Polyglutamine-Expanded Androgen Receptor Alteration of Skeletal Muscle Homeostasis and Myonuclear Aggregation Are Affected by Sex, Age and Muscle Metabolism

 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Gene Copy Number Determination
2.3. Histological Analysis
2.4. Biochemical Analysis
2.5. Immunofluorescence Analysis
2.6. Quantitative Real-Time PCR Analysis
2.7. Statistical Analysis
3. Results
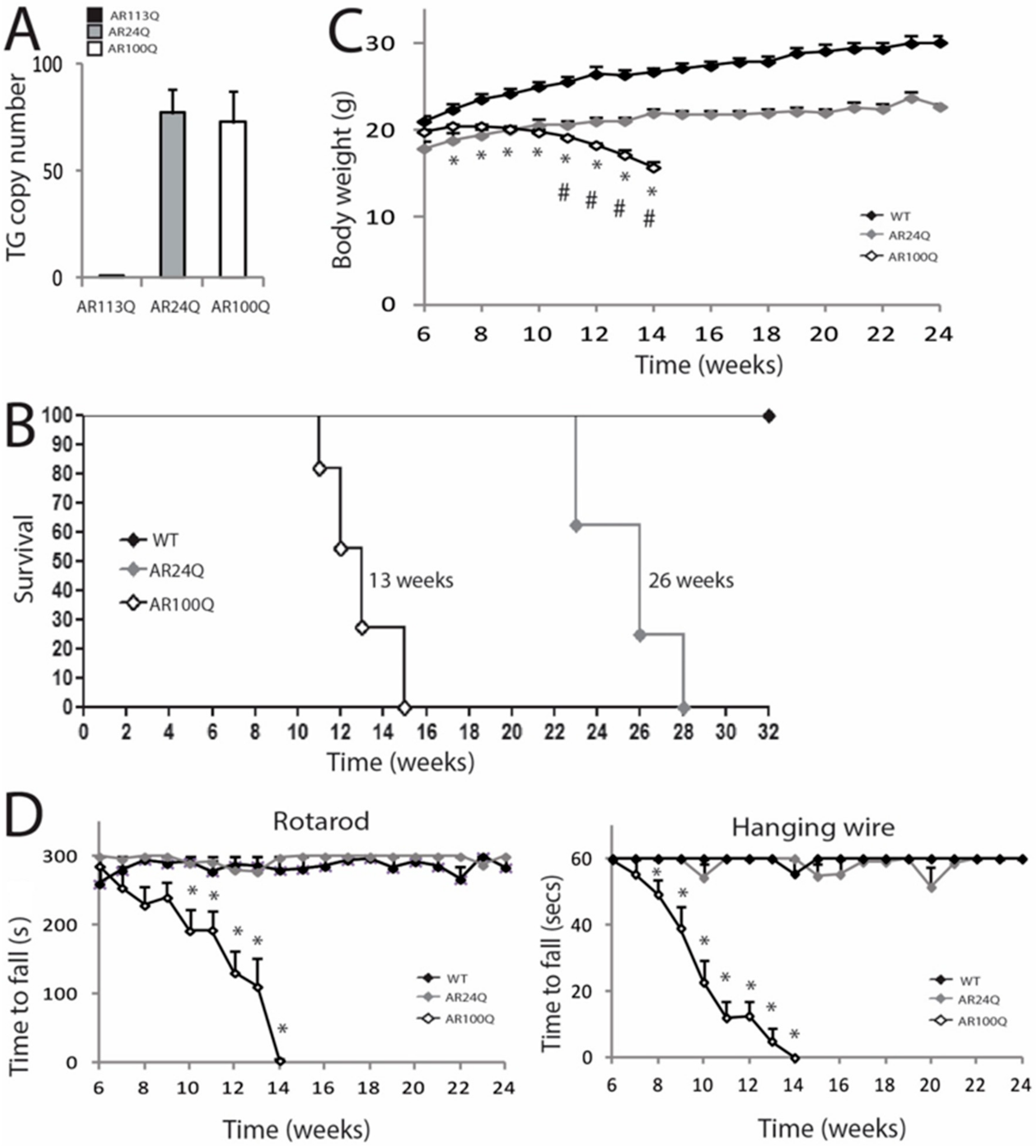
3.1. PolyQ-Expanded AR, but not Non-Expanded AR, Causes Motor Dysfunction
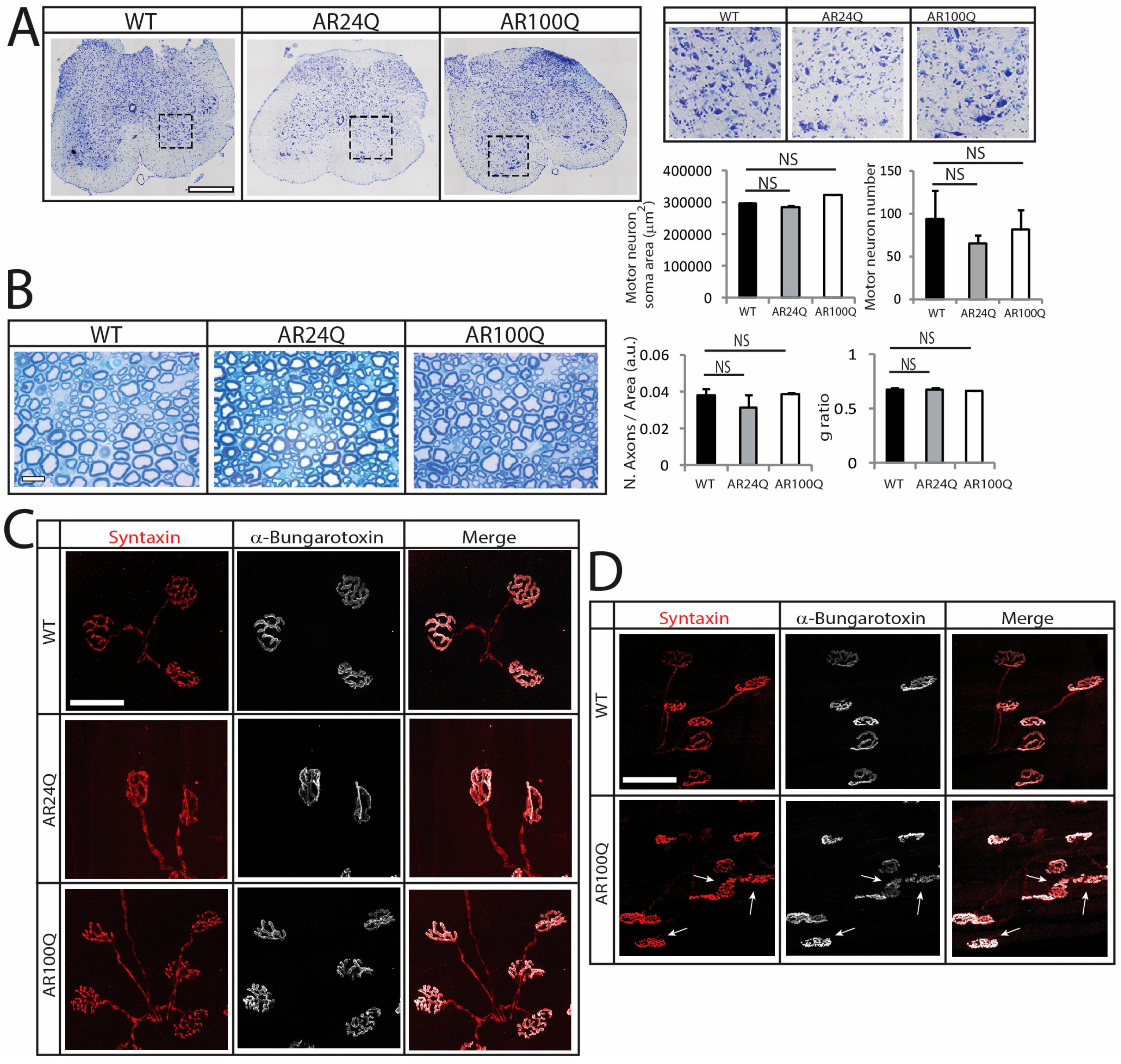
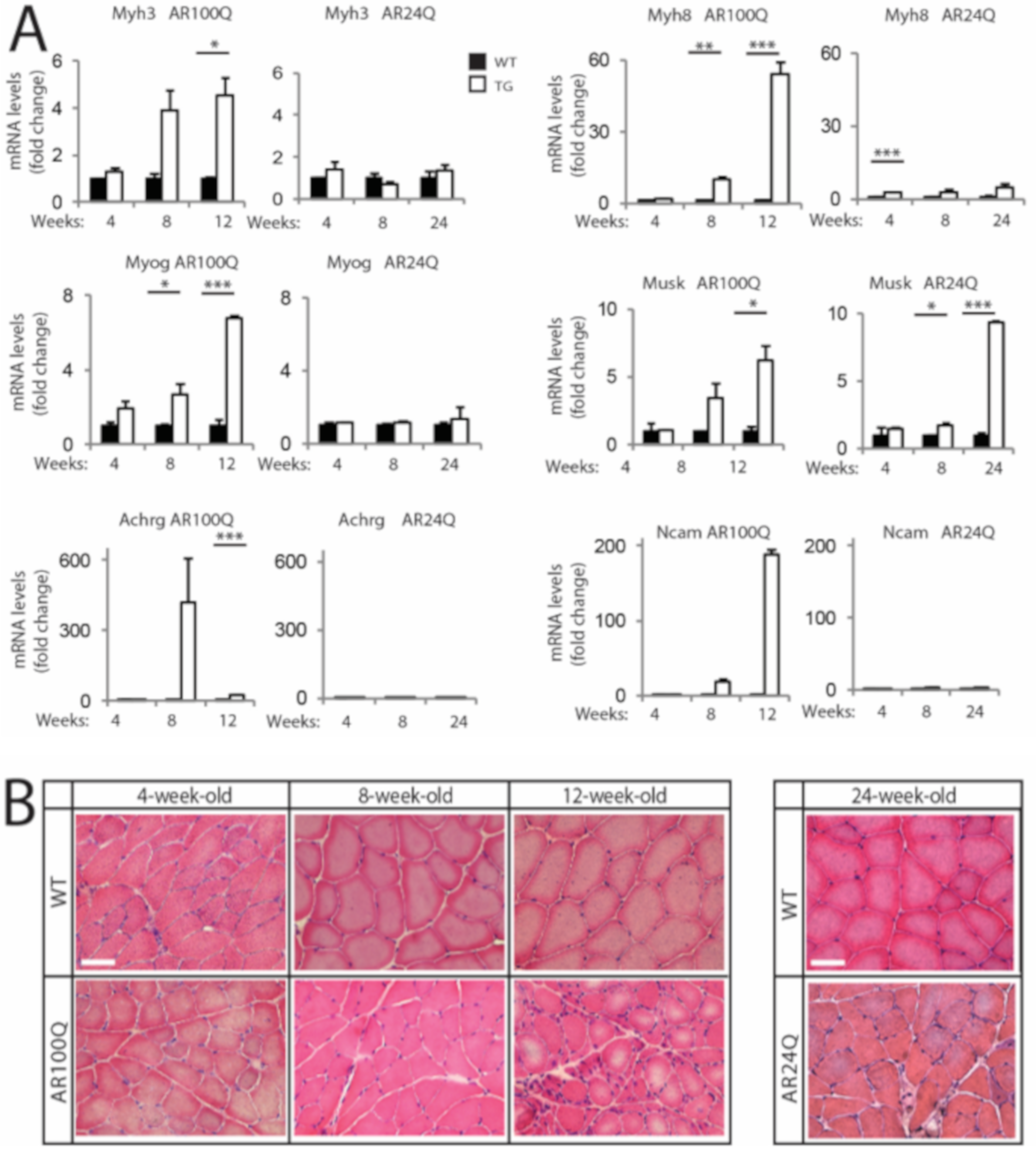
3.2. Denervation is a Late Event in SBMA Muscle
3.3. PolyQ-Expanded AR Alters Skeletal Muscle Homeostasis and Metabolism and Causes Mitochondrial Dysfunction
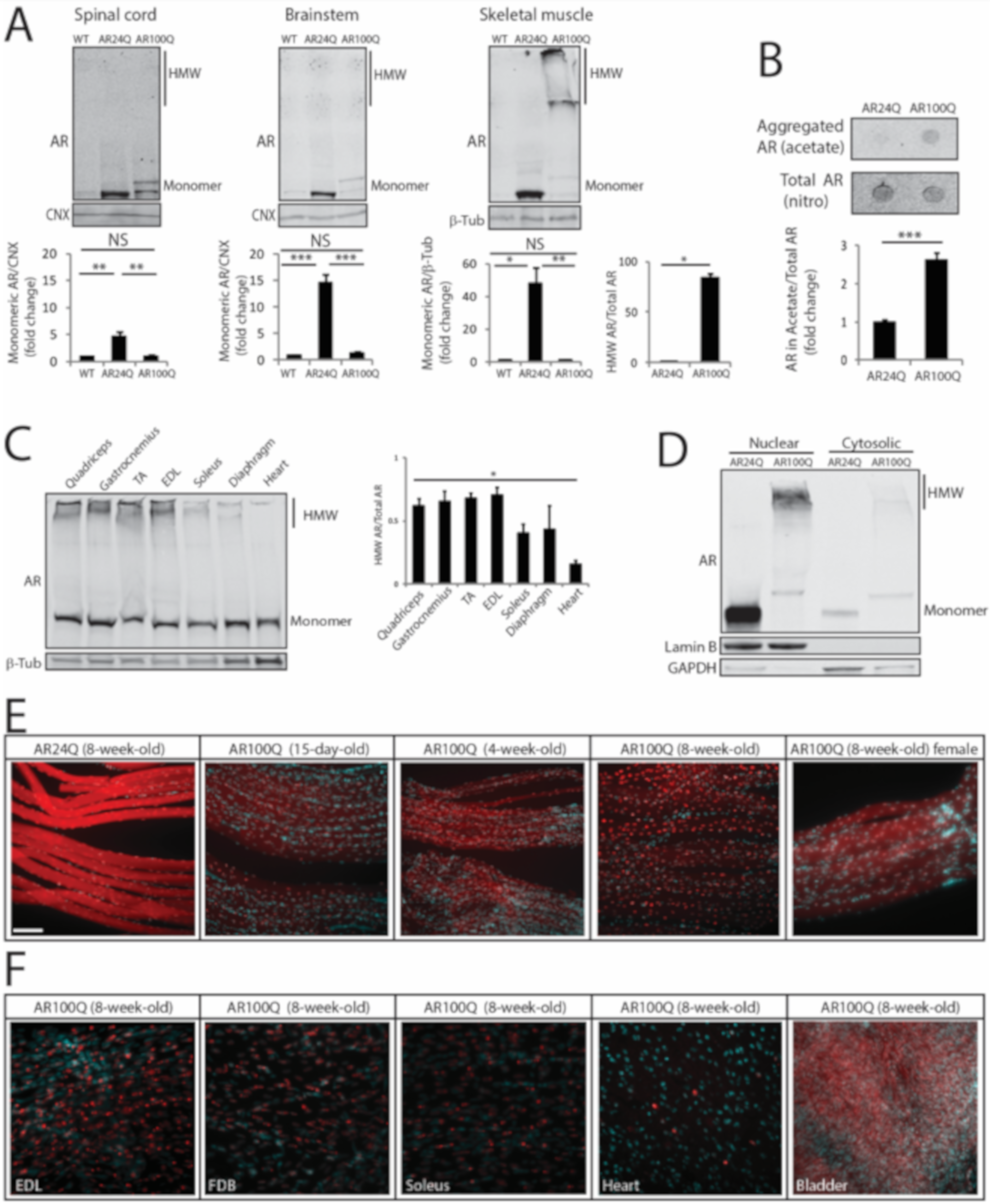
3.4. PolyQ Expansion Leads to 2% SDS-Resistant Aggregate and Inclusion Body Formation Selectively in Skeletal Muscle
3.5. Expression of PolyQ-Expanded AR in the Adulthood Elicits Some, but not All Aspects of Disease Manifestations in Mouse
4. Discussion
 IIa IIa/IIx IIx IIx/IIb) is endowed with mixed properties. The more pronounced fiber-type switch in MyHC types may contribute to the loss of muscle force and performance of SBMA mice. A major determinant for the plastic remodeling of myosin isoforms in adult muscles is denervation [48]. In aging and ALS, fiber-type switch correlates with the progressive death of fast-fatigable MNs. Loss of fast-fatigable MNs is compensated, at least at initial stages, through reinnervation of orphan fibers by slow fatigue-resistant MNs, a process achieved via compensatory sprouting leading to muscle fiber-type grouping [49,50]. Notably, although we did not find physical denervation of the myofibers, NMJ morphology progressively deteriorated in SBMA mice, indicating NMJ instability at the late stage of disease. Overall, this scenario supports a functional, rather than a structural, impairment of the motor unit likely causing aberrant communication between muscle and MN, ultimately contributing to the loss of motor performance. Nonetheless, in AR100Q mice fiber-type changes preceded denervation. These observations imply that all these pathological processes may result from intrinsic processes occurring in muscle and in other peripheral tissues, the nature of which remains to be clarified
IIa IIa/IIx IIx IIx/IIb) is endowed with mixed properties. The more pronounced fiber-type switch in MyHC types may contribute to the loss of muscle force and performance of SBMA mice. A major determinant for the plastic remodeling of myosin isoforms in adult muscles is denervation [48]. In aging and ALS, fiber-type switch correlates with the progressive death of fast-fatigable MNs. Loss of fast-fatigable MNs is compensated, at least at initial stages, through reinnervation of orphan fibers by slow fatigue-resistant MNs, a process achieved via compensatory sprouting leading to muscle fiber-type grouping [49,50]. Notably, although we did not find physical denervation of the myofibers, NMJ morphology progressively deteriorated in SBMA mice, indicating NMJ instability at the late stage of disease. Overall, this scenario supports a functional, rather than a structural, impairment of the motor unit likely causing aberrant communication between muscle and MN, ultimately contributing to the loss of motor performance. Nonetheless, in AR100Q mice fiber-type changes preceded denervation. These observations imply that all these pathological processes may result from intrinsic processes occurring in muscle and in other peripheral tissues, the nature of which remains to be clarifiedSupplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kennedy, W.R.; Alter, M.; Sung, J.H. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 1968, 18, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Guber, R.D.; Takyar, V.; Kokkinis, A.; Fox, D.A.; Alao, H.; Kats, I.; Bakar, D.; Remaley, A.T.; Hewitt, S.M.; Kleiner, D.E.; et al. Nonalcoholic fatty liver disease in spinal and bulbar muscular atrophy. Neurology 2017, 89, 2481–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, A.E.; Thomas, P.K.; Baraitser, M.; Bradbury, P.G.; Morgan-Hughes, J.A.; Ponsford, J.R. X-linked recessive bulbospinal neuronopathy: A report of ten cases. J. Neurol Neurosurg Psychiatry 1982, 45, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Katsuno, M.; Adachi, H.; Doyu, M.; Minamiyama, M.; Sang, C.; Kobayashi, Y.; Inukai, A.; Sobue, G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat. Med. 2003, 9, 768–773. [Google Scholar] [CrossRef]
- Katsuno, M.; Adachi, H.; Kume, A.; Li, M.; Nakagomi, Y.; Niwa, H.; Sang, C.; Kobayashi, Y.; Doyu, M.; Sobue, G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 2002, 35, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.J.; Greenberg, C.R.; Allingham-Hawkins, D.J.; Spriggs, E.L. Expression of X-linked bulbospinal muscular atrophy (Kennedy disease) in two homozygous women. Neurology 2002, 59, 770–772. [Google Scholar] [CrossRef]
- Yu, Z.; Dadgar, N.; Albertelli, M.; Gruis, K.; Jordan, C.; Robins, D.M.; Lieberman, A.P. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J. Clin. Invest. 2006, 116, 2663–2672. [Google Scholar] [CrossRef]
- Chevalier-Larsen, E.S.; O’Brien, C.J.; Wang, H.; Jenkins, S.C.; Holder, L.; Lieberman, A.P.; Merry, D.E. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J. Neurosci. 2004, 24, 4778–4786. [Google Scholar] [CrossRef] [Green Version]
- Querin, G.; Bertolin, C.; Da Re, E.; Volpe, M.; Zara, G.; Pegoraro, E.; Caretta, N.; Foresta, C.; Silvano, M.; Corrado, D.; et al. Non-neural phenotype of spinal and bulbar muscular atrophy: Results from a large cohort of Italian patients. J. Neurol. Neurosurg. Psychiatry 2016, 87, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yokota, K.; Amao, R.; Maeno, T.; Haga, N.; Taguri, M.; Ohtsu, H.; Ichikawa, Y.; Goto, J.; Tsuji, S. An open trial of long-term testosterone suppression in spinal and bulbar muscular atrophy. Muscle Nerve. 2013, 47, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Miwa, S.; Kobayashi, Y.; Merry, D.E.; Yamamoto, M.; Tanaka, F.; Doyu, M.; Hashizume, Y.; Fischbeck, K.H.; Sobue, G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann. Neurol. 1998, 44, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, M.; Adachi, H.; Waza, M.; Banno, H.; Suzuki, K.; Tanaka, F.; Doyu, M.; Sobue, G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA). Exp. Neurol. 2006, 200, 8–18. [Google Scholar] [CrossRef]
- Walcott, J.L.; Merry, D.E. Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J. Biol. Chem. 2002, 277, 50855–50859. [Google Scholar] [CrossRef] [Green Version]
- Palazzolo, I.; Nedelsky, N.B.; Askew, C.E.; Harmison, G.G.; Kasantsev, A.G.; Taylor, J.P.; Fischbeck, K.H.; Pennuto, M. B2 attenuates polyglutamine-expanded androgen receptor toxicity in cell and fly models of spinal and bulbar muscular atrophy. J. Neurosci. Res. 2010, 88, 2207–2216. [Google Scholar] [CrossRef] [Green Version]
- Pennuto, M.; Rinaldi, C. From gene to therapy in spinal and bulbar muscular atrophy: Are we there yet? Mol. Cell Endocrinol. 2018, 465, 113–121. [Google Scholar] [CrossRef]
- Sambataro, F.; Pennuto, M. Cell-autonomous and non-cell-autonomous toxicity in polyglutamine diseases. Prog. Neurobiol. 2012, 97, 152–172. [Google Scholar] [CrossRef]
- Cortes, C.J.; Ling, S.C.; Guo, L.T.; Hung, G.; Tsunemi, T.; Ly, L.; Tokunaga, S.; Lopez, E.; Sopher, B.L.; Bennett, C.F.; et al. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 2014, 82, 295–307. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, A.P.; Yu, Z.; Murray, S.; Peralta, R.; Low, A.; Guo, S.; Yu, X.X.; Cortes, C.J.; Bennett, C.F.; Monia, B.P.; et al. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014, 7, 774–784. [Google Scholar] [CrossRef] [Green Version]
- Rosenbohm, A.; Hirsch, S.; Volk, A.E.; Grehl, T.; Grosskreutz, J.; Hanisch, F.; Herrmann, A.; Kollewe, K.; Kress, W.; Meyer, T.; et al. The metabolic and endocrine characteristics in spinal and bulbar muscular atrophy. J. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Manzano, R.; Soraru, G.; Grunseich, C.; Fratta, P.; Zuccaro, E.; Pennuto, M.; Rinaldi, C. Beyond motor neurons: Expanding the clinical spectrum in Kennedy’s disease. J. Neurol Neurosurg Psychiatry 2018, 89, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Palazzolo, I.; Stack, C.; Kong, L.; Musaro, A.; Adachi, H.; Katsuno, M.; Sobue, G.; Taylor, J.P.; Sumner, C.J.; Fischbeck, K.H.; et al. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 2009, 63, 316–328. [Google Scholar] [CrossRef] [Green Version]
- Polanco, M.J.; Parodi, S.; Piol, D.; Stack, C.; Chivet, M.; Contestabile, A.; Miranda, H.C.; Lievens, P.M.; Espinoza, S.; Jochum, T.; et al. Adenylyl cyclase activating polypeptide reduces phosphorylation and toxicity of the polyglutamine-expanded androgen receptor in spinobulbar muscular atrophy. Sci. Transl. Med. 2016, 8, 370ra181. [Google Scholar] [CrossRef] [PubMed]
- Milioto, C.; Malena, A.; Maino, E.; Polanco, M.J.; Marchioretti, C.; Borgia, D.; Pereira, M.G.; Blaauw, B.; Lieberman, A.P.; Venturini, R.; et al. Beta-agonist stimulation ameliorates the phenotype of spinal and bulbar muscular atrophy mice and patient-derived myotubes. Sci. Rep. 2017, 7, 41046. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Rhodes, L.E.; Kokkinis, A.D.; White, M.J.; Watts, C.A.; Auh, S.; Jeffries, N.O.; Shrader, J.A.; Lehky, T.J.; Li, L.; Ryder, J.E.; et al. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: A randomised placebo-controlled trial. Lancet. Neurol. 2011, 10, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Pennuto, M.; Basso, M. In Vitro and In Vivo Modeling of Spinal and Bulbar Muscular Atrophy. J. Mol. Neurosci. 2016, 58, 365–373. [Google Scholar] [CrossRef]
- Rocchi, A.; Milioto, C.; Parodi, S.; Armirotti, A.; Borgia, D.; Pellegrini, M.; Urciuolo, A.; Molon, S.; Morbidoni, V.; Marabita, M.; et al. Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta. Neuropathol. 2016, 132, 127–144. [Google Scholar] [CrossRef]
- D’Haene, B.; Vandesompele, J.; Hellemans, J. Accurate and objective copy number profiling using real-time quantitative PCR. Methods 2010, 50, 262–270. [Google Scholar] [CrossRef]
- Borgia, D.; Malena, A.; Spinazzi, M.; Desbats, M.A.; Salviati, L.; Russell, A.P.; Miotto, G.; Tosatto, L.; Pegoraro, E.; Soraru, G.; et al. Increased mitophagy in the skeletal muscle of spinal and bulbar muscular atrophy patients. Hum. Mol. Genet. 2017, 26, 1087–1103. [Google Scholar] [CrossRef] [Green Version]
- Soraru, G.; D’Ascenzo, C.; Polo, A.; Palmieri, A.; Baggio, L.; Vergani, L.; Gellera, C.; Moretto, G.; Pegoraro, E.; Angelini, C. Spinal and bulbar muscular atrophy: Skeletal muscle pathology in male patients and heterozygous females. J. Neurol Sci. 2008, 264, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, E.; Yu, Z.; Chua, J.P.; Shimamura, R.; Zhao, L.; Zhu, F.; Venneti, S.; Pennuto, M.; Guan, Y.; Hung, G.; et al. Rescue of Metabolic Alterations in AR113Q Skeletal Muscle by Peripheral Androgen Receptor Gene Silencing. Cell Rep. 2016, 17, 125–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef]
- Yu, Z.; Wang, A.M.; Adachi, H.; Katsuno, M.; Sobue, G.; Yue, Z.; Robins, D.M.; Lieberman, A.P. Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet. 2011, 7, e1002321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, J.P.; Reddy, S.L.; Merry, D.E.; Adachi, H.; Katsuno, M.; Sobue, G.; Robins, D.M.; Lieberman, A.P. Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum. Mol. Genet. 2014, 23, 1376–1386. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, S.; Harmison, G.G.; Meyertholen, K.; Pennuto, M.; Burnett, B.G.; Fischbeck, K.H. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009, 18, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.J.; Knutson, T.M.; Colomer Gould, V.F.; Paulson, H.L. In vivo suppression of polyglutamine neurotoxicity by C-terminus of Hsp70-interacting protein (CHIP) supports an aggregation model of pathogenesis. Neurobiol. Dis. 2009, 33, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, T.; Polanco, M.J.; Scaramuzzino, C.; Rocchi, A.; Milioto, C.; Emionite, L.; Ognio, E.; Sambataro, F.; Galbiati, M.; Poletti, A.; et al. Androgens affect muscle, motor neuron and survival in a mouse model of SOD1-related amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 1929–1938. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kaneko, K.; Matsumoto, G.; Kurosawa, M.; Nukina, N. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J. Neurosci. 2009, 29, 5153–5162. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.R.; Montie, H.L.; Jain, P.; Legleiter, J.; Merry, D.E. Identification of novel polyglutamine-expanded aggregation species in spinal and bulbar muscular atrophy. Brain Res. 2015, 1628, 254–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochum, T.; Ritz, M.E.; Schuster, C.; Funderburk, S.F.; Jehle, K.; Schmitz, K.; Brinkmann, F.; Hirtz, M.; Moss, D.; Cato, A.C. Toxic and non-toxic aggregates from the SBMA and normal forms of androgen receptor have distinct oligomeric structures. Biochim. Biophys. Acta 2012, 1822, 1070–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, H.; Katsuno, M.; Minamiyama, M.; Waza, M.; Sang, C.; Nakagomi, Y.; Kobayashi, Y.; Tanaka, F.; Doyu, M.; Inukai, A.; et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain 2005, 128, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Kistner, A.; Gossen, M.; Zimmermann, F.; Jerecic, J.; Ullmer, C.; Lubbert, H.; Bujard, H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA 1996, 93, 10933–10938. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, C.; Bott, L.C.; Chen, K.L.; Harmison, G.G.; Katsuno, M.; Sobue, G.; Pennuto, M.; Fischbeck, K.H. Insulinlike growth factor (IGF)-1 administration ameliorates disease manifestations in a mouse model of spinal and bulbar muscular atrophy. Mol. Med. 2012, 18, 1261–1268. [Google Scholar] [CrossRef] [Green Version]
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423. [Google Scholar] [CrossRef]
- Schiaffino, S.; Reggiani, C. Fiber types in mammalian skeletal muscles. Physiol. Rev. 2011, 91, 1447–1531. [Google Scholar] [CrossRef] [Green Version]
- Gordon, T.; Hegedus, J.; Tam, S.L. Adaptive and maladaptive motor axonal sprouting in aging and motoneuron disease. Neurol. Res. 2004, 26, 174–185. [Google Scholar] [CrossRef]
- Hepple Russell, T.; Rice Charles, L. Innervation and neuromuscular control in ageing skeletal muscle. J. Physiol. 2015, 594, 1965–1978. [Google Scholar] [CrossRef]
- Cutress, M.L.; Whitaker, H.C.; Mills, I.G.; Stewart, M.; Neal, D.E. Structural basis for the nuclear import of the human androgen receptor. J. Cell Sci. 2008, 121, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Daigle, J.G.; Cunningham, K.M.; Coyne, A.N.; Ruan, K.; Grima, J.C.; Bowen, K.E.; Wadhwa, H.; Yang, P.; Rigo, F.; et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell 2018, 173, 958–971 e917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Kim, H.J.; Wang, H.; Monaghan, J.; Freyermuth, F.; Sung, J.C.; O’Donovan, K.; Fare, C.M.; Diaz, Z.; Singh, N.; et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell 2018, 173, 677–692 e620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, F.J.; Pluciennik, A.; Merry, D.E. Impaired Nuclear Export of Polyglutamine-Expanded Androgen Receptor in Spinal and Bulbar Muscular Atrophy. Sci. Rep. 2019, 9, 119. [Google Scholar] [CrossRef] [PubMed]
- Nedelsky, N.B.; Pennuto, M.; Smith, R.B.; Palazzolo, I.; Moore, J.; Nie, Z.; Neale, G.; Taylor, J.P. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron 2010, 67, 936–952. [Google Scholar] [CrossRef] [Green Version]
- Montie, H.L.; Cho, M.S.; Holder, L.; Liu, Y.; Tsvetkov, A.S.; Finkbeiner, S.; Merry, D.E. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009, 18, 1937–1950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzolo, I.; Burnett, B.G.; Young, J.E.; Brenne, P.L.; La Spada, A.R.; Fischbeck, K.H.; Howell, B.W.; Pennuto, M. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum. Mol. Genet. 2007, 16, 1593–1603. [Google Scholar] [CrossRef]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.B.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 1998, 95, 41–53. [Google Scholar] [CrossRef] [Green Version]
- Saudou, F.; Finkbeiner, S.; Devys, D.; Greenberg, M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 1998, 95, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Gidalevitz, T.; Ben-Zvi, A.; Ho, K.H.; Brignull, H.R.; Morimoto, R.I. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science 2006, 311, 1471–1474. [Google Scholar] [CrossRef] [Green Version]
- Van Oosten-Hawle, P.; Porter, R.S.; Morimoto, R.I. Regulation of organismal proteostasis by transcellular chaperone signaling. Cell 2013, 153, 1366–1378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, C.; Mager, I.; Wood, M.J. Proteostasis and Diseases of the Motor Unit. Front. Mol. Neurosci. 2016, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeoni, S.; Mancini, M.A.; Stenoien, D.L.; Marcelli, M.; Weigel, N.L.; Zanisi, M.; Martini, L.; Poletti, A. Motoneuronal cell death is not correlated with aggregate formation of androgen receptors containing an elongated polyglutamine tract. Hum. Mol. Genet. 2000, 9, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, H.; Kume, A.; Li, M.; Nakagomi, Y.; Niwa, H.; Do, J.; Sang, C.; Kobayashi, Y.; Doyu, M.; Sobue, G. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum. Mol. Genet. 2001, 10, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Abel, A.; Walcott, J.; Woods, J.; Duda, J.; Merry, D.E. Expression of expanded repeat androgen receptor produces neurologic disease in transgenic mice. Hum. Mol. Genet. 2001, 10, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Tompkins, M.M.; Basgall, E.J.; Zamrini, E.; Hill, W.D. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am. J. Pathol. 1997, 150, 119–131. [Google Scholar]
- Bondareff, W.; Mountjoy, C.Q.; Roth, M.; Hauser, D.L. Neurofibrillary degeneration and neuronal loss in Alzheimer’s disease. Neurobiol. Aging 1989, 10, 709–715. [Google Scholar] [CrossRef]
- Kuemmerle, S.; Gutekunst, C.A.; Klein, A.M.; Li, X.J.; Li, S.H.; Beal, M.F.; Hersch, S.M.; Ferrante, R.J. Huntington aggregates may not predict neuronal death in Huntington’s disease. Ann. Neurol. 1999, 46, 842–849. [Google Scholar] [CrossRef]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, A.; Del Mar, N.; Deng, Y.P.; Meade, C.A.; Sun, Z.; Goldowitz, D. R6/2 neurons with intranuclear inclusions survive for prolonged periods in the brains of chimeric mice. J. Comp. Neurol. 2007, 505, 603–629. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chivet, M.; Marchioretti, C.; Pirazzini, M.; Piol, D.; Scaramuzzino, C.; Polanco, M.J.; Romanello, V.; Zuccaro, E.; Parodi, S.; D’Antonio, M.; et al. Polyglutamine-Expanded Androgen Receptor Alteration of Skeletal Muscle Homeostasis and Myonuclear Aggregation Are Affected by Sex, Age and Muscle Metabolism. Cells 2020, 9, 325. https://doi.org/10.3390/cells9020325
Chivet M, Marchioretti C, Pirazzini M, Piol D, Scaramuzzino C, Polanco MJ, Romanello V, Zuccaro E, Parodi S, D’Antonio M, et al. Polyglutamine-Expanded Androgen Receptor Alteration of Skeletal Muscle Homeostasis and Myonuclear Aggregation Are Affected by Sex, Age and Muscle Metabolism. Cells. 2020; 9(2):325. https://doi.org/10.3390/cells9020325
Chicago/Turabian StyleChivet, Mathilde, Caterina Marchioretti, Marco Pirazzini, Diana Piol, Chiara Scaramuzzino, Maria Josè Polanco, Vanina Romanello, Emanuela Zuccaro, Sara Parodi, Maurizio D’Antonio, and et al. 2020. "Polyglutamine-Expanded Androgen Receptor Alteration of Skeletal Muscle Homeostasis and Myonuclear Aggregation Are Affected by Sex, Age and Muscle Metabolism" Cells 9, no. 2: 325. https://doi.org/10.3390/cells9020325