Animal, Fungi, and Plant Genome Sequences Harbor Different Non-Canonical Splice Sites



Abstract
:1. Introduction
2. Materials and Methods
2.1. Analysis and Validation of Splice Site Combinations
2.2. Calculation of the Splice Site Diversity
2.3. Investigation of a Common Non-Canonical Splice Site in Fungal Genome Sequences
2.4. Detection of Spliceosomal Components
2.5. Correlation between the GC Content of the Genome and the GC Content of the Splice Sites
2.6. Phylogeny of Non-Canonical Splice Sites
2.7. Usage of Non-Canonical Splice Sites
2.8. Genomic Read Mapping and Variant Calling
2.9. Data Availability
3. Results
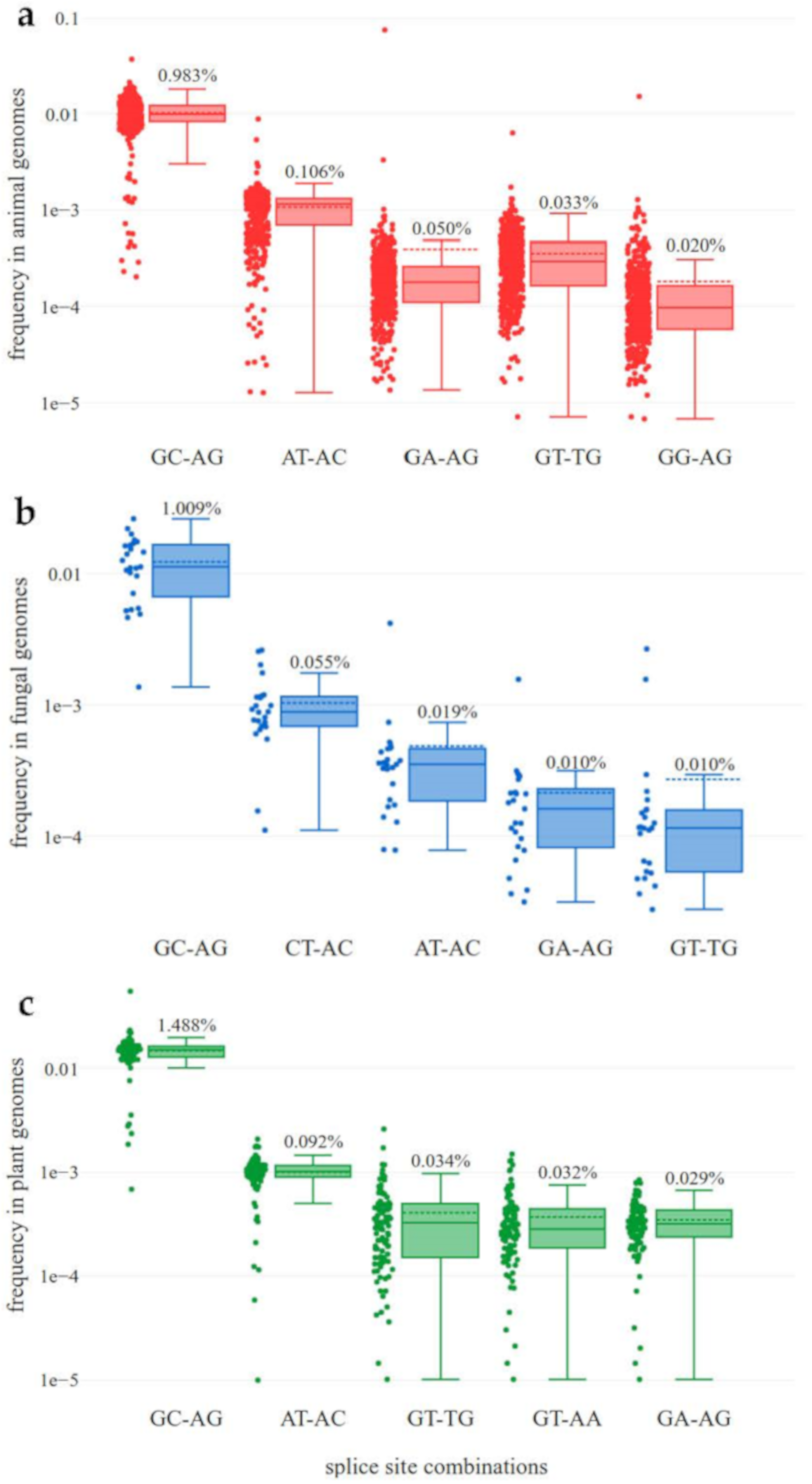
3.1. Analysis of Non-Canonical Splice Sites
3.2. High Diversity of Non-Canonical Splice Sites in Animals
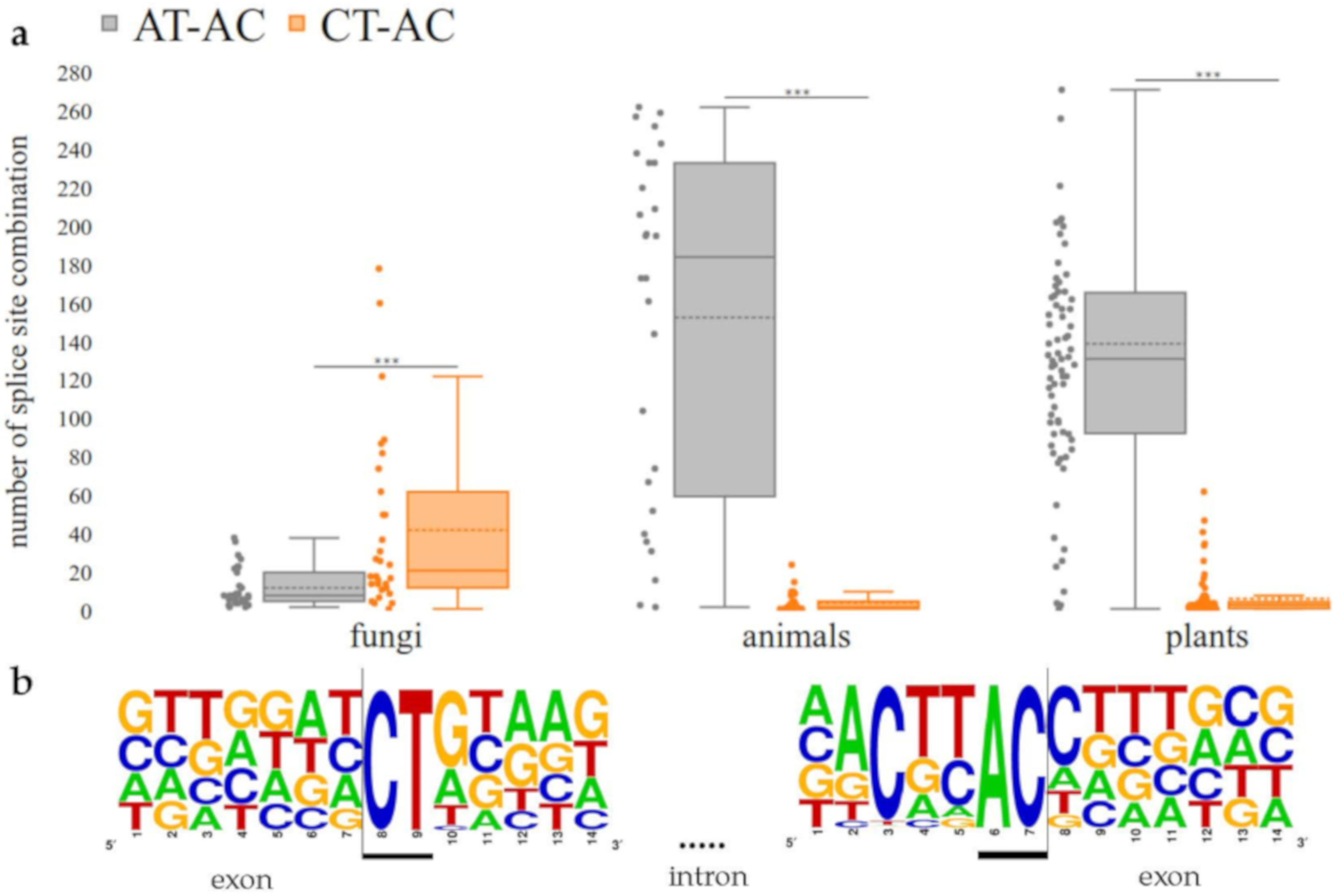
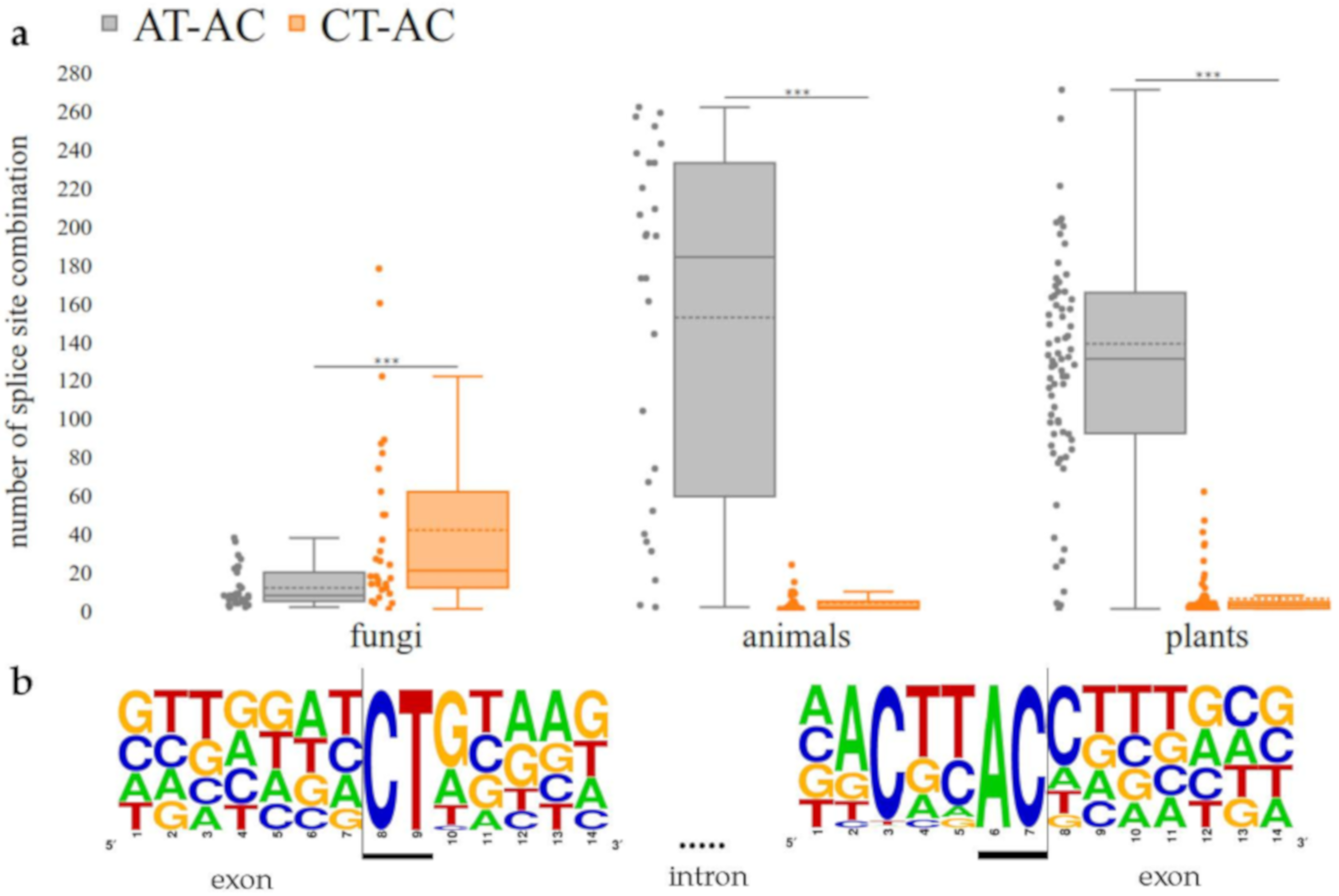
3.3. CT-AC is a Frequent Splice Site Combination in Fungal Annotations
3.4. Intron Size Analysis
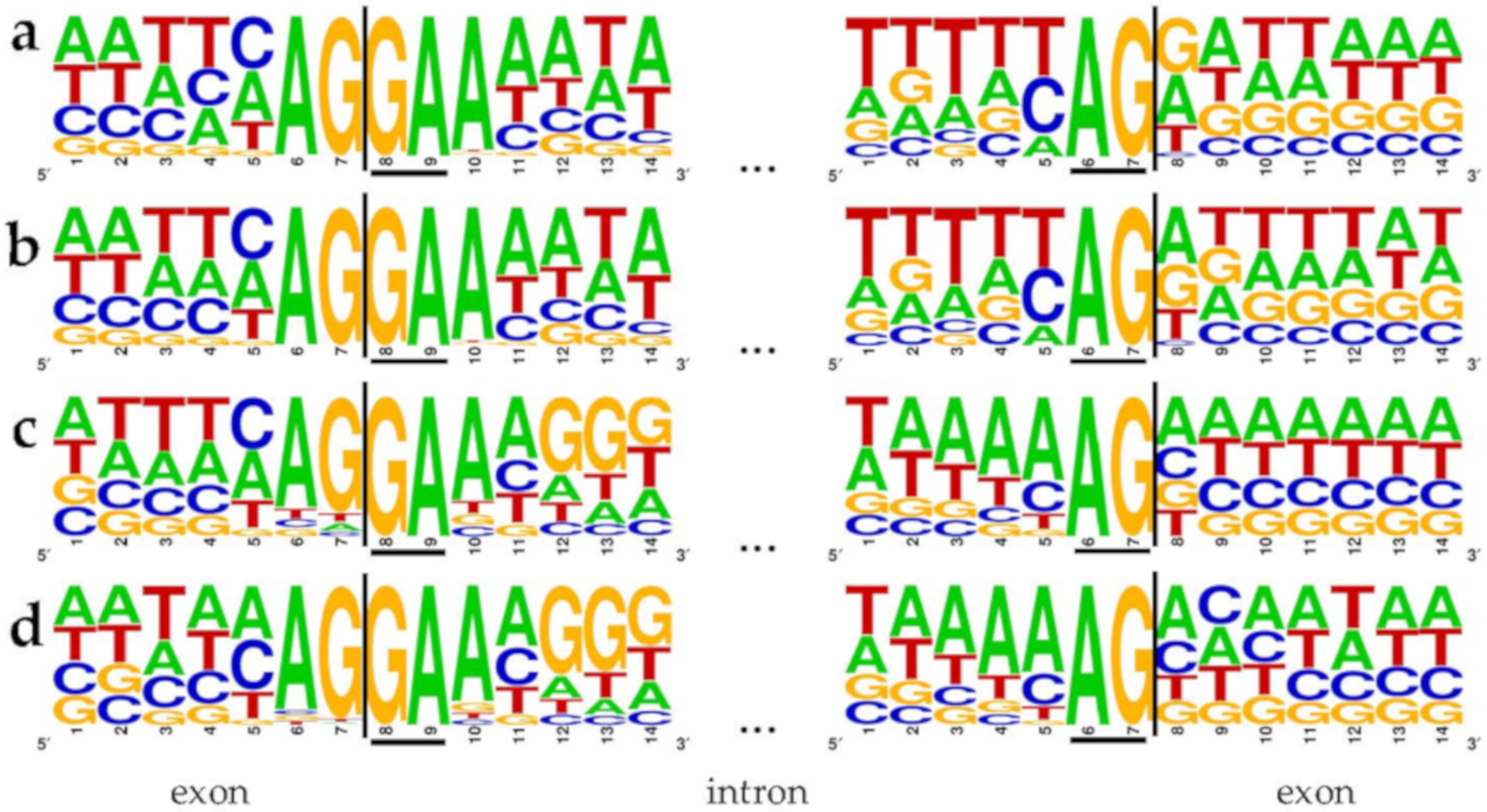
3.5. Conservation of Non-Canonical Splice Site Combinations across Species
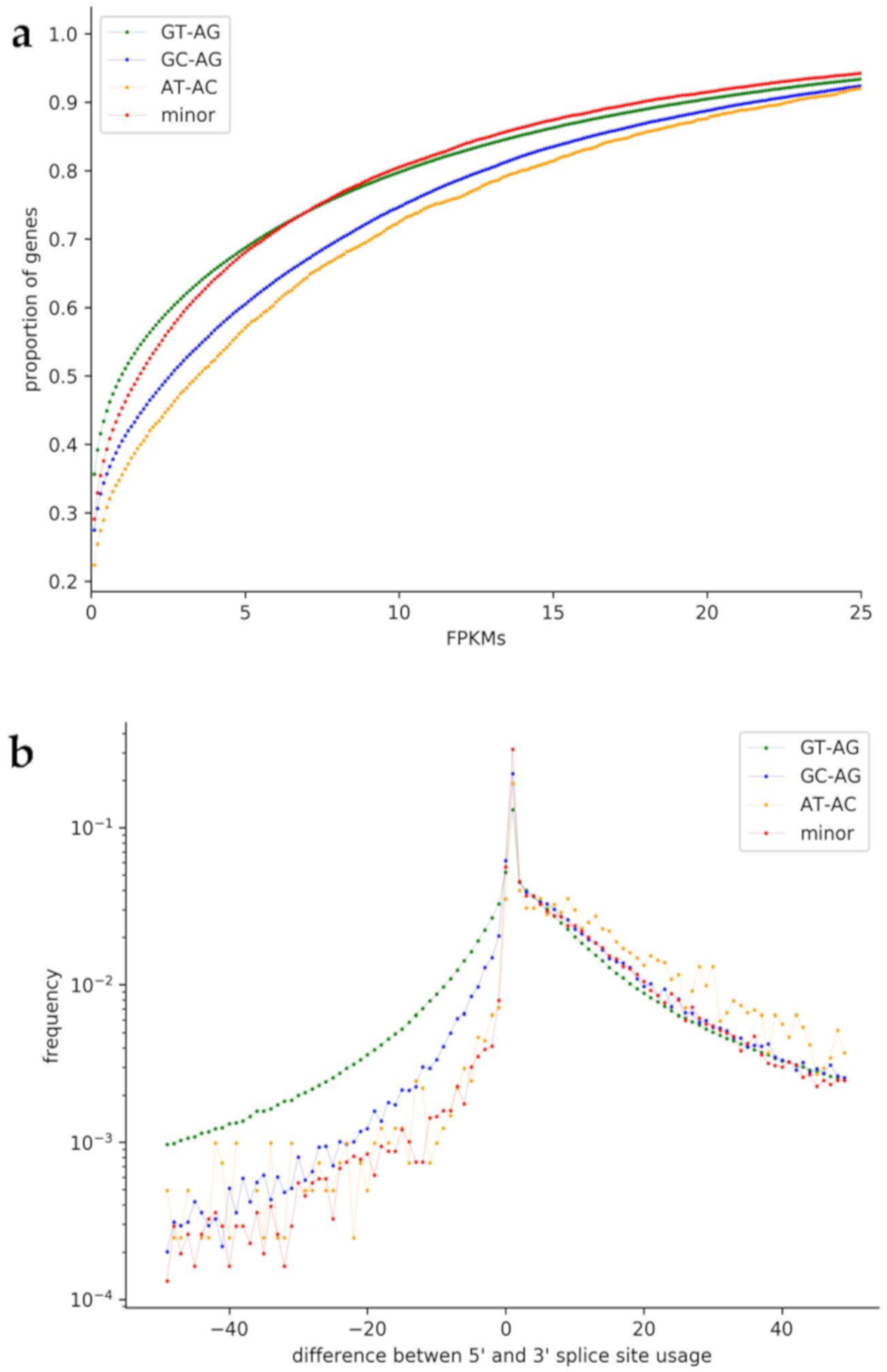
3.6. Usage of Non-Canonical Splice Sites
4. Discussion
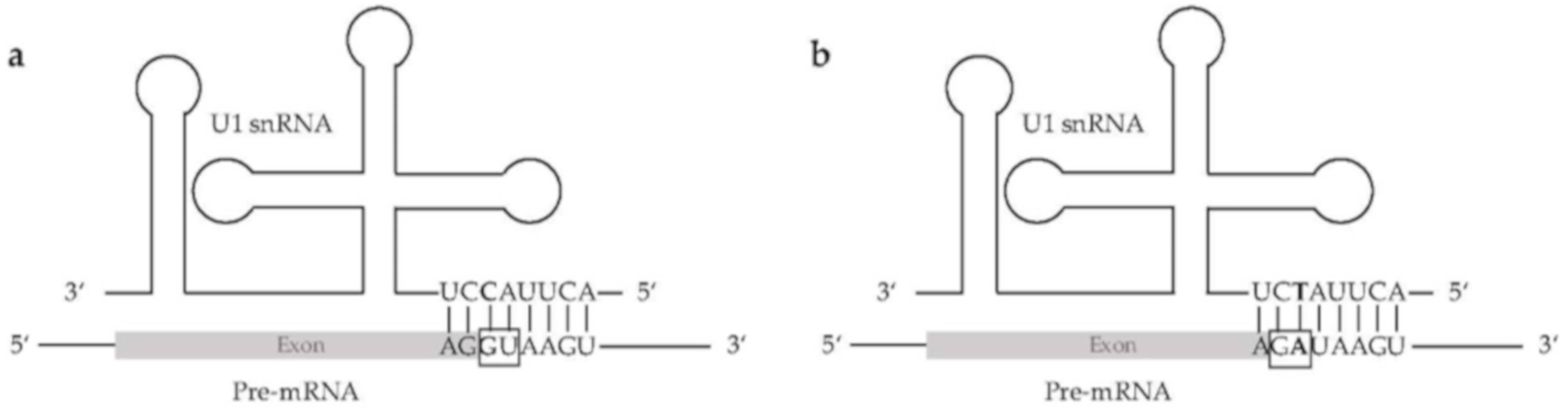
4.1. Analysis of Non-Canonical Splice Sites
4.2. High Diversity of Non-Canonical Splice Sites in Animals
4.3. CT-AC is a Frequent Splice Site Combination in Fungal Annotations
4.4. Conservation of Non-Canonical Splice Site Combinations Across Species
4.5. Usage of Non-Canonical Splice Sites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moore, M.J.; Sharp, P.A. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science 1992, 256, 992–997. [Google Scholar] [CrossRef]
- Barbosa-Morais, N.L.; Irimia, M.; Pan, Q.; Xiong, H.Y.; Gueroussov, S.; Lee, L.J.; Slobodeniuc, V.; Kutter, C.; Watt, S.; Çolak, R.; et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012, 338, 1587–1593. [Google Scholar] [CrossRef] [Green Version]
- Ben-Dov, C.; Hartmann, B.; Lundgren, J.; Valcárcel, J. Genome-wide analysis of alternative pre-mRNA splicing. J. Biol. Chem. 2008, 283, 1229–1233. [Google Scholar] [CrossRef] [Green Version]
- Matlin, A.J.; Clark, F.; Smith, C.W. Understanding alternative splicing: Towards a cellular code. Nat. Rev. Mol. Cell Biol. 2005, 6, 386–398. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.R.; Blazquez, L.; Ule, J. Lessons from non-canonical splicing. Nat. Rev. Genet. 2016, 17, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Chen, B.; Ye, Q.; Shao, J.; Lyu, Z.; Wen, J. Sense-antisense gene overlap causes evolutionary retention of the few introns in Giardia genome and the implications. bioRxiv 2018, 333310. [Google Scholar] [CrossRef] [Green Version]
- Chorev, M.; Carmel, L. The function of introns. Front. Genet. 2012, 3, 55. [Google Scholar] [CrossRef] [Green Version]
- Burset, M.; Seledtsov, I.A.; Solovyev, V.V. Analysis of canonical and non-canonical splice sites in mammalian genomes. Nucleic Acids Res. 2000, 28, 4364–4375. [Google Scholar] [CrossRef]
- Pucker, B.; Brockington, S.F. Genome-wide analyses supported by RNA-Seq reveal non-canonical splice sites in plant genomes. BMC Genom. 2018, 19, 980. [Google Scholar] [CrossRef] [Green Version]
- Bon, E.; Casaregola, S.; Blandin, G.; Llorente, B.; Neuvéglise, C.; Munsterkotter, M.; Guldener, U.; Mewes, H.W.; Helden, J.V.; Dujon, B.; et al. Molecular evolution of eukaryotic genomes: Hemiascomycetous yeast spliceosomal introns. Nucleic Acids Res. 2003, 31, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, J.M., Jr. The recent origins of spliceosomal introns revisited. Curr. Opin. Genet. Dev. 1998, 8, 637–648. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA1. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.A.; Grate, L.; Spingola, M.; Ares, M., Jr. Test of intron predictions reveals novel splice sites, alternatively spliced mRNAs and new introns in meiotically regulated genes of yeast. Nucleic Acids Res. 2000, 28, 1700–1706. [Google Scholar] [CrossRef] [PubMed]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.A.; Burge, C.B. Classification of introns: U2-type or U12-type. Cell 1997, 91, 875–879. [Google Scholar] [CrossRef] [Green Version]
- Hall, S.L.; Padgett, R.A. Requirement of U12 snRNA for in vivo splicing of a minor class of eukaryotic nuclear pre-mRNA introns. Science 1996, 271, 1716–1718. [Google Scholar] [CrossRef]
- Turunen, J.J.; Niemelä, E.H.; Verma, B.; Frilander, M.J. The significant other: Splicing by the minor spliceosome. Wiley Interdiscip. Rev. RNA 2013, 4, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, R.C.; Incorvaia, R.; Padgett, R.A. Terminal intron dinucleotide sequences do not distinguish between U2-and U12-dependent introns. Mol. Cell 1997, 1, 151–160. [Google Scholar] [CrossRef]
- Sheth, N.; Roca, X.; Hastings, M.L.; Roeder, T.; Krainer, A.R.; Sachidanandam, R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006, 34, 3955–3967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parada, G.E.; Munita, R.; Cerda, C.A.; Gysling, K. A comprehensive survey of non-canonical splice sites in the human transcriptome. Nucleic Acids Res. 2014, 42, 10564–10578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Krainer, A.R. Splicing of a divergent subclass of AT-AC introns requires the major spliceosomal snRNAs. RNA 1997, 3, 586–601. [Google Scholar] [PubMed]
- Kubota, T.; Roca, X.; Kimura, T.; Kokunai, Y.; Nishino, I.; Sakoda, S.; Krainer, A.R.; Takahashi, M.P. A mutation in a rare type of intron in a sodium-channel gene results in aberrant splicing and causes myotonia. Hum. Mutat. 2011, 32, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, M.E.; Fica, S.M.; Galej, W.P.; Norman, C.M.; Newman, A.J.; Nagai, K. Postcatalytic spliceosome structure reveals mechanism of 3′–splice site selection. Science 2017, 358, 1283–1288. [Google Scholar] [CrossRef] [Green Version]
- Burge, C.B.; Tuschl, T.; Sharp, P.A. Splicing of precursors to mRNAs by the spliceosomes. Cold Spring Harb. Monogr. Ser. 1999, 37, 525–560. [Google Scholar]
- Roca, X.; Krainer, A.R.; Eperon, I.C. Pick one, but be quick: 5′ splice sites and the problems of too many choices. Genes Dev. 2013, 27, 129–144. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y. The spliceosome: A protein-directed metalloribozyme. J. Mol. Biol. 2017, 429, 2640–2653. [Google Scholar] [CrossRef]
- Kupfer, D.M.; Drabenstot, S.D.; Buchanan, K.L.; Lai, H.; Zhu, H.; Dyer, D.W.; Roe, B.A.; Murphy, J.W. Introns and splicing elements of five diverse fungi. Eukaryot. Cell 2004, 3, 1088–1100. [Google Scholar] [CrossRef] [Green Version]
- Kitamura–Abe, S.; Itoh, H.; Washio, T.; Tsutsumi, A.; Tomita, M. Characterization of the splice sites in GT–AG and GC–AG introns in higher eukaryotes using full-length cDNAs. J. Bioinform. Comput. Biol. 2004, 2, 309–331. [Google Scholar] [CrossRef]
- Michael, D.; Manyuan, L. Intron—Exon structures of eukaryotic model organisms. Nucleic Acids Res. 1999, 27, 3219–3228. [Google Scholar] [CrossRef] [Green Version]
- Modrek, B.; Resch, A.; Grasso, C.; Lee, C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001, 29, 2850–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pucker, B.; Holtgräwe, D.; Weisshaar, B. Consideration of non-canonical splice sites improves gene prediction on the Arabidopsis thaliana Niederzenz-1 genome sequence. BMC Res. Notes 2017, 10, 667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparks, M.E.; Brendel, V. Incorporation of splice site probability models for non-canonical introns improves gene structure prediction in plants. Bioinformatics 2005, 21, iii20–iii30. [Google Scholar] [CrossRef] [Green Version]
- Dubrovina, A.S.; Kiselev, K.V.; Zhuravlev, Y.N. The role of canonical and noncanonical pre-mRNA splicing in plant stress responses. BioMed Res. Int. 2013, 2013, 264314. [Google Scholar] [CrossRef]
- Alexandrov, N.N.; Troukhan, M.E.; Brover, V.V.; Tatarinova, T.; Flavell, R.B.; Feldmann, K.A. Features of Arabidopsis genes and genome discovered using full-length cDNAs. Plant Mol. Biol. 2006, 60, 69–85. [Google Scholar] [CrossRef]
- Niu, X.; Luo, D.; Gao, S.; Ren, G.; Chang, L.; Zhou, Y.; Luo, X.; Li, Y.; Hou, P.; Tang, W.; et al. A conserved unusual posttranscriptional processing mediated by short, direct repeated (SDR) sequences in plants. J. Genet. Genom. 2010, 37, 85–99. [Google Scholar] [CrossRef]
- Erkelenz, S.; Theiss, S.; Kaisers, W.; Ptok, J.; Walotka, L.; Müller, L.; Hillebrand, F.; Brillen, A.L.; Sladek, M.; Schaal, H. Ranking noncanonical 5′ splice site usage by genome-wide RNA-seq analysis and splicing reporter assays. Genome Res. 2018, 28, 1826–1840. [Google Scholar] [CrossRef]
- Grützmann, K.O.; Szafranski, K.A.; Pohl, M.A.; Voigt, K.E.; Petzold, A.N.; Schuster, S.T. Fungal alternative splicing is associated with multicellular complexity and virulence: A genome-wide multi-species study. DNA Res. 2013, 21, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Kriventseva, E.V.; Tegenfeldt, F.; Petty, T.J.; Waterhouse, R.M.; Simao, F.A.; Pozdnyakov, I.A.; Ioannidis, P.; Zdobnov, E.M. OrthoDB v8: Update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2014, 43, D250–D256. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M.; International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pucker, B. Updates for Genome-Wide Investigation of Non-Canonical Splice Sites in Eukaryotes. Zenodo 2019. [Google Scholar] [CrossRef]
- Pucker, B.; Frey, K. RNA-Seq Read Coverage Depth of Splice Sites in Animals; Bielefeld University: Bielefeld, Germany, 2019. [Google Scholar]
- Pucker, B.; Frey, K. RNA-Seq Read Coverage Depth of Splice Sites in Fungi; Bielefeld University: Bielefeld, Germany, 2019. [Google Scholar]
- Dobin, A.; Gingeras, T.R. Comment on “TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions” by Kim et al. Available online: https://www.biorxiv.org/content/10.1101/000851v1.full?%3Fcollection (accessed on 15 February 2020).
- Ho, J.; Tumkaya, T.; Aryal, S.; Choi, H.; Claridge-Chang, A. Moving beyond P values: Everyday data analysis with estimation plots. bioRxiv 2018, 377978. [Google Scholar] [CrossRef] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Heip, C. A new index measuring evenness. J. Mar. Biol. Assoc. UK 1974, 54, 555–557. [Google Scholar] [CrossRef]
- Breslow, N. A generalized Kruskal-Wallis test for comparing K samples subject to unequal patterns of censorship. Biometrika 1970, 57, 579–594. [Google Scholar] [CrossRef]
- Jones, E.; Oliphant, T.; Peterson, P. SciPy: Open Source Scientific Tools for Python. 2001. Available online: https://www.bibsonomy.org/bibtex/21b37d2cc741af879d7958f2f7c23c420/microcuts (accessed on 18 February 2020).
- Plotly Technologies Inc. Collaborative Data Science. Available online: https://plot.ly (accessed on 15 February 2020).
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Stark, H.; Dube, P.; Lührmann, R.; Kastner, B. Arrangement of RNA and proteins in the spliceosomal U1 small nuclear ribonucleoprotein particle. Nature 2001, 409, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Moore, M.J.; Brockington, S.F.; Soltis, D.E.; Wong, G.K.-S.; Carpenter, E.J.; Zhang, Y.; Chen, L.; Yan, Z.; Xie, Y.; et al. Dissecting Molecular Evolution in the Highly Diverse Plant Clade Caryophyllales Using Transcriptome Sequencing. Mol. Biol. Evol. 2015, 32, 2001–2014. [Google Scholar] [CrossRef] [PubMed]
- Schilbert, H.M.; Pellegrinelli, V.; Rodriguez-Cuenca, S.; Vidal-Puig, A.; Pucker, B. Harnessing natural diversity to identify key amino acid residues in prolidase. BioRxiv 2018, 423475. [Google Scholar] [CrossRef]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [Green Version]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Baasner, J.-S.; Howard, D.; Pucker, B. Influence of neighboring small sequence variants on functional impact prediction. BioRxiv 2019. [Google Scholar] [CrossRef]
- Frey, K.; Pucker, B. Animal, fungi, and plant genome sequences harbour different non-canonical splice sites. BioRxiv 2019, 616565. [Google Scholar] [CrossRef]
- Jackson, I.J. A reappraisal of non-consensus mRNA splice sites. Nucleic Acids Res. 1991, 19, 3795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, H.M. Non-canonical GA and GG 5′Intron Donor Splice Sites Are Common in the Copepod Eurytemora affinis. G3 Genes Genomes Genet. 2017, 7, 3967–3969. [Google Scholar]
- Lee, C.E. Evolutionary mechanisms of habitat invasions, using the copepod Eurytemora affinis as a model system. Evol. Appl. 2016, 9, 248–270. [Google Scholar] [CrossRef]
- Seo, H.-C.; Kube, M.; Edvardsen, R.B.; Jensen, M.F.; Beck, A.; Spriet, E.; Gorsky, G.; Thompson, E.M.; Lehrach, H.; Reinhardt, R.; et al. Miniature genome in the marine chordate Oikopleura dioica. Science 2001, 294, 2506. [Google Scholar] [CrossRef]
- Brackenridge, S.; Wilkie, A.O.; Screaton, G.R. Efficient use of a ‘dead-end’GA 5′ splice site in the human fibroblast growth factor receptor genes. EMBO J. 2003, 22, 1620–1631. [Google Scholar] [CrossRef] [Green Version]
- Mount, S.M. A catalogue of splice junction sequences. Nucleic Acids Res. 1982, 10, 459–472. [Google Scholar] [CrossRef]
- Tyler, B.M. Phytophthora sojae: Root rot pathogen of soybean and model oomycete. Mol. Plant Pathol. 2007, 8, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Förster, H.; Coffey, M.O.; Elwood, H.; Sogin, M.L. Sequence analysis of the small subunit ribosomal RNAs of three zoosporic fungi and implications for fungal evolution. Mycologia 1990, 82, 306–312. [Google Scholar] [CrossRef]
- Shen, D.; Ye, W.; Dong, S.; Wang, Y.; Dou, D. Characterization of intronic structures and alternative splicing in Phytophthora sojae by comparative analysis of expressed sequence tags and genomic sequences. Can. J. Microbiol. 2011, 57, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Garalde, D.R.; Snell, E.A.; Jachimowicz, D.; Sipos, B.; Lloyd, J.H.; Bruce, M.; Pantic, N.; Admassu, T.; James, P.; Warland, A.; et al. Highly parallel direct RNA sequencing on an array of nanopores. Nat. Methods 2018, 15, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Aebi, M.; Hornig, H.; Padgett, R.A.; Reiser, J.; Weissmann, C. Sequence requirements for splicing of higher eukaryotic nuclear pre-mRNA. Cell 1986, 47, 555–565. [Google Scholar] [CrossRef]
- Yan, X.; Sablok, G.; Feng, G.; Ma, J.; Zhao, H.; Sun, X. nagnag: Identification and quantification of NAGNAG alternative splicing using RNA-Seq data. FEBS Lett. 2015, 589, 1766–1770. [Google Scholar] [CrossRef] [Green Version]
- Talerico, M.E.; Berget, S.M. Effect of 5′ splice site mutations on splicing of the preceding intron. Mol. Cell. Biol. 1990, 10, 6299–6305. [Google Scholar] [CrossRef] [Green Version]
- Berget, S.M. Exon recognition in vertebrate splicing. J. Biol. Chem. 1995, 270, 2411–2414. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, J.M.; Breton, G.; Schroeder, J.I. mRNA metabolism of flowering-time regulators in wild-type Arabidopsis revealed by a nuclear cap binding protein mutant, abh1. Plant J. 2007, 50, 1049–1062. [Google Scholar] [CrossRef]
- Donaldson, M.E.; Saville, B.J. Natural antisense transcripts in fungi: Natural antisense transcripts in fungi. Mol. Microbiol. 2012, 85, 405–417. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GT-AG | GC-AG | AT-AC | Others | |
|---|---|---|---|---|
| Animals | 98.334% | 0.983% | 0.106% | 0.577% |
| Fungi | 98.715% | 1.009% | 0.019% | 0.257% |
| Plants | 97.886% | 1.488% | 0.092% | 0.534% |
| All | 98.265% | 1.074% | 0.101% | 0.560% |
| Splice Site Combination | Introns Divisible by Three and without In-Frame Stop Codon | Number of Introns Divisible by Three and without In-Frame Stop Codon | |
|---|---|---|---|
| Animals | GT-AG | 1.4% | n = 922561 |
| AT-AC | 2.8% | n = 1945 | |
| GC-AG | 3.0% | n = 19166 | |
| Others | 4.1% | n = 15554 | |
| Fungi | GT-AG | 9.2% | n = 208522 |
| AT-AC | 10.7% | n = 46 | |
| GC-AG | 13.3% | n = 3096 | |
| Others | 13.1% | n = 785 | |
| Plants | GT-AG | 2.7% | n = 382538 |
| AT-AC | 3.6% | n = 10356 | |
| GC-AG | 4.8% | n = 478 | |
| Others | 5.8% | n = 4481 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frey, K.; Pucker, B. Animal, Fungi, and Plant Genome Sequences Harbor Different Non-Canonical Splice Sites. Cells 2020, 9, 458. https://doi.org/10.3390/cells9020458
Frey K, Pucker B. Animal, Fungi, and Plant Genome Sequences Harbor Different Non-Canonical Splice Sites. Cells. 2020; 9(2):458. https://doi.org/10.3390/cells9020458
Chicago/Turabian StyleFrey, Katharina, and Boas Pucker. 2020. "Animal, Fungi, and Plant Genome Sequences Harbor Different Non-Canonical Splice Sites" Cells 9, no. 2: 458. https://doi.org/10.3390/cells9020458