y.; An, Y.; Chen, J.; Tu, M.; Lu, Z.; Wei, J.; et al. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int. J. Cancer 2013, 132, 993–1003. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).