Mitophagy in Acute Kidney Injury and Kidney Repair


Abstract
:1. Introduction
2. Mitochondrial Pathology in AKI Development and Abnormal Repair
3. Overviews of Autophagy
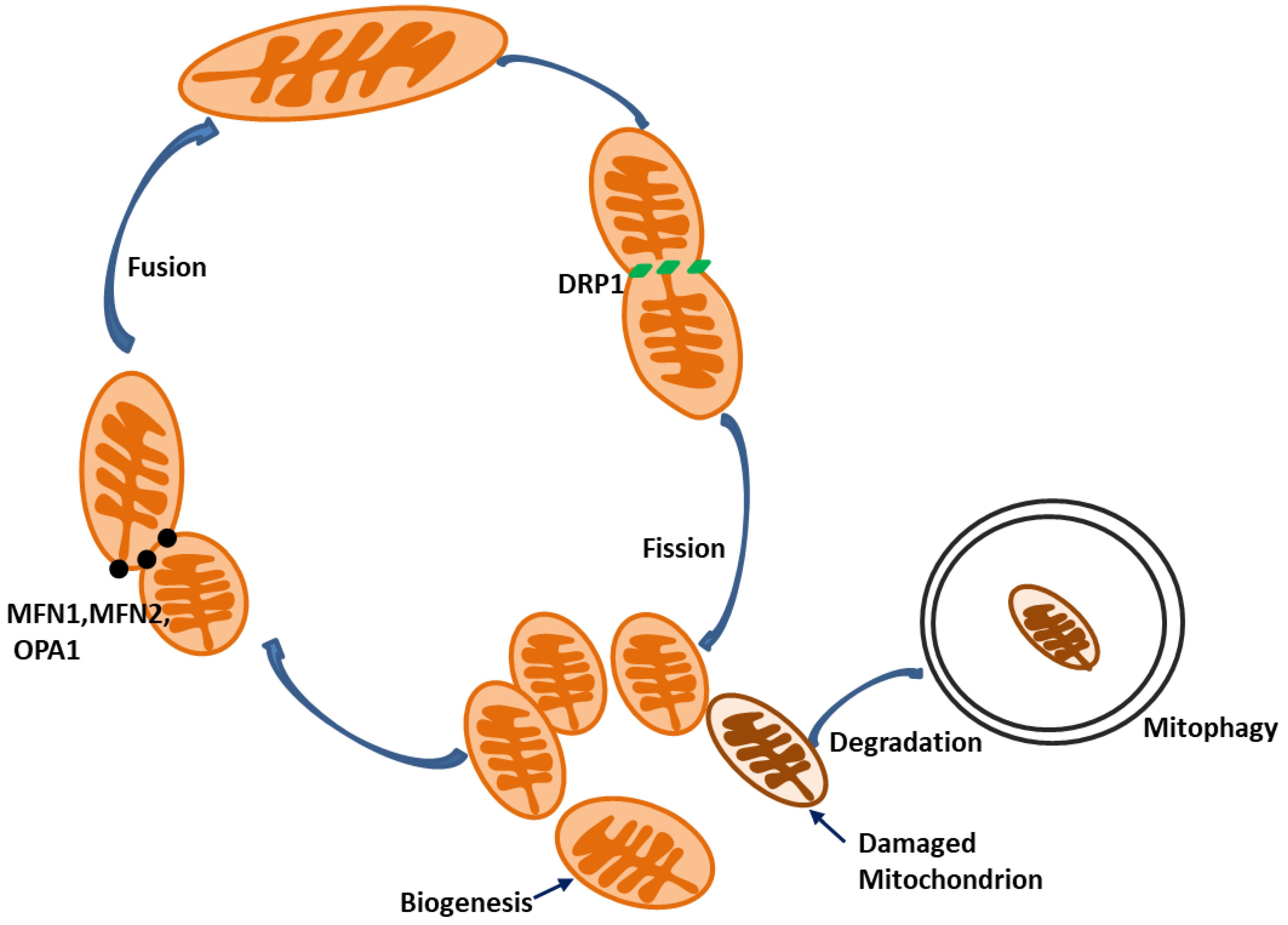
4. Mitophagy
4.1. PINK1-PARK2 Pathway of Mitophagy
4.2. BNIP3 and NIX Mediated Mitophagy
4.3. FUNDC1-Mediated Mitophagy
5. Mitochondrial Fission and Fusion
6. Mitochondrial Biogenesis
7. Mitophagy in AKI
7.1. Mitophagy in Renal Ischemia–Reperfusion Injury
7.2. Mitophagy in Nephrotoxin-Induced AKI
7.3. Mitophagy in Sepsis-Associated AKI
7.4. Mitophagy in Other Types of AKI
8. Mitophagy in Kidney Repair and Renal Fibrosis
9. Conclusions
Funding
Conflicts of Interest
References
- Schneider, A.G.; Bellomo, R.; Bagshaw, S.M.; Glassford, N.J.; Lo, S.; Jun, M.; Cass, A.; Gallagher, M. Choice of renal replacement therapy modality and dialysis dependence after acute kidney injury: A systematic review and meta-analysis. Intensiv. Care Med. 2013, 39, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Bucaloiu, I.D.; Kirchner, H.L.; Norfolk, E.R.; Hartle, J.E., 2nd; Perkins, R.M. Increased risk of death and de novo chronic kidney disease following reversible acute kidney injury. Kidney Int. 2012, 81, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Li, X.; Zhang, D.; Chen, J.K.; Su, Y.; Smith, S.B.; Dong, Z. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int. 2015, 87, 137–150. [Google Scholar] [CrossRef]
- Sancho-Martinez, S.M.; Lopez-Novoa, J.M.; Lopez-Hernandez, F.J. Pathophysiological role of different tubular epithelial cell death modes in acute kidney injury. Clin. Kidney J. 2015, 8, 548–559. [Google Scholar] [CrossRef]
- Ferenbach, D.A.; Bonventre, J.V. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 2015, 11, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Bravi, C.A.; Vertosick, E.; Benfante, N.; Tin, A.; Sjoberg, D.; Hakimi, A.A.; Touijer, K.; Montorsi, F.; Eastham, J.; Russo, P.; et al. Impact of Acute Kidney Injury and Its Duration on Long-term Renal Function After Partial Nephrectomy. Eur. Urol. 2019, 76, 398–403. [Google Scholar] [CrossRef]
- Forni, L.G.; Darmon, M.; Ostermann, M.; Oudemans-van Straaten, H.M.; Pettila, V.; Prowle, J.R.; Schetz, M.; Joannidis, M. Renal recovery after acute kidney injury. Care Med. 2017, 43, 855–866. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Li, L.; Ying, Z.; Cao, Z.; Ma, Y.; Mao, X.; Li, J.; Qi, X.; Zhang, Z.; Wang, X. A small molecule protects mitochondrial integrity by inhibiting mTOR activity. Proc. Natl. Acad. Sci. USA 2019, 116, 23332–23338. [Google Scholar] [CrossRef]
- Chen, H.H.; Chen, Y.T.; Yang, C.C.; Chen, K.H.; Sung, P.H.; Chiang, H.J.; Chen, C.H.; Chua, S.; Chung, S.Y.; Chen, Y.L.; et al. Melatonin pretreatment enhances the therapeutic effects of exogenous mitochondria against hepatic ischemia-reperfusion injury in rats through suppression of mitochondrial permeability transition. J. Pineal Res. 2016, 61, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Meka, D.P.; Muller-Rischart, A.K.; Nidadavolu, P.; Mohammadi, B.; Motori, E.; Ponna, S.K.; Aboutalebi, H.; Bassal, M.; Annamneedi, A.; Finckh, B.; et al. Parkin cooperates with GDNF/RET signaling to prevent dopaminergic neuron degeneration. J. Clin. Investig. 2015, 125, 1873–1885. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jedrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; He, L.; Liu, J.; Dong, Z. Mitophagy: Basic Mechanism and Potential Role in Kidney Diseases. Kidney Dis. (Basel) 2015, 1, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef]
- Busquets-Cortes, C.; Capo, X.; Bibiloni, M.D.M.; Martorell, M. Peripheral Blood Mononuclear Cells Antioxidant Adaptations to Regular Physical Activity in Elderly People. Nutrients 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Harper, J.W.; Ordureau, A.; Heo, J.M. Building and decoding ubiquitin chains for mitophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 93–108. [Google Scholar] [CrossRef]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 2019, 16, 99–111. [Google Scholar] [CrossRef]
- Xiao, X.; Hu, Y.; Quiros, P.M.; Wei, Q.; Lopez-Otin, C.; Dong, Z. OMA1 mediates OPA1 proteolysis and mitochondrial fragmentation in experimental models of ischemic kidney injury. Am. J. Physiol. Renal Physiol. 2014, 306, F1318–F1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parikh, S.M.; Yang, Y.; He, L.; Tang, C.; Zhan, M.; Dong, Z. Mitochondrial function and disturbances in the septic kidney. Semin Nephrol. 2015, 35, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar] [PubMed]
- Yang, Y.; Liu, H.; Liu, F.; Dong, Z. Mitochondrial dysregulation and protection in cisplatin nephrotoxicity. Arch. Toxicol. 2014, 88, 1249–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, M.; Brooks, C.; Liu, F.; Sun, L.; Dong, Z. Mitochondrial dynamics: Regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int. 2013, 83, 568–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.; Dong, Z.; Harris, R.; Murray, P.; Parikh, S.M.; Rosner, M.H.; Kellum, J.A.; Ronco, C. Cellular and Molecular Mechanisms of AKI. J. Am. Soc. Nephrol. 2016, 27, 1288–1299. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Funk, J.A.; Schnellmann, R.G. Persistent disruption of mitochondrial homeostasis after acute kidney injury. Am. J. Physiol. Renal Physiol. 2012, 302, F853–F864. [Google Scholar] [CrossRef] [Green Version]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Investig. 2009, 119, 1275–1285. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Tran, M.; Tam, D.; Bardia, A.; Bhasin, M.; Rowe, G.C.; Kher, A.; Zsengeller, Z.K.; Akhavan-Sharif, M.R.; Khankin, E.V.; Saintgeniez, M.; et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Investig. 2011, 121, 4003–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, H.M.; Huang, L.; Wilson, R.J.; Bajwa, A.; Sesaki, H.; Yan, Z.; Rosin, D.L.; Kashatus, D.F.; Okusa, M.D. Dynamin-Related Protein 1 Deficiency Promotes Recovery from AKI. J. Am. Soc. Nephrol. 2018, 29, 194–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, H.H.; Liu, S.; Soong, Y.; Seshan, S.V.; Cohen-Gould, L.; Manichev, V.; Feldman, L.C.; Gustafsson, T. Mitochondria Protection after Acute Ischemia Prevents Prolonged Upregulation of IL-1beta and IL-18 and Arrests CKD. J. Am. Soc. Nephrol. 2017, 28, 1437–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, W.S.; Collier, J.B.; Morris, M.; Beeson, C.C.; Megyesi, J.; Schnellmann, R.G. 5-HT1F receptor regulates mitochondrial homeostasis and its loss potentiates acute kidney injury and impairs renal recovery. Am. J. Physiol. Renal Physiol. 2018, 315, F1119–F1128. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Zhong, X.; He, J.; Zhang, X.; Li, C.; Tian, X.; Xia, W.; Gan, H.; Xia, Y. UCP2 alleviates tubular epithelial cell apoptosis in lipopolysaccharide-induced acute kidney injury by decreasing ROS production. Biomed. Pharmacother. 2019, 115, 108914. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, E.Y.; Pevzner, I.B.; Zorova, L.D.; Chernikov, V.P.; Prusov, A.N.; Kireev, I.I.; Silachev, D.N.; Skulachev, V.P.; Zorov, D.B. Mitochondrial Damage and Mitochondria-Targeted Antioxidant Protection in LPS-Induced Acute Kidney Injury. Antioxidants 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Kong, M.J.; Bak, S.H.; Han, K.H.; Kim, J.I.; Park, J.W.; Park, K.M. Fragmentation of kidney epithelial cell primary cilia occurs by cisplatin and these cilia fragments are excreted into the urine. Redox Biol. 2019, 20, 38–45. [Google Scholar] [CrossRef]
- Dare, A.J.; Bolton, E.A.; Pettigrew, G.J.; Bradley, J.A.; Saeb-Parsy, K.; Murphy, M.P. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015, 5, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Jin, F.; Shu, G.; Xu, X.; Qi, J.; Kang, X.; Yu, H.; Lu, K.; Jiang, S.; Han, F.; et al. Enhanced efficiency of mitochondria-targeted peptide SS-31 for acute kidney injury by pH-responsive and AKI-kidney targeted nanopolyplexes. Biomaterials 2019, 211, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.K.; Kondeti, V.K.; Sharma, I.; Chandel, N.S.; Quaggin, S.E.; Kanwar, Y.S. Beneficial Effects of Myo-Inositol Oxygenase Deficiency in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2017, 28, 1421–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.C.; Liang, C.J.; Huang, T.M.; Liu, C.W.; Wang, S.H.; Young, G.H.; Tsai, J.S.; Tseng, Y.C.; Peng, Y.S.; Wu, V.C.; et al. Indoxyl sulfate enhances IL-1beta-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFkappaB/AP-1 pathway. Arch. Toxicol. 2016, 90, 2779–2792. [Google Scholar] [CrossRef] [PubMed]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-kappaB pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Han, H.; Yan, M.; Zhu, S.; Liu, J.; Liu, Z.; He, L.; Tan, J.; Liu, Y.; Liu, H.; et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018, 14, 880–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotzer, V.L.; Blum, J.S. Autophagy and adaptive immunity. Immunology 2010, 131, 9–17. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Wong, P.M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 complex: Sensing nutrient signals for autophagy activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Klionsky, D.J. An overview of the molecular mechanism of autophagy. Curr. Top MicroBiol. Immunol. 2009, 335, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. NeuroBiol. 2013, 106-107, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Sanchez, R.; Rose, J.; Guimaraes, R.; Mari, M.; Papinski, D.; Rieter, E.; Geerts, W.J.; Hardenberg, R.; Kraft, C. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell Biol. 2018, 217, 2743–2763. [Google Scholar] [CrossRef]
- Choi, J.; Park, S.; Biering, S.B.; Selleck, E.; Liu, C.Y.; Zhang, X.; Fujita, N.; Saitoh, T.; Akira, S.; Yoshimori, T.; et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity 2014, 40, 924–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury. Semin Nephrol. 2014, 34, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Knuppertz, L.; Osiewacz, H.D. Orchestrating the network of molecular pathways affecting aging: Role of nonselective autophagy and mitophagy. Mech. Ageing Dev. 2016, 153, 30–40. [Google Scholar] [CrossRef]
- Guimaraes, R.S.; Delorme-Axford, E.; Klionsky, D.J.; Reggiori, F. Assays for the biochemical and ultrastructural measurement of selective and nonselective types of autophagy in the yeast Saccharomyces cerevisiae. Methods 2015, 75, 141–150. [Google Scholar] [CrossRef]
- Mizumura, K.; Choi, A.M.; Ryter, S.W. Emerging role of selective autophagy in human diseases. Front. Pharmacol. 2014, 5, 244. [Google Scholar] [CrossRef] [Green Version]
- Yoshii, S.R.; Mizushima, N. Autophagy machinery in the context of mammalian mitophagy. Biochim. Biophys. Acta 2015, 1853, 2797–2801. [Google Scholar] [CrossRef] [Green Version]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Li, G.; Liu, L.; Feng, L.; Wang, X.; Jin, H. Regulation and function of mitophagy in development and cancer. Autophagy 2013, 9, 1720–1736. [Google Scholar] [CrossRef] [PubMed]
- Diwan, A.; Wansapura, J.; Syed, F.M.; Matkovich, S.J.; Lorenz, J.N.; Dorn, G.W., 2nd. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation 2008, 117, 396–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015, 25, 158–170. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Wang, M.; Ruan, Y.; Tan, J.; Wang, H.; Yang, T.; Li, J.; Zhou, Q. Ginkgolic Acids Impair Mitochondrial Function by Decreasing Mitochondrial Biogenesis and Promoting FUNDC1-Dependent Mitophagy. J. Agric. Food Chem. 2019, 67, 10097–10106. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Riley, B.E.; Lougheed, J.C.; Callaway, K.; Velasquez, M.; Brecht, E.; Nguyen, L.; Shaler, T.; Walker, D.; Yang, Y.; Regnstrom, K.; et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 2013, 4, 1982. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Cizeau, J.; Vande Velde, C.; Park, J.H.; Bozek, G.; Bolton, J.; Shi, L.; Dubik, D.; Greenberg, A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J. Biol. Chem. 1999, 274, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweers, R.L.; Zhang, J.; Randall, M.S.; Loyd, M.R.; Li, W.; Dorsey, F.C.; Kundu, M.; Opferman, J.T.; Cleveland, J.L.; Miller, J.L.; et al. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc. Natl. Acad. Sci. USA 2007, 104, 19500–19505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ney, P.A. NIX induces mitochondrial autophagy in reticulocytes. Autophagy 2008, 4, 354–356. [Google Scholar] [CrossRef]
- Esteban-Martinez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2018, 14, 915–917. [Google Scholar] [CrossRef]
- Hu, L.; Wang, H.; Huang, L.; Zhao, Y.; Wang, J. The Protective Roles of ROS-Mediated Mitophagy on (125)I Seeds Radiation Induced Cell Death in HCT116 Cells. Oxidative Med. Cell. Longev. 2016, 2016, 9460462. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.Y.; Swanson, J.; You, M.; Guan, K.L. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Siraj, S.; Zhang, R.; Chen, Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy 2017, 13, 1080–1081. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Wang, C.; Li, F.; Peng, J.; Wen, B.; Gong, Q.; Shi, Y.; Tang, Y. Structural insights into the recognition of phosphorylated FUNDC1 by LC3B in mitophagy. Protein Cell 2017, 8, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Zhang, D.; Zhou, L.; Pei, Y.; Zhuang, Y.; Cui, W.; Chen, J. FUN14 domain-containing 1 promotes breast cancer proliferation and migration by activating calcium-NFATC1-BMI1 axis. EBioMedicine 2019, 41, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Chen, Z.; Wang, Y.; Tan, Z.; Zhu, C.; Li, Y.; Han, Z.; Chen, L.; Gao, R.; Liu, L.; et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy 2016, 12, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Sugo, M.; Kimura, H.; Arasaki, K. Syntaxin 17 regulates the localization and function of PGAM5 in mitochondrial division and mitophagy. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Putti, R.; Sica, R.; Migliaccio, V.; Lionetti, L. Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol. 2015, 6, 109. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Zhao, Y.; Gong, J.; Huang, W.; Su, H.; Yuan, F.; Fang, K.; Wang, D.; Li, J.; Zou, X.; et al. Berberine Protects Glomerular Podocytes via Inhibiting Drp1-Mediated Mitochondrial Fission and Dysfunction. Theranostics 2019, 9, 1698–1713. [Google Scholar] [CrossRef]
- Toda, C.; Kim, J.D.; Impellizzeri, D.; Cuzzocrea, S.; Liu, Z.W.; Diano, S. UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell 2016, 164, 872–883. [Google Scholar] [CrossRef]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Ong, S.B.; Kalkhoran, S.B.; Hernandez-Resendiz, S.; Samangouei, P.; Ong, S.G.; Hausenloy, D.J. Mitochondrial-Shaping Proteins in Cardiac Health and Disease—The Long and the Short of It! Cardiovasc. Drugs Ther. 2017, 31, 87–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samangouei, P.; Crespo-Avilan, G.E.; Cabrera-Fuentes, H.; Hernandez-Resendiz, S.; Ismail, N.I.; Katwadi, K.B.; Boisvert, W.A.; Hausenloy, D.J. MiD49 and MiD51: New mediators of mitochondrial fission and novel targets for cardioprotection. Cond. Med. 2018, 1, 239–246. [Google Scholar] [PubMed]
- Zhao, C.; Chen, Z.; Qi, J.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zeng, M.; Zhang, B.; Wang, N.; et al. Drp1-dependent mitophagy protects against cisplatin-induced apoptosis of renal tubular epithelial cells by improving mitochondrial function. Oncotarget 2017, 8, 20988–21000. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Wang, H.; Jiang, C.; Zhang, M. Renal ischemia/reperfusion-induced mitophagy protects against renal dysfunction via Drp1-dependent-pathway. Exp. Cell Res. 2018, 369, 27–33. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W., 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Bockler, S.; Westermann, B. ER-mitochondria contacts as sites of mitophagosome formation. Autophagy 2014, 10, 1346–1347. [Google Scholar] [CrossRef] [Green Version]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Yang, M.; Linn, B.S.; Zhang, Y.; Ren, J. Mitophagy and mitochondrial integrity in cardiac ischemia-reperfusion injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2293–2302. [Google Scholar] [CrossRef]
- Duann, P.; Lin, P.H. Mitochondria Damage and Kidney Disease. Adv. Exp. Med. Biol. 2017, 982, 529–551. [Google Scholar] [CrossRef]
- Nah, J.; Miyamoto, S.; Sadoshima, J. Mitophagy as a Protective Mechanism against Myocardial Stress. Compr. Physiol. 2017, 7, 1407–1424. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.; Parikh, S.M. Mitochondrial biogenesis in the acutely injured kidney. Nephron. Clin. Pract. 2014, 127, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Zhang, J.; Wang, J.; Yan, T.; Xu, F.; Wu, B.; Xiao, F.; Bi, K.; Niu, J.; Jia, Y. The anti-nephritic activity of a polysaccharide from okra (Abelmoschus esculentus (L.) Moench) via modulation of AMPK-Sirt1-PGC-1alpha signaling axis mediated anti-oxidative in type 2 diabetes model mice. Int. J. Biol. Macromol. 2019, 140, 568–576. [Google Scholar] [CrossRef]
- Li, S.Y.; Susztak, K. The Role of Peroxisome Proliferator-Activated Receptor gamma Coactivator 1alpha (PGC-1alpha) in Kidney Disease. Semin Nephrol. 2018, 38, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Cheng, X.-T.; Lin, M.-Y.; Huang, N.; Sheng, Z.-H. Defending stressed mitochondria: Uncovering the role of MUL1 in suppressing neuronal mitophagy. Autophagy 2019, 16, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Renal Physiol. 2013, 305, F495–F509. [Google Scholar] [CrossRef]
- Livingston, M.J.; Wang, J.; Zhou, J. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy 2019, 15, 2142–2162. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.; Han, H.; Liu, Z.; Liu, Y.; Yin, L.; Cai, J.; He, L.; Liu, Y.; Chen, G.; Zhang, Z.; et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019, 10, 677. [Google Scholar] [CrossRef]
- Feng, J.; Li, H.; Zhang, Y.; Wang, Q.; Zhao, S.; Meng, P.; Li, J. Mammalian STE20-Like Kinase 1 Deletion Alleviates Renal Ischaemia-Reperfusion Injury via Modulating Mitophagy and the AMPK-YAP Signalling Pathway. Cell Physiol. Biochem. 2018, 51, 2359–2376. [Google Scholar] [CrossRef]
- Zhao, C.; Chen, Z.; Xu, X.; An, X.; Duan, S.; Huang, Z.; Zhang, C.; Wu, L.; Zhang, B.; Zhang, A.; et al. Pink1/Parkin-mediated mitophagy play a protective role in cisplatin induced renal tubular epithelial cells injury. Exp. Cell Res. 2017, 350, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, C.; Cai, J.; Chen, G.; Zhang, D.; Zhang, Z.; Dong, Z. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018, 9, 1113. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zhang, L.; Zhang, Y.; Yu, X.; Sun, X.; Zhu, T.; Li, X.; Liang, W.; Han, Y.; Qin, C. PINK1 Deficiency Ameliorates Cisplatin-Induced Acute Kidney Injury in Rats. Front. Physiol. 2019, 10, 1225. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Yang, Y.; Huang, Z.; Zhou, J.; Li, Y.; Zhong, X. Panax notoginseng saponins mitigate cisplatin induced nephrotoxicity by inducing mitophagy via HIF-1alpha. Oncotarget 2017, 8, 102989–103003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.G.; Xu, W.; Li, T.; Lu, J.Y.; Yang, Y.; Li, Q.; Zeng, Z.H.; Ai, Y.H. Involvement of phosphatase and tensin homolog-induced putative kinase 1-Parkin-mediated mitophagy in septic acute kidney injury. Chin. Med. J. 2019, 132, 2340–2347. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yan, X.; Yang, D.; Zhou, J.; Song, J.; Yang, D. Rapamycin attenuates mitochondrial injury and renal tubular cell apoptosis in experimental contrast-induced acute kidney injury in rats. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Y.; Xu, J.; Tian, F.; Hu, S.; Chen, Y. Melatonin attenuates myocardial ischemia-reperfusion injury via improving mitochondrial fusion/mitophagy and activating the AMPK-OPA1 signaling pathways. J. Pineal Res. 2019, 66, e12542. [Google Scholar] [CrossRef]
- Bastin, A.J.; Ostermann, M.; Slack, A.J.; Diller, G.P.; Finney, S.J.; Evans, T.W. Acute kidney injury after cardiac surgery according to Risk/Injury/Failure/Loss/End-stage, Acute Kidney Injury Network, and Kidney Disease: Improving Global Outcomes classifications. J. Crit. Care 2013, 28, 389–396. [Google Scholar] [CrossRef]
- Wei, L.; Qin, Y.; Jiang, L.; Yu, X.; Xi, Z. PPARgamma and mitophagy are involved in hypoxia/reoxygenation-induced renal tubular epithelial cells injury. J. Recept. Signal Transduct. Res. 2019, 39, 235–242. [Google Scholar] [CrossRef]
- Doi, K. Role of kidney injury in sepsis. J. Intensive Care 2016, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Takasu, O.; Gaut, J.P.; Watanabe, E.; To, K.; Fagley, R.E.; Sato, B.; Jarman, S.; Efimov, I.R.; Janks, D.L.; Srivastava, A.; et al. Mechanisms of cardiac and renal dysfunction in patients dying of sepsis. Am. J. Respir. Crit. Care Med. 2013, 187, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.X.; Yang, C.; Zhang, W.H.; Su, H.Y.; Liu, Z.J.; Pan, Q.; Liu, H.F. Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. 2019, 235, 116828. [Google Scholar] [CrossRef] [PubMed]
- Fahling, M.; Seeliger, E.; Patzak, A.; Persson, P.B. Understanding and preventing contrast-induced acute kidney injury. Nat. Rev. Nephrol. 2017, 13, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Zhao, F.; Tang, C.Y.; Luo, M.; Yang, S.K.; Cheng, W.; Li, X.W.; Duan, S.B. Mitophagy Plays a Protective Role in Iodinated Contrast-Induced Acute Renal Tubular Epithelial Cells Injury. Cell Physiol. Biochem. 2018, 46, 975–985. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Zhao, F.; Tang, C.Y.; Li, X.W.; Luo, M.; Duan, S.B. Comparison of iohexol and iodixanol induced nephrotoxicity, mitochondrial damage and mitophagy in a new contrast-induced acute kidney injury rat model. Arch. Toxicol. 2018, 92, 2245–2257. [Google Scholar] [CrossRef]
- Gong, X.; Duan, Y.; Zheng, J.; Ye, Z.; Hei, T.K. Tetramethylpyrazine Prevents Contrast-Induced Nephropathy via Modulating Tubular Cell Mitophagy and Suppressing Mitochondrial Fragmentation, CCL2/CCR2-Mediated Inflammation, and Intestinal Injury. Oxidative Med. Cell. Longev. 2019, 2019, 7096912. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Trejo, O.E.; Reyes-Fermin, L.M.; Briones-Herrera, A.; Tapia, E.; Leon-Contreras, J.C.; Hernandez-Pando, R.; Sanchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef]
- Ward, D.B.; Brown, K.C.; Valentovic, M.A. Radiocontrast Agent Diatrizoic Acid Induces Mitophagy and Oxidative Stress via Calcium Dysregulation. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Peng, Z.Z.; Li, Q.; Wen, R.; Tao, L.J. Renal Fibrosis and Mitochondrial Damage. Chin. Med. J. 2018, 131, 2769–2772. [Google Scholar] [CrossRef]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, D.; Chung, K.P.; Nakahira, K.; Patino, E.; Rice, M.C.; Torres, L.K.; Muthukumar, T.; Choi, A.M.; Akchurin, O.M.; Choi, M.E. Mitophagy-dependent macrophage reprogramming protects against kidney fibrosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| AKI Categories | Pathways that Regulate Mitophagy | Roles on AKI | Mechanisms | References |
|---|---|---|---|---|
| Renal Ischemia- Reperfusion | P53/sestrin-2 pathway, HIF-1α/BNIP3 pathway | Protect against AKI | Modulate cell apoptosis | [107] |
| DRP1-dependent pathway | Protect against AKI | Eliminate damaged mitochondria | [94] | |
| PINK1/PARK2 pathway | Protect against AKI | 1. Eliminate damaged mitochondria | [45,108] | |
| 2. Modulate the removal of ROS | ||||
| 3. Relieve inflammatory response | ||||
| 4. Suppress mitochondrial depolarization | ||||
| 5. Improve ATP production | ||||
| BNIP3-mediated pathway | Protect against AKI | 1. Remove damaged mitochondria | [109] | |
| 2. Modulate the elimination of ROS | ||||
| 3. Relieve inflammatory response | ||||
| OPA1-related pathway | Protect against AKI | 1. Degrade the damaged mitochondria | [110] | |
| 2. Interrupt the mitochondrial damage signalling | ||||
| Cisplatin-induced AKI | PINK1/PARK2 pathway | Protect against AKI | 1. Suppress apoptosis | [111,112,113] |
| 2. Protect mitochondrial function | ||||
| 3. Inhibit Drp1-mediated mitochondrial fission | ||||
| DRP1-dependent pathway | Protect against AKI | Inhibit mitochondrial dysfunction | [93] | |
| HIF-1α/BNIP3/BCEN-1 pathway | Protect against AKI | Alleviate apoptosis | [114] | |
| Spesis-induced AKI | PINK1/PARK2 pathway | Protect against AKI | Prevent cell apoptosis | [115] |
| Contrast media-Induced AKI | PINK1/PARK2 pathway | Protect against AKI | 1. Reduce mitochondrial ROS | [35,116] |
| 2. Inhibit NLRP3 inflammasome activation |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Cai, J.; Tang, C.; Dong, Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells 2020, 9, 338. https://doi.org/10.3390/cells9020338
Wang Y, Cai J, Tang C, Dong Z. Mitophagy in Acute Kidney Injury and Kidney Repair. Cells. 2020; 9(2):338. https://doi.org/10.3390/cells9020338
Chicago/Turabian StyleWang, Ying, Juan Cai, Chengyuan Tang, and Zheng Dong. 2020. "Mitophagy in Acute Kidney Injury and Kidney Repair" Cells 9, no. 2: 338. https://doi.org/10.3390/cells9020338
APA StyleWang, Y., Cai, J., Tang, C., & Dong, Z. (2020). Mitophagy in Acute Kidney Injury and Kidney Repair. Cells, 9(2), 338. https://doi.org/10.3390/cells9020338