Mitochondrial Calcium Regulation of Redox Signaling in Cancer

 , ,
, ,  and
and
Abstract
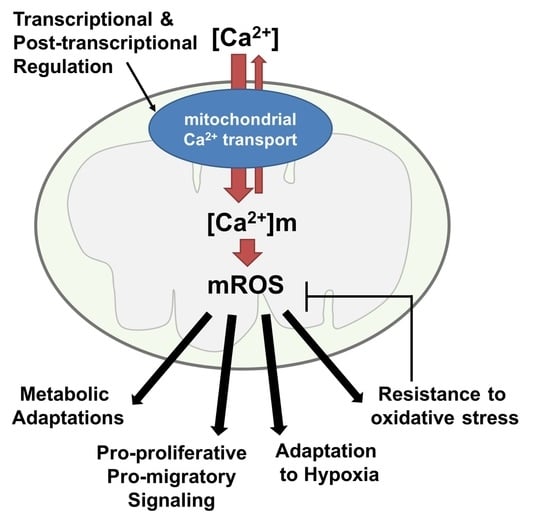
:
1. Introduction
2. Mitochondrial Calcium (Ca2+) Transport
2.1. Mitochondrial Ca2+ Uptake
2.2. Mitochondrial Ca2+ Extrusion
3. Mitochondrial Ca2+, Reactive Oxygen Species (mROS) and Cancer
3.1. Mitochondrial Reactive Oxygen Species (mROS)
3.2. Mitochondrial Ca2+ Regulates Mitochondrial Metabolism and Reactive Oxygen Species (mROS) Generation
3.3. Reactive Oxygen Species (mROS) and Mitochondrial Redox Signaling in Cancer
4. Interplay between Mitochondrial Ca2+ and Reactive Oxygen Species (mROS) in Cancer
4.1. Voltage-Dependent Anion Channel (VDAC)
4.2. Mitochondrial Ca2+ Uniporter (MCU)
4.3. Mitochondrial Ca2+ Uniporter (MCU) Regulators
4.4. Mitochondrial Na+/ Ca2+/Li+ Exchanger (NCLX (SLC8B1))
5. Perspectives and Conclusion
Conflicts of Interest
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Park, M.K.; Ashby, M.C.; Erdemli, G.; Petersen, O.H.; Tepikin, A.V. Perinuclear, perigranular and sub-plasmalemmal mitochondria have distinct functions in the regulation of cellular calcium transport. EMBO J. 2001, 20, 1863–1874. [Google Scholar] [CrossRef] [Green Version]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium dynamics as a machine for decoding signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef]
- Mammucari, C.; Raffaello, A.; Vecellio Reane, D.; Gherardi, G.; De Mario, A.; Rizzuto, R. Mitochondrial calcium uptake in organ physiology: From molecular mechanism to animal models. Pflügers Arch. 2018, 470, 1165–1179. [Google Scholar] [CrossRef] [Green Version]
- Hempel, N.; Trebak, M. Crosstalk between calcium and reactive oxygen species signaling in cancer. Cell Calcium 2017, 63, 70–96. [Google Scholar] [CrossRef] [Green Version]
- Boehning, D.; Patterson, R.L.; Sedaghat, L.; Glebova, N.O.; Kurosaki, T.; Snyder, S.H. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 2003, 5, 1051–1061. [Google Scholar] [CrossRef]
- Herrera-Cruz, M.S.; Simmen, T. Cancer: Untethering mitochondria from the endoplasmic reticulum? Front. Oncol. 2017, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Morciano, G.; Marchi, S.; Morganti, C.; Sbano, L.; Bittremieux, M.; Kerkhofs, M.; Corricelli, M.; Danese, A.; Karkucinska-Wieckowska, A.; Wieckowski, M.R.; et al. Role of mitochondria-associated er membranes in calcium regulation in cancer-specific settings. Neoplasia 2018, 20, 510–523. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Gincel, D. The voltage-dependent anion channel. Cell Biochem. Biophys. 2003, 39, 279–292. [Google Scholar] [CrossRef]
- Messina, A.; Reina, S.; Guarino, F.; De Pinto, V. Vdac isoforms in mammals. Biochim. Biophys. Acta 2012, 1818, 1466–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Mizrachi, D. Vdac1: From structure to cancer therapy. Front. Oncol. 2012, 2, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies mcu as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabò, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Raffaello, A.; De Stefani, D.; Sabbadin, D.; Teardo, E.; Merli, G.; Picard, A.; Checchetto, V.; Moro, S.; Szabò, I.; Rizzuto, R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013, 32, 2362–2376. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.F.; Phillips, C.B.; Ranaghan, M.; Tsai, C.W.; Wu, Y.; Williams, C.; Miller, C. Dual functions of a small regulatory subunit in the mitochondrial calcium uniporter complex. ELife. 2016, 5, e15545. [Google Scholar] [CrossRef]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovács-Bogdán, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. Emre is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Perocchi, F.; Gohil, V.M.; Girgis, H.S.; Bao, X.R.; McCombs, J.E.; Palmer, A.E.; Mootha, V.K. Micu1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature 2010, 467, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, M.; Muallem, S. The gatekeepers of mitochondrial calcium influx: Micu1 and micu2. EMBO Rep. 2014, 15, 205–206. [Google Scholar] [CrossRef]
- Mallilankaraman, K.; Doonan, P.; Cárdenas, C.; Chandramoorthy, H.C.; Müller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgó, J.; Birnbaum, M.J.; et al. Micu1 is an essential gatekeeper for mcu-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Csordás, G.; Golenár, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. Micu1 controls both the threshold and cooperative activation of the mitochondrial ca²⁺ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.C.; Liu, J.; Holmström, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. Micu1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [Green Version]
- Kamer, K.J.; Grabarek, Z.; Mootha, V.K. High-affinity cooperative Ca2+ binding by micu1-micu2 serves as an on-off switch for the uniporter. EMBO Rep. 2017, 18, 1397–1411. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. Micu3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. Micu2, a paralog of micu1, resides within the mitochondrial uniporter complex to regulate calcium handling. PloS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [Green Version]
- Vecellio Reane, D.; Vallese, F.; Checchetto, V.; Acquasaliente, L.; Butera, G.; De Filippis, V.; Szabò, I.; Zanotti, G.; Rizzuto, R.; Raffaello, A. A micu1 splice variant confers high sensitivity to the mitochondrial Ca2+ uptake machinery of skeletal muscle. Mol. Cell 2016, 64, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Mallilankaraman, K.; Cárdenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenár, T.; Csordás, G.; Madireddi, P.; Yang, J.; Müller, M.; et al. Mcur1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S. J.; Breves, S.L.; Corbally, D.P.; et al. Mcur1 is a scaffold factor for the mcu complex function and promotes mitochondrial bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef]
- De Marchi, U.; Santo-Domingo, J.; Castelbou, C.; Sekler, I.; Wiederkehr, A.; Demaurex, N. Nclx protein, but not letm1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced nad(p)h production and modulating matrix redox state. J. Biol. Chem. 2014, 289, 20377–20385. [Google Scholar] [CrossRef] [Green Version]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Ohana, E.; Hershfinkel, M.; Volokita, M.; Elgazar, V.; Beharier, O.; Silverman, W.F.; Argaman, M.; Sekler, I. Lithium-calcium exchange is mediated by a distinct potassium-independent sodium-calcium exchanger. J. Biol. Chem. 2004, 279, 25234–25240. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. Nclx is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Doering, A.E.; Nicoll, D.A.; Lu, Y.; Lu, L.; Weiss, J.N.; Philipson, K.D. Topology of a functionally important region of the cardiac Na+/Ca2+ exchanger. J. Biol. Chem. 1998, 273, 778–783. [Google Scholar] [CrossRef] [Green Version]
- Sekler, I. Standing of giants shoulders the story of the mitochondrial Na+ Ca2+ exchanger. Biochem. Biophys. Res. Commun. 2015, 460, 50–52. [Google Scholar] [CrossRef]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. Pink1-associated parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Kostic, M.; Ludtmann, M.H.; Bading, H.; Hershfinkel, M.; Steer, E.; Chu, C.T.; Abramov, A.Y.; Sekler, I. Pka phosphorylation of nclx reverses mitochondrial calcium overload and depolarization, promoting survival of pink1-deficient dopaminergic neurons. Cell Rep. 2015, 13, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Eisner, D.A. Regulation of intracellular and mitochondrial sodium in health and disease. Circ. Res. 2009, 104, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Nita, I.I.; Hershfinkel, M.; Lewis, E.C.; Sekler, I. A crosstalk between Na+ channels, Na+/K+ pump and mitochondrial Na+ transporters controls glucose-dependent cytosolic and mitochondrial Na+ signals. Cell Calcium 2015, 57, 69–75. [Google Scholar] [CrossRef]
- Shao, J.; Fu, Z.; Ji, Y.; Guan, X.; Guo, S.; Ding, Z.; Yang, X.; Cong, Y.; Shen, Y. Leucine zipper-EF-hand containing transmembrane protein 1 (letm1) forms a Ca2+/H+ antiporter. Sci. Rep. 2016, 6, 34174. [Google Scholar] [CrossRef] [Green Version]
- Tamai, S.; Iida, H.; Yokota, S.; Sayano, T.; Kiguchiya, S.; Ishihara, N.; Hayashi, J.; Mihara, K.; Oka, T. Characterization of the mitochondrial protein letm1, which maintains the mitochondrial tubular shapes and interacts with the aaa-atpase bcs1l. J. Cell Sci. 2008, 121, 2588–2600. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Pinton, P. The mitochondrial calcium uniporter complex: Molecular components, structure and physiopathological implications. J. Physiol. 2014, 592, 829–839. [Google Scholar] [CrossRef]
- Pathak, T.; Trebak, M. Mitochondrial ca2+ signaling. Pharmacol. Therapeutics 2018, 192, 112–123. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria central role of complex iii. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef]
- Nishikawa, T.; Araki, E. Impact of mitochondrial ROS production in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal. 2007, 9, 343–353. [Google Scholar] [CrossRef]
- Aldosari, S.; Awad, M.; Harrington, E.O.; Sellke, F.W.; Abid, M.R. Subcellular reactive oxygen species (ROS) in cardiovascular pathophysiology. Antioxidants (Basel) 2018, 7, E14. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Aykin-Burns, N.; Ahmad, I.M.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and hydrogen peroxide mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar]
- Hempel, N.; Ye, H.; Abessi, B.; Mian, B.; Melendez, J.A. Altered redox status accompanies progression to metastatic human bladder cancer. Free Radic. Biol. Med. 2009, 46, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Cao, X.; Chen, Y. Mitochondria and calcium signaling in embryonic development. Semin. Cell Dev. Biol. 2009, 20, 337–345. [Google Scholar] [CrossRef]
- Kim, M.S.; Usachev, Y.M. Mitochondrial Ca2+ cycling facilitates activation of the transcription factor nfat in sensory neurons. J. Neurosci. 2009, 29, 12101–12114. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Denton, R.M.; Richards, D.A.; Chin, J.G. Calcium ions and the regulation of NAD+-linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem. J. 1978, 176, 899–906. [Google Scholar] [CrossRef]
- Hansford, R.G. Physiological role of mitochondrial Ca2+ transport. J. Bioenerg. Biomembr. 1994, 26, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Rajendran, G.; Harris, R.A.; Taylor, J.A., 3rd. Metabolic flexibility in cancer: Targeting the pyruvate dehydrogenase kinase:Pyruvate dehydrogenase axis. Mol. Cancer Ther. 2019, 18, 1673–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, G.A.; Denton, R.M. Regulation of NAD+-linked isocitrate dehydrogenase and 2-oxoglutarate dehydrogenase by Ca2+ ions within toluene-permeabilized rat heart mitochondria. Interactions with regulation by adenine nucleotides and NADH/ NAD+ ratios. Biochem. J. 1988, 252, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Al-Khallaf, H. Isocitrate dehydrogenases in physiology and cancer: Biochemical and molecular insight. Cell Biosci. 2017, 7, 37. [Google Scholar] [CrossRef]
- May, J.L.; Kouri, F.M.; Hurley, L.A.; Liu, J.; Tommasini-Ghelfi, S.; Ji, Y.; Gao, P.; Calvert, A.E.; Lee, A.; Chandel, N.S.; et al. Idh3alpha regulates one-carbon metabolism in glioblastoma. Sci. Adv. 2019, 5, eaat0456. [Google Scholar] [CrossRef] [Green Version]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- McCormack, J.G.; Denton, R.M. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef]
- Rutter, G.A.; Denton, R.M. The binding of Ca2+ ions to pig heart NAD+-isocitrate dehydrogenase and the 2-oxoglutarate dehydrogenase complex. Biochem. J. 1989, 263, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, C.T.; Anderson, J.L.; Denton, R.M. Studies on the regulation of the human e1 subunit of the 2-oxoglutarate dehydrogenase complex, including the identification of a novel calcium-binding site. Biochem. J. 2014, 459, 369–381. [Google Scholar] [CrossRef]
- Cardenas, C.; Muller, M.; McNeal, A.; Lovy, A.; Jana, F.; Bustos, G.; Urra, F.; Smith, N.; Molgo, J.; Diehl, J.A.; et al. Selective vulnerability of cancer cells by inhibition of Ca(2+) transfer from endoplasmic reticulum to mitochondria. Cell Rep. 2016, 15, 219–220. [Google Scholar] [CrossRef] [Green Version]
- Feissner, R.F.; Skalska, J.; Gaum, W.E.; Sheu, S.S. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. (Landmark Ed.) 2009, 14, 1197–1218. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex iii releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Sohal, R.S.; Allen, R.G. Relationship between metabolic rate, free radicals, differentiation and aging: A unified theory. Basic Life Sci. 1985, 35, 75–104. [Google Scholar]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Bienert, G.P.; Møller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef]
- Zhang, H.; Go, Y.M.; Jones, D.P. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial gsh system in protection against oxidative stress. Arch. Biochem. Biophys. 2007, 465, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Izakovic, M.; Mazur, M.; Rhodes, C.J.; Telser, J. Role of oxygen radicals in DNA damage and cancer incidence. Mol. Cell. Biochem. 2004, 266, 37–56. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Sato, H.; Sato, M.; Kanai, H.; Uchiyama, T.; Iso, T.; Ohyama, Y.; Sakamoto, H.; Tamura, J.; Nagai, R.; Kurabayashi, M. Mitochondrial reactive oxygen species and c-src play a critical role in hypoxic response in vascular smooth muscle cells. Cardiovasc. Res. 2005, 67, 714–722. [Google Scholar] [CrossRef] [Green Version]
- Porporato, P.E.; Payen, V.L.; Pérez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Sporn, M.B.; Liby, K.T. Nrf2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gal, K.; Ibrahim, M.X.; Wiel, C.; Sayin, V.I.; Akula, M.K.; Karlsson, C.; Dalin, M.G.; Akyürek, L.M.; Lindahl, P.; Nilsson, J.; et al. Antioxidants can increase melanoma metastasis in mice. Sci. Transl. Med. 2015, 7, 308re8. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, hif1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsang, T.M.; Teahan, O.; et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 2008, 283, 22700–22708. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. Microrna-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins iscu1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microrna-210 modulates mitochondrial function and decreases iscu and cox10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [Green Version]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase m2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitosugi, T.; Kang, S.; Vander Heiden, M.G.; Chung, T.W.; Elf, S.; Lythgoe, K.; Dong, S.; Lonial, S.; Wang, X.; Chen, G.Z.; et al. Tyrosine phosphorylation inhibits pkm2 to promote the warburg effect and tumor growth. Sci. Signal. 2009, 2, ra73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. MnSOD upregulation sustains the warburg effect via mitochondrial ROS and AMPK-dependent signalling in cancer. Nat. Commun. 2015, 6, 6053. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Pinton, P. Alterations of calcium homeostasis in cancer cells. Curr. Opin. Pharmacol. 2016, 29, 1–6. [Google Scholar] [CrossRef]
- Mazure, N.M. Vdac in cancer. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 665–673. [Google Scholar] [CrossRef]
- Ko, J.H.; Gu, W.; Lim, I.; Zhou, T.; Bang, H. Expression profiling of mitochondrial voltage-dependent anion channel-1 associated genes predicts recurrence-free survival in human carcinomas. PloS ONE 2014, 9, e110094. [Google Scholar] [CrossRef] [Green Version]
- Magri, A.; Reina, S.; De Pinto, V. Vdac1 as pharmacological target in cancer and neurodegeneration: Focus on its role in apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Zakar, M.; Rosenthal, K.; Abu-Hamad, S. Key regions of vdac1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 2009, 1787, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Lemeshko, V.V. Vdac electronics: 1. Vdac-hexo(gluco)kinase generator of the mitochondrial outer membrane potential. Biochim. Biophys. Acta 2014, 1838, 1362–1371. [Google Scholar] [CrossRef] [Green Version]
- Abu-Hamad, S.; Zaid, H.; Israelson, A.; Nahon, E.; Shoshan-Barmatz, V. Hexokinase-i protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: Mapping the site of binding. J. Biol. Chem. 2008, 283, 13482–13490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewski, N.; Nogueira, V.; Bhaskar, P.; Coy, P.E.; Skeen, J.E.; Gottlob, K.; Chandel, N.S.; Thompson, C.B.; Robey, R.B.; Hay, N. Hexokinase-mitochondria interaction mediated by akt is required to inhibit apoptosis in the presence or absence of bax and bak. Mol. Cell. 2004, 16, 819–830. [Google Scholar] [CrossRef] [PubMed]
- da-Silva, W.S.; Gomez-Puyou, A.; de Gomez-Puyou, M.T.; Moreno-Sanchez, R.; De Felice, F.G.; de Meis, L.; Oliveira, M.F.; Galina, A. Mitochondrial bound hexokinase activity as a preventive antioxidant defense: Steady-state ADP formation as a regulatory mechanism of membrane potential and reactive oxygen species generation in mitochondria. J. Biol. Chem. 2004, 279, 39846–39855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathupala, S.P.; Ko, Y.H.; Pedersen, P.L. Hexokinase ii: Cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene 2006, 25, 4777–4786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, D.; Antunes, F.; Canali, R.; Rettori, D.; Cadenas, E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 2003, 278, 5557–5563. [Google Scholar] [CrossRef] [Green Version]
- Vultur, A.; Gibhardt, C.S.; Stanisz, H.; Bogeski, I. The role of the mitochondrial calcium uniporter (MCU) complex in cancer. Pflugers Arch. 2018, 470, 1149–1163. [Google Scholar] [CrossRef]
- Tosatto, A.; Sommaggio, R.; Kummerow, C.; Bentham, R.B.; Blacker, T.S.; Berecz, T.; Duchen, M.R.; Rosato, A.; Bogeski, I.; Szabadkai, G.; et al. The mitochondrial calcium uniporter regulates breast cancer progression via hif-1α. EMBO Mol. Med. 2016, 8, 569–585. [Google Scholar] [CrossRef]
- Hall, D.D.; Wu, Y.; Domann, F.E.; Spitz, D.R.; Anderson, M.E. Mitochondrial calcium uniporter activity is dispensable for mda-mb-231 breast carcinoma cell survival. PloS ONE 2014, 9, e96866. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Wang, X.; Shen, Q.; Yang, X.; Yu, C.; Cai, C.; Cai, G.; Meng, X.; Zou, F. Mitochondrial Ca2+ uniporter is critical for store-operated Ca2+ entry-dependent breast cancer cell migration. Biochem. Biophys. Res. Commun. 2015, 458, 186–193. [Google Scholar] [CrossRef]
- Ren, T.; Zhang, H.; Wang, J.; Zhu, J.; Jin, M.; Wu, Y.; Guo, X.; Ji, L.; Huang, Q.; Zhang, H.; et al. Mcu-dependent mitochondrial Ca2+ inhibits NAD+/SIRT3/SOD2 pathway to promote ROS production and metastasis of HCC cells. Oncogene 2017, 36, 5897–5909. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Shanmughapriya, S.; Tomar, D.; Siddiqui, N.; Lynch, S.; Nemani, N.; Breves, S.L.; Zhang, X.; Tripathi, A.; Palaniappan, P.; et al. Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol. Cell 2017, 65, 1014.e7–1028.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, M.C.; Peters, A.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 695–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, T.; Wang, J.; Zhang, H.; Yuan, P.; Zhu, J.; Wu, Y.; Huang, Q.; Guo, X.; Zhang, J.; Ji, L.; et al. MCUR1-mediated mitochondrial calcium signaling facilitates cell survival of hepatocellular carcinoma via reactive oxygen species-dependent p53 degradation. Antioxid. Redox Signal. 2018, 28, 1120–1136. [Google Scholar] [CrossRef]
- Chen, L.; Sun, Q.; Zhou, D.; Song, W.; Yang, Q.; Ju, B.; Zhang, L.; Xie, H.; Zhou, L.; Hu, Z.; et al. HINT2 triggers mitochondrial Ca2+ influx by regulating the mitochondrial Ca2+ uniporter (MCU) complex and enhances gemcitabine apoptotic effect in pancreatic cancer. Cancer Lett. 2017, 411, 106–116. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Mustafi, S.B.; Xiong, X.; Dwivedi, S.K.D.; Nesin, V.; Saha, S.; Zhang, M.; Dhanasekaran, D.; Jayaraman, M.; Mannel, R.; et al. MICU1 drives glycolysis and chemoresistance in ovarian cancer. Nat. Commun. 2017, 8, 14634. [Google Scholar] [CrossRef]
- Gottlieb, T.M.; Leal, J.F.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef]
- Jin, M.; Wang, J.; Ji, X.; Cao, H.; Zhu, J.; Chen, Y.; Yang, J.; Zhao, Z.; Ren, T.; Xing, J. MCUR1 facilitates epithelial-mesenchymal transition and metastasis via the mitochondrial calcium dependent ROS/Nrf2/Notch pathway in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 136. [Google Scholar] [CrossRef] [Green Version]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Wang, W.; Xie, Q.; Zhou, X.; Yao, J.; Zhu, X.; Huang, P.; Zhang, L.; Wei, J.; Xie, H.; Zhou, L.; et al. Mitofusin-2 triggers mitochondria Ca2+ influx from the endoplasmic reticulum to induce apoptosis in hepatocellular carcinoma cells. Cancer Lett. 2015, 358, 47–58. [Google Scholar] [CrossRef]
- Jadiya, P.; Kolmetzky, D.W.; Tomar, D.; Di Meco, A.; Lombardi, A.A.; Lambert, J.P.; Luongo, T.S.; Ludtmann, M.H.; Pratico, D.; Elrod, J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer’s disease. Nat. Commun. 2019, 10, 3885. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; O’Rourke, B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J. Bioenerg. Biomembr. 2009, 41, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, Y.; Wan, L.J.; Cui, S.Y.; Gong, Y.P.; Li, C.L. The mitochondrial Na+/Ca2+ exchanger may reduce high glucose-induced oxidative stress and nucleotide-binding oligomerization domain receptor 3 inflammasome activation in endothelial cells. J. Geriatr. Cardiol. 2015, 12, 270–278. [Google Scholar] [PubMed]
- Ben-Kasus Nissim, T.; Zhang, X.; Elazar, A.; Roy, S.; Stolwijk, J.A.; Zhou, Y.; Motiani, R.K.; Gueguinou, M.; Hempel, N.; Hershfinkel, M.; et al. Mitochondria control store-operated Ca2+ entry through Na+ and redox signals. EMBO J. 2017, 36, 797–815. [Google Scholar] [CrossRef] [Green Version]
- Kaddour-Djebbar, I.; Lakshmikanthan, V.; Shirley, R.B.; Ma, Y.; Lewis, R.W.; Kumar, M.V. Therapeutic advantage of combining calcium channel blockers and trail in prostate cancer. Mol. Cancer Ther. 2006, 5, 1958–1966. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, Y.; Takata, N.; Suzuki-Karasaki, M.; Yoshida, Y.; Tokuhashi, Y.; Suzuki-Karasaki, Y. Disrupting mitochondrial Ca2+ homeostasis causes tumor-selective trail sensitization through mitochondrial network abnormalities. Int. J. Oncol. 2017, 51, 1146–1158. [Google Scholar] [CrossRef]
- Choudhary, V.; Kaddour-Djebbar, I.; Alaisami, R.; Kumar, M.V.; Bollag, W.B. Mitofusin 1 degradation is induced by a disruptor of mitochondrial calcium homeostasis, CGP37157: A role in apoptosis in prostate cancer cells. Int. J. Oncol. 2014, 44, 1767–1773. [Google Scholar] [CrossRef] [Green Version]
- Kaddour-Djebbar, I.; Choudhary, V.; Brooks, C.; Ghazaly, T.; Lakshmikanthan, T.; Dong, Z.; Kumar, M.V. Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells. Int. J. Oncol. 2010, 36, 1437–1444. [Google Scholar]
- Scherzed, A.; Hackenberg, S.; Froelich, K.; Rak, K.; Ginzkey, C.; Hagen, R.; Schendzielorz, P.; Kleinsasser, N. Effects of salinomycin and cgp37157 on head and neck squamous cell carcinoma cell lines in vitro. Mol. Med. Rep. 2015, 12, 4455–4461. [Google Scholar] [CrossRef]
- Bae, J.H.; Park, J.W.; Kwon, T.K. Ruthenium red, inhibitor of mitochondrial Ca2+ uniporter, inhibits curcumin-induced apoptosis via the prevention of intracellular Ca2+ depletion and cytochrome c release. Biochem. Biophys. Res. Commun. 2003, 303, 1073–1079. [Google Scholar] [CrossRef]
- Cui, C.; Yang, J.; Fu, L.; Wang, M.; Wang, X. Progress in understanding mitochondrial calcium uniporter complex-mediated calcium signalling: A potential target for cancer treatment. Br. J. Pharmacol. 2019, 176, 1190–1205. [Google Scholar] [CrossRef] [PubMed]
- Arduino, D.M.; Wettmarshausen, J.; Vais, H.; Navas-Navarro, P.; Cheng, Y.; Leimpek, A.; Ma, Z.; Delrio-Lorenzo, A.; Giordano, A.; Garcia-Perez, C.; et al. Systematic identification of MCU modulators by orthogonal interspecies chemical screening. Mol. Cell. 2017, 67, 711.e7–723.e7. [Google Scholar] [CrossRef] [PubMed]
- Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Phillips, D.R.; Cutts, S.M. Mitoxantrone, more than just another topoisomerase II poison. Med. Res. Rev. 2016, 36, 248–299. [Google Scholar] [CrossRef] [PubMed]
- Santo-Domingo, J.; Vay, L.; Hernandez-Sanmiguel, E.; Lobaton, C.D.; Moreno, A.; Montero, M.; Alvarez, J. The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br. J. Pharmacol. 2007, 151, 647–654. [Google Scholar] [CrossRef] [Green Version]
- Kon, N.; Murakoshi, M.; Isobe, A.; Kagechika, K.; Miyoshi, N.; Nagayama, T. Ds16570511 is a small-molecule inhibitor of the mitochondrial calcium uniporter. Cell Death Discov. 2017, 3, 17045. [Google Scholar] [CrossRef]
- Ren, J.; Lu, Y.; Qian, Y.; Chen, B.; Wu, T.; Ji, G. Recent progress regarding kaempferol for the treatment of various diseases. Exp. Ther. Med. 2019, 18, 2759–2776. [Google Scholar] [CrossRef] [Green Version]
- Montero, M.; Lobaton, C.D.; Hernandez-Sanmiguel, E.; Santodomingo, J.; Vay, L.; Moreno, A.; Alvarez, J. Direct activation of the mitochondrial calcium uniporter by natural plant flavonoids. Biochem. J. 2004, 384, 19–24. [Google Scholar] [CrossRef] [Green Version]
- Vay, L.; Hernandez-Sanmiguel, E.; Santo-Domingo, J.; Lobaton, C. D.; Moreno, A.; Montero, M.; Alvarez, J. Modulation of Ca2+ release and Ca2+ oscillations in hela cells and fibroblasts by mitochondrial Ca2+ uniporter stimulation. J. Physiol. 2007, 580, 39–49. [Google Scholar] [CrossRef]
- Chiesi, M.; Schwaller, R.; Eichenberger, K. Structural dependency of the inhibitory action of benzodiazepines and related compounds on the mitochondrial Na+- Ca2+ exchanger. Biochem. Pharmacol. 1988, 37, 4399–4403. [Google Scholar] [CrossRef]
- Wingrove, D.E.; Gunter, T.E. Kinetics of mitochondrial calcium transport. II. A kinetic description of the sodium-dependent calcium efflux mechanism of liver mitochondria and inhibition by ruthenium red and by tetraphenylphosphonium. J. Biol. Chem. 1986, 261, 15166–15171. [Google Scholar]
- Wei, A.C.; Liu, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. Mitochondrial Ca2+ influx and efflux rates in guinea pig cardiac mitochondria: Low and high affinity effects of cyclosporine a. Biochim. Biophys. Acta 2011, 1813, 1373–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Hershfinkel, M.; Sekler, I. Molecular identity and functional properties of the mitochondrial Na+/Ca2+ exchanger. J. Biol. Chem. 2012, 287, 31650–31657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurkowitz, M.S.; Altschuld, R.A.; Brierley, G.P.; Cragoe, E.J., Jr. Inhibition of Na+-dependent Ca2+ efflux from heart mitochondria by amiloride analogues. FEBS Lett. 1983, 162, 262–265. [Google Scholar] [CrossRef] [Green Version]
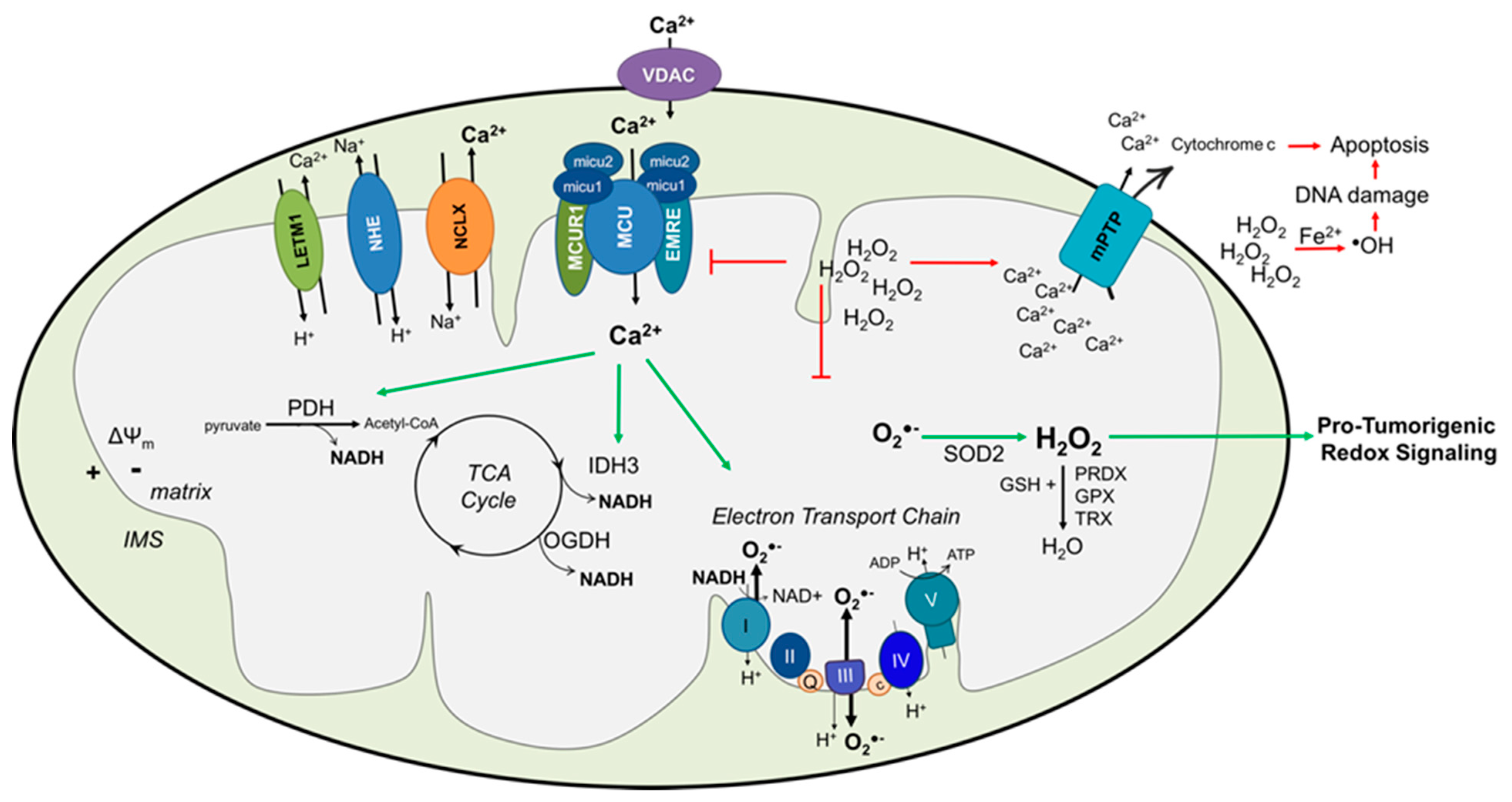

{kind=link}
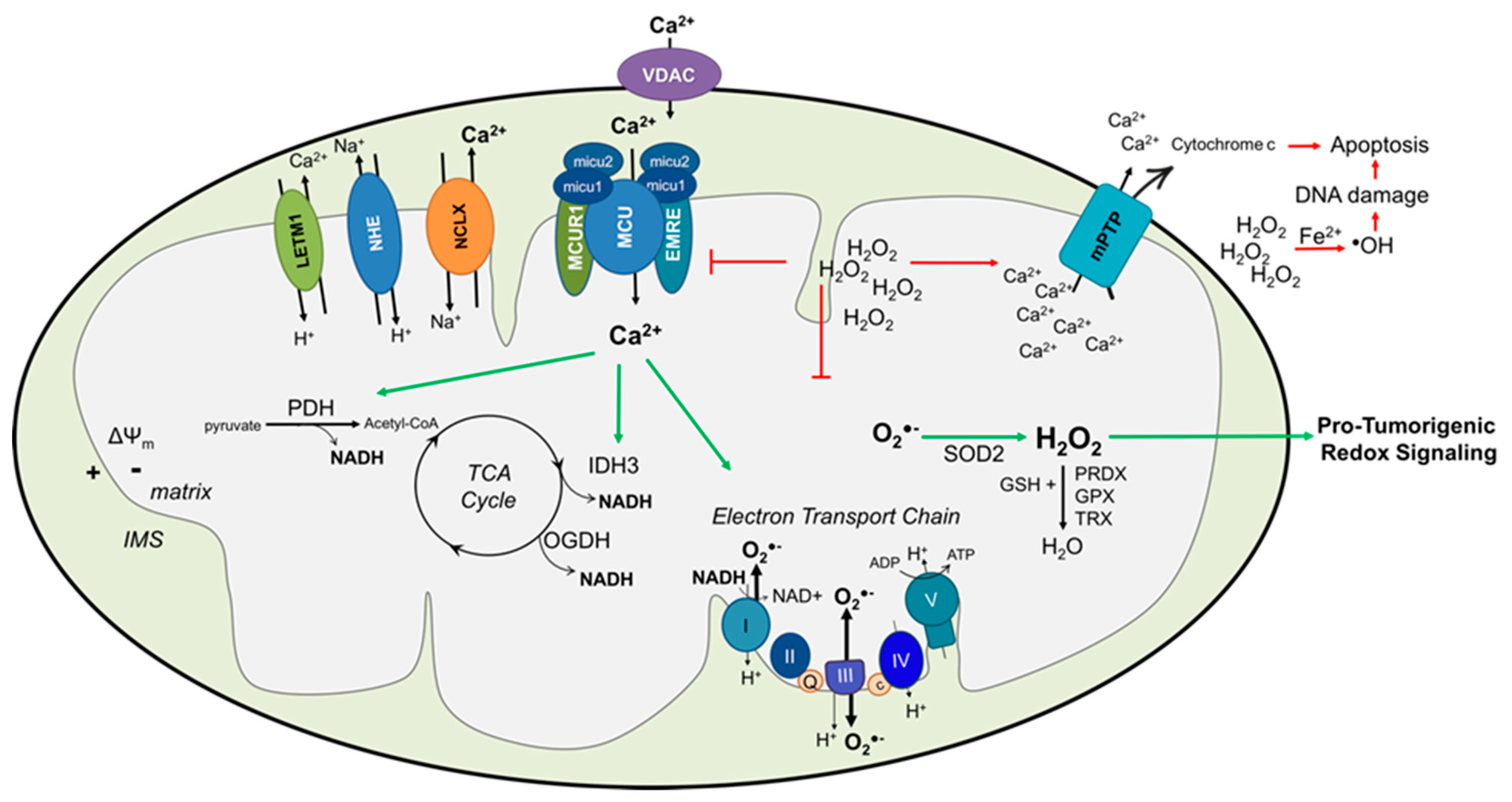{kind=link}
| Cancer Type | [mCa2+] Regulator (Expression in Tumor Specimens) | Cell line (Normal/Control Cells) | [Ca2+]m Phenotype | mROS Phenotype | Cellular Phenotype | Reference |
|---|---|---|---|---|---|---|
| Breast Cancer | MCU (elevated in TNBC) | TNBC cell lines: MDA-MB-231 MDA-MB-468 BT-549 (none) | MCU knock-down results in decreased mCa2+ uptake induced by ATP; MCU expression increases [Ca2+]m transients and cytosolic Ca2+ buffering by mitochondria. | MCU knock-down results in decreased mito O2∙− mitochondrial GSSG/GSH, and H2O2, leading to decreased HIF-1α activation. | MCU knock-down results in decreased migration, invasion, clonogenic potential, in vivo tumor growth and metastasis; no effect on proliferation, cell cycle or cell death. | [118] |
| MCU (elevated in ER negative and basal-like breast cancers) | MDA-MB-231 | Effects on [Ca2+]m not tested. MCU knock-down had no major effect on cytosolic Ca2+. | Not investigated | MCU knock-down results in potentiation of cell death by ionomycin; no effect on proliferation. | [123] | |
| MCU, MICU1 (high MCU, low MICU1 associated with poor survival of breast cancer patients) | MDA-MB-231 | MCU knock-down results in decreased mCa2+ uptake induced by ATP; MCU dominant negative (DN) expression decreases the integrated area of response induced by ATP. | No effect on MitoSox following MCU or MICU1 knock-down in MDA-MB-231 | MCU knock-down increases AMPK activation. MCU or MICU1 knock-down, or MCU-DN had no effect on clonogenic survival in response to therapy-related stress in MDA-MB-231. | [119] | |
| (HMEC) | MICU1 knock-down increases peak amplitude of [Ca2+]m uptake; increases integrated area of response induced by ATP. | MCU knock-down in HeLa affects cell viability in response to ceramide. MCU and MICU1 knock-down in HMEC affects cell viability in response to ceramide. | ||||
| MCU (elevated expression correlates with metastatic disease) | MDA-MB-231 | MCU inhibition by RuR or MCU knock-down decreases [Ca2+]m induced by serum; decreases SOCE induced by thapsigargin. | Not tested | MCU inhibition by RuR or MCU knock-down decreases migration. | [120] | |
| Cervical Cancer | MICU1 | HeLa (human endothelial cells [HEC]) | MICU1 knockdown in HeLa and HEC results in increased [Ca2+]m under resting conditions; no effect on the peak [Ca2+]m or [Ca2+]cyto induced by histamine. | MICU1 knockdown in HeLa and HEC results in increased basal mROS. | MICU1 knockdown in HeLa increases ceramide induced cell death; no effect on proliferation. | [20] |
| MICU1 knockdown in HEC increases LPS and cycloheximide induced cell death; decreases migration | ||||||
| MCU | HeLa | Not tested | Not tested | MCU knock-down in HeLa affects cell viability in response to ceramide | [119] | |
| Liver Cancer | MCU, MICU1 (high MCU, low MICU1 expression in HCC compared to matched normal) | MHCC97H, SMMC7721, BEL7402 (Normal hepatocyte HL-7702) | HCC have increased basal [Ca2+]m compared to normal hepatocyte. | MCU knock-down in HCC decreased mROS and total ROS. | MCU knock-down in HCC decreased migration, invasion and in vivo metastasis. | [121] |
| MCU knock-down in HCC decreased [Ca2+]m in response to histamine. | ||||||
| MCU expression in HCC increased [Ca2+]m in response to histamine. | MCU expression in HCC increases mROS and total ROS, leading to ROS-dependent JNK activation. | MCU over expression in HCC increased migration, invasion and in vivo metastasis. | ||||
| MICU1 knock-down in HCC increased [Ca2+]m in response to histamine. | ||||||
| MCUR1 (increased expression in HCC compared to matched normal) | BEL7402, MHCC97H (none) | MCUR1 knock-down decreased [Ca2+]m in response to histamine. | MCUR1 knock-down decreased mROS and total ROS. | MCUR1 knock-down increased apoptosis; decreased proliferation, clonogenic potential, and in vivo tumor growth. | [124] | |
| MCUR1 overexpression in HCC increased [Ca2+]m in response to histamine; which is abrogated by MCU inhibition with Ru360. | MCUR1 expression in HCC increases mROS and total ROS, leading to p53 inactivation via Akt/MDM2 pathway. | MCUR1 over-expression decreased apoptosis; increased proliferation, clonogenic potential, and in vivo tumor growth. | ||||
| Pancreatic Cancer | HINT2: regulator of MICU1/2, EMRE (HINT2 downregulated in Panc, decrease associated with poor prognosis) | BxPC-3, L3.6pl | HINT2 overexpression decreases MICU1 and MICU2 expression; increases EMRE. | HINT2 overexpression increased mROS. | HINT2 overexpression increased apoptosis, decreased migration, invasion, clonogenic potential and tumor growth in vivo. | [125] |
| HINT2 overexpression increased [Ca2+]m. | ||||||
| Ovarian Cancer | MICU1 (high MICU1 expression associated with decreased patient survival; increased expression in chemoresistant OVCA specimens) | CP20, OV90 (normal fallopian tube and surface eptilelium cell lines: FTE188, OSE) | MICU1 knock-down increased [Ca2+]m in response to cisplatin. | MICU1 knock-down increased mROS. | MICU1 knock-down decreases glycolysis in OVCA cells. | [126] |
| Increased MICU1 expression correlates with pPDH. MICU1 knock-down restores PDH activity. | MICU1 overexpression enhances glycolysis in FTE188 and OSE. |
| Target | Compound | Cellular Effects (Cancer Cells Tested) | Reference |
|---|---|---|---|
| MCU Inhibitor | Ruthenium Red/Ru360 | Targets DXXE motif of MCU. Lacks specificity. Enhanced cytotoxicity in leukemia, HCC, breast and pancreatic cancer cells. | [120,124,140,141] |
| Mitoxantrone (and analogs) | Targets DXXE motif of MCU. Not specific to MCU, has DNA intercalating activity, inhibits DNA topoisomerase II and cell proliferation. Used to treat prostate cancer, metastatic breast cancer, some leukemias | [141,142,143] | |
| KB-R7943 | Inhibits Na+/Ca2+ exchanger (NCX1). Has anti-tumor activity, but not tested in context of MCU expression. | [144] | |
| DS16570511 | MCU inhibitor—unclear mechanism. Not tested in cancer cells. | [145] | |
| MCU Activator | Kaempferol | Lacks specificity/Mechanisms of MCU activation unclear. Pro-apoptotic. Anticancer properties observed in various tumor types. | [146,147,148] |
| NCLX Inhibitor | CGP37157 (benzothiazepine) | Off-target effects on other Ca2+ channels observed. Sensitizes tumor melanoma, osteosarcoma, and prostate cancer cells to pro-apoptotic stimuli. | [135,136,149] |
| tetraphenylphosphonium | Demonstrated to inhibit mitochondrial Na+/Ca2+ exchange. Lacks demonstrated specificity toward NCLX. | [150] | |
| Cyclosporin A | NCLX inhibition at higher concentrations (IC50 = 2μM) than those required to inhibit mPTP. | [151] | |
| Verapamil | Ca2+ channel blocker, inhibits mitochondrial Na+ Ca2+ exchange, not specific toward NCLX. | [152] | |
| Amiloride analogs | Na+ channel blocker, not specific toward NCLX. | [153] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delierneux, C.; Kouba, S.; Shanmughapriya, S.; Potier-Cartereau, M.; Trebak, M.; Hempel, N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells 2020, 9, 432. https://doi.org/10.3390/cells9020432
Delierneux C, Kouba S, Shanmughapriya S, Potier-Cartereau M, Trebak M, Hempel N. Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells. 2020; 9(2):432. https://doi.org/10.3390/cells9020432
Chicago/Turabian StyleDelierneux, Céline, Sana Kouba, Santhanam Shanmughapriya, Marie Potier-Cartereau, Mohamed Trebak, and Nadine Hempel. 2020. "Mitochondrial Calcium Regulation of Redox Signaling in Cancer" Cells 9, no. 2: 432. https://doi.org/10.3390/cells9020432
APA StyleDelierneux, C., Kouba, S., Shanmughapriya, S., Potier-Cartereau, M., Trebak, M., & Hempel, N. (2020). Mitochondrial Calcium Regulation of Redox Signaling in Cancer. Cells, 9(2), 432. https://doi.org/10.3390/cells9020432