CD38 in Neurodegeneration and Neuroinflammation


Abstract
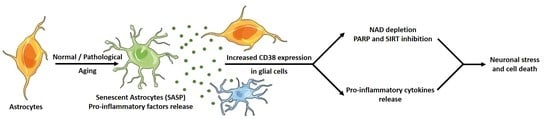
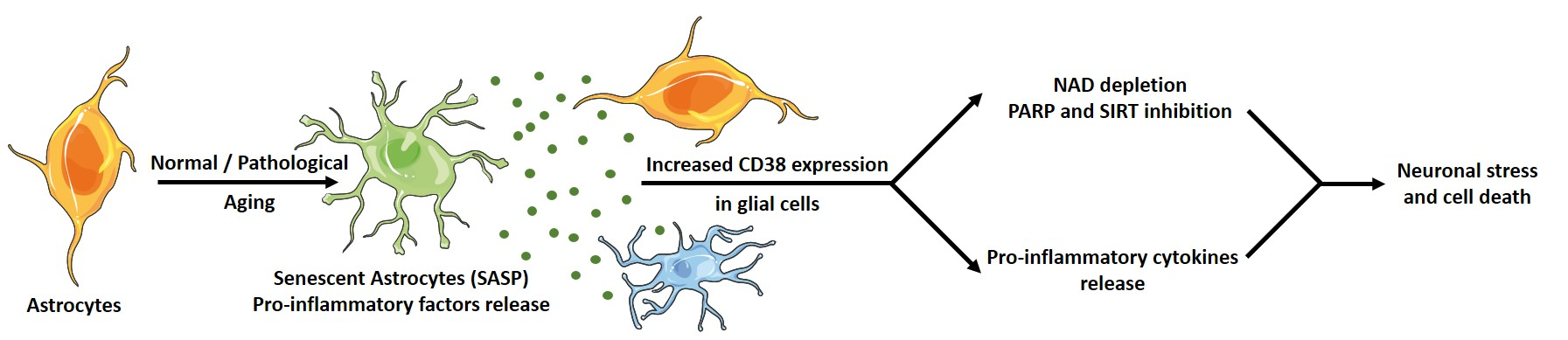
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Complex Biology of CD38
3. Physiological Role of CD38 in the Brain
3.1. CD38 Expression in the Brain
3.2. Physiological Role of CD38 Enzyme Function in the Brain
3.3. Physiological Role of CD38 Receptor Function
3.4. CD38 Role in Neuroinflammation
4. CD38 Involvement in Neurodegeneration and Neuroinflammation: Evidences
5. Aging, the Missing Link between CD38 and Neurodegeneration/Inflammation?
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Mehta, D.; Jackson, R.; Paul, G.; Shi, J.; Sabbagh, M. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs 2017, 26, 735–739. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Clement, J.; Wong, M.; Poljak, A.; Sachdev, P.; Braidy, N. The Plasma NAD + Metabolome Is Dysregulated in “Normal” Aging. Rejuvenation Res. 2018, 22, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.-H.; Lu, M.; Lee, B.-Y.; Ugurbil, K.; Chen, W. In vivo NAD assay reveals the intracellular NAD contents and redox state in healthy human brain and their age dependences. Proc. Natl. Acad. Sci. USA 2015, 112, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Guest, J.; Grant, R.; Mori, T.A.; Croft, K.D. Changes in oxidative damage, inflammation and [NAD(H)] with age in cerebrospinal fluid. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science (80-) 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Chini, E.N. CD38 as a regulator of cellular NAD: a novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 Protein as a Source of Extracellular Precursors for Sustained NAD + Biosynthesis in FK866-treated Tumor Cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef] [Green Version]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [Green Version]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar]
- Horenstein, L.A.; Stockinger, H.; Imhof, A.B.; Malavasi, F. CD38 binding to human myeloid cells is mediated by mouse and human CD31. Biochem. J. 1998, 330, 1129–1135. [Google Scholar] [CrossRef]
- Newman, P.J.; Newman, D.K. Signal Transduction Pathways Mediated by PECAM-1. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 953–964. [Google Scholar] [CrossRef]
- Kalinowska, A.; Losy, J. PECAM-1, a key player in neuroinflammation. Eur. J. Neurol. 2006, 13, 1284–1290. [Google Scholar] [CrossRef]
- Fang, C.; Li, T.; Li, Y.; Xu, G.J.; Deng, Q.W.; Chen, Y.J.; Hou, Y.N.; Lee, H.C.; Zhao, Y.J. CD38 produces nicotinic acid adenosine dinucleotide phosphate in the lysosome. J. Biol. Chem. 2018, 293, 8151–8160. [Google Scholar] [CrossRef] [Green Version]
- Zubiaur, M.; Fernández, O.; Ferrero, E.; Salmerón, J.; Malissen, B.; Malavasi, F.; Sancho, J. CD38 Is Associated with Lipid Rafts and upon Receptor Stimulation Leads to Akt/Protein Kinase B and Erk Activation in the Absence of the CD3-ζ Immune Receptor Tyrosine-based Activation Motifs. J. Biol. Chem. 2002, 277, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Ceni, C.; Pochon, N.; Brun, V.; Muller-Steffner, H.; Andrieux, A.; Grunwald, D.; Schuber, F.; De Waard, M.; Lund, F.; Villaz, M.; et al. CD38-dependent ADP-ribosyl cyclase activity in developing and adult mouse brain. Biochem. J. 2003, 370, 175–183. [Google Scholar] [CrossRef]
- Yamada, M.; Mizuguchi, M.; Otsuka, N.; Ikeda, K.; Takahashi, H. Ultrastructural localization of CD38 immunoreactivity in rat brain. Brain Res. 1997, 756, 52–60. [Google Scholar] [CrossRef]
- Braidy, N.; Poljak, A.; Grant, R.; Jayasena, T.; Mansour, H.; Chan-Ling, T.; Guillemin, G.J.; Smythe, G.; Sachdev, P. Mapping NAD+ metabolism in the brain of ageing Wistar rats: potential targets for influencing brain senescence. Biogerontology 2014, 15, 177–198. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Otsuka, N.; Sato, M.; Ishii, Y.; Kon, S.; Yamada, M.; Nishina, H.; Katada, T.; Ikeda, K. Neuronal localization of CD38 antigen in the human brain. Brain Res. 1995, 697, 235–240. [Google Scholar] [CrossRef]
- Quintana, D.S.; Rokicki, J.; van der Meer, D.; Alnæs, D.; Kaufmann, T.; Córdova-Palomera, A.; Dieset, I.; Andreassen, O.A.; Westlye, L.T. Oxytocin pathway gene networks in the human brain. Nat. Commun. 2019, 10, 668. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Wu, D.; Ding, X.; Ying, W. CD38 plays key roles in both antioxidation and cell survival of H2O2-treated primary rodent astrocytes. Int. J. Physiol. Pathophysiol. Pharmacol. 2014, 6, 102–108. [Google Scholar]
- Mamik, M.K.; Banerjee, S.; Walseth, T.F.; Hirte, R.; Tang, L.; Borgmann, K.; Ghorpade, A. HIV-1 and IL-1β regulate astrocytic CD38 through mitogen-activated protein kinases and nuclear factor-κB signaling mechanisms. J. Neuroinflamm. 2011, 8, 145. [Google Scholar] [CrossRef] [Green Version]
- Kou, W.; Banerjee, S.; Eudy, J.; Smith, L.M.; Persidsky, R.; Borgmann, K.; Wu, L.; Sakhuja, N.; Deshpande, M.S.; Walseth, T.F.; et al. CD38 regulation in activated astrocytes: Implications for neuroinflammation and HIV-1 brain infection. J. Neurosci. Res. 2009, 87, 2326–2339. [Google Scholar] [CrossRef]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef]
- Ma, Y.; Jiang, J.; Wang, L.; Nie, H.; Xia, W.; Liu, J.; Ying, W. CD38 is a key enzyme for the survival of mouse microglial BV2 cells. Biochem. Biophys. Res. Commun. 2012, 418, 714–719. [Google Scholar] [CrossRef]
- Ma, Y.; Cao, W.; Wang, L.; Jiang, J.; Nie, H.; Wang, B.; Wei, X.; Ying, W. Basal CD38/cyclic ADP-ribose-dependent signaling mediates ATP release and survival of microglia by modulating connexin 43 hemichannels. Glia 2014, 62, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Jacob-Hirsch, J.; Amariglio, N.; Rechavi, G.; Moutin, M.-J.; Lund, F.E.; Stein, R. Dual Role of CD38 in Microglial Activation and Activation-Induced Cell Death. J. Immunol. 2008, 181, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Hattori, T.; Kaji, M.; Ishii, H.; Jureepon, R.; Takarada-Iemata, M.; Minh Ta, H.; Manh Le, T.; Konno, A.; Hirai, H.; Shiraishi, Y.; et al. CD38 positively regulates postnatal development of astrocytes cell-autonomously and oligodendrocytes non-cell-autonomously. Glia 2017, 65, 974–989. [Google Scholar] [CrossRef] [PubMed]
- Nelissen, T.P.; Bamford, R.A.; Tochitani, S.; Akkus, K.; Kudzinskas, A.; Yokoi, K.; Okamoto, H.; Yamamoto, Y.; Burbach, J.P.H.; Matsuzaki, H.; et al. CD38 is Required for Dendritic Organization in Visual Cortex and Hippocampus. Neuroscience 2018, 372, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Higashida, H.; Salmina, A.B.; Olovyannikova, R.Y.; Hashii, M.; Yokoyama, S.; Koizumi, K.; Jin, D.; Liu, H.-X.; Lopatina, O.; Amina, S.; et al. Cyclic ADP-ribose as a universal calcium signal molecule in the nervous system. Neurochem. Int. 2007, 51, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Ceni, C.; Muller-Steffner, H.; Lund, F.; Pochon, N.; Schweitzer, A.; De Waard, M.; Schuber, F.; Villaz, M.; Moutin, M.-J. Evidence for an Intracellular ADP-ribosyl Cyclase/NAD+-glycohydrolase in Brain from CD38-deficient Mice. J. Biol. Chem. 2003, 278, 40670–40678. [Google Scholar] [CrossRef] [Green Version]
- Soares, S.; Thompson, M.; White, T.; Isbell, A.; Yamasaki, M.; Prakash, Y.; Lund, F.E.; Galione, A.; Chini, E.N. NAADP as a second messenger: neither CD38 nor base-exchange reaction are necessary for in vivo generation of NAADP in myometrial cells. AJP Cell Physiol. 2006, 292, C227–C239. [Google Scholar] [CrossRef] [Green Version]
- Jin, D.; Liu, H.-X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [Google Scholar] [CrossRef]
- Martucci, L.L.; Amar, M.; Chaussenot, R.; Benet, G.; Bauer, O.; de Zélicourt, A.; Nosjean, A.; Launay, J.-M.; Callebert, J.; Sebrié, C.; et al. A multiscale analysis in CD38 −/− mice unveils major prefrontal cortex dysfunctions. FASEB J. 2019, 33, 5823–5835. [Google Scholar] [CrossRef]
- Lee, M.R.; Shin, J.H.; Deschaine, S.; Daurio, A.M.; Stangl, B.L.; Yan, J.; Ramchandani, V.A.; Schwandt, M.L.; Grodin, E.N.; Momenan, R.; et al. A role for the CD38 rs3796863 polymorphism in alcohol and monetary reward: evidence from CD38 knockout mice and alcohol self-administration, [11C]-raclopride binding, and functional MRI in humans. Am. J. Drug Alcohol Abuse 2019, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Demeule, M.; Labelle, M.; Rgina, A.; Berthelet, F.; Béliveau, R. Isolation of endothelial cells from brain, lung, and kidney: Expression of the multidrug resistance p-glycoprotein isoforms. Biochem. Biophys. Res. Commun. 2001, 281, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Lertkiatmongkol, P.; Liao, D.; Mei, H.; Hu, Y.; Newman, P.J. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr. Opin. Hematol. 2016, 23, 253–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilan, N.; Mohsenin, A.; Cheung, L.; Madri, J.A. PECAM-1 shedding during apoptosis generates a membrane-anchored truncated molecule with unique signaling characteristics. FASEB J. 2001, 15, 362–372. [Google Scholar] [CrossRef]
- Goldberger, A.; Middleton, K.A.; Oliver, J.A.; Paddock, C.; Yan, H.C.; DeLisser, H.M.; Albelda, S.M.; Newman, P.J. Biosynthesis and processing of the cell adhesion molecule PECAM-1 includes production of a soluble form. J. Biol. Chem. 1994, 269, 17183–17191. [Google Scholar]
- Zaremba, J.; Losy, J. sPECAM-1 in serum and CSF of acute ischaemic stroke patients. Acta Neurol. Scand. 2002, 106, 292–298. [Google Scholar] [CrossRef]
- Zaremba, J.; Losy, J. Soluble platelet endothelial cell adhesion molecule-1 in ischaemic stroke patients is related to the extent of early brain damage. Cent. J. Immunol. 2002, 27, 90–96. [Google Scholar]
- Banerjee, S.; Walseth, T.F.; Borgmann, K.; Wu, L.; Bidasee, K.R.; Kannan, M.S.; Ghorpade, A. CD38/Cyclic ADP-Ribose Regulates Astrocyte Calcium Signaling: Implications for Neuroinflammation and HIV-1-Associated Dementia. J. Neuroimmune Pharmacol. 2008, 3, 154–164. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Blacher, E.; Vaknine, H.; Lund, F.E.; Stein, R.; Mayo, L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro. Oncol. 2012, 14, 1037–1049. [Google Scholar] [CrossRef]
- Blacher, E.; Ben Baruch, B.; Levy, A.; Geva, N.; Green, K.D.; Garneau-Tsodikova, S.; Fridman, M.; Stein, R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int. J. Cancer 2015, 136, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Liu, Z.-Y.; Ai, Y.-H.; Zhang, L.-N.; Zou, Y.; Peng, Q.-Y. Blocking the CD38/cADPR pathway plays a double-edged role in LPS stimulated microglia. Neuroscience 2017, 361, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, K.-C.; Ryu, W.-I.; Amirault, K.M.; Healy, R.A.; Siegel, A.J.; McPhie, D.L.; Forester, B.; Cohen, B.M. Late-onset Alzheimer’s disease is associated with inherent changes in bioenergetics profiles. Sci. Rep. 2017, 7, 14038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakade, C.; Chong, R.; Bradley, E.; Thomas, B.; Morgan, J. Upregulation of GPR109A in Parkinson’s Disease. PLoS ONE 2014, 9, e109818. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, Q.; Bao, R.; Zhang, N.; Wang, Y.; Polo-Parada, L.; Tarim, A.; Alemifar, A.; Han, X.; Wilkins, H.M.; et al. Deletion of Nampt in Projection Neurons of Adult Mice Leads to Motor Dysfunction, Neurodegeneration, and Death. Cell Rep. 2017, 20, 2184–2200. [Google Scholar] [CrossRef] [Green Version]
- Braidy, N.; Lim, C.K.; Grant, R.; Brew, B.J.; Guillemin, G.J. Serum nicotinamide adenine dinucleotide levels through disease course in multiple sclerosis. Brain Res. 2013, 1537, 267–272. [Google Scholar] [CrossRef]
- Otsuka, K.; Mizuguchi, M.; Aizawa, T.; Haga, S.; Sato, M.; Inoya, H.; Namba, Y.; Machinami, R. Immunoreactivity in Alzheimer’s neurofibrillary tangles (abstract). Brain Pathol. 1994, 4, 558. [Google Scholar]
- Jude, J.A.; Dileepan, M.; Subramanian, S.; Solway, J.; Panettieri, R.A.; Walseth, T.F.; Kannan, M.S. miR-140-3p regulation of TNF-α-induced CD38 expression in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L460–L468. [Google Scholar] [CrossRef] [Green Version]
- Dileepan, M.; Jude, J.A.; Rao, S.P.; Walseth, T.F.; Panettieri, R.A.; Subramanian, S.; Kannan, M.S. MicroRNA-708 regulates CD38 expression through signaling pathways JNK MAP kinase and PTEN/AKT in human airway smooth muscle cells. Respir. Res. 2014, 15, 107. [Google Scholar] [CrossRef] [Green Version]
- Denk, J.; Boelmans, K.; Siegismund, C.; Lassner, D.; Arlt, S.; Jahn, H. MicroRNA Profiling of CSF Reveals Potential Biomarkers to Detect Alzheimer‘s Disease. PLoS ONE 2015, 10, e0126423. [Google Scholar] [CrossRef] [Green Version]
- Saad, M.; Lesage, S.; Saint-Pierre, A.; Corvol, J.-C.; Zelenika, D.; Lambert, J.-C.; Vidailhet, M.; Mellick, G.D.; Lohmann, E.; Durif, F.; et al. Genome-wide association study confirms BST1 and suggests a locus on 12q24 as the risk loci for Parkinson’s disease in the European population. Hum. Mol. Genet. 2011, 20, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Choe, C.; Lardong, K.; Gelderblom, M.; Ludewig, P.; Leypoldt, F.; Koch-Nolte, F.; Gerloff, C.; Magnus, T. CD38 Exacerbates Focal Cytokine Production, Postischemic Inflammation and Brain Injury after Focal Cerebral Ischemia. PLoS ONE 2011, 6, e19046. [Google Scholar] [CrossRef]
- Long, A.; Park, J.H.; Klimova, N.; Fowler, C.; Loane, D.J.; Kristian, T. CD38 Knockout Mice Show Significant Protection Against Ischemic Brain Damage Despite High Level Poly-ADP-Ribosylation. Neurochem. Res. 2017, 42, 283–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blacher, E.; Dadali, T.; Bespalko, A.; Haupenthal, V.J.; Grimm, M.O.W.; Hartmann, T.; Lund, F.E.; Stein, R.; Levy, A. Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann. Neurol. 2015, 78, 88–103. [Google Scholar] [CrossRef]
- Kim, S.; Kim, T.; Lee, H.-R.; Jang, E.-H.; Ryu, H.-H.; Kang, M.; Rah, S.-Y.; Yoo, J.; Lee, B.; Kim, J.-I.; et al. Impaired learning and memory in CD38 null mutant mice. Mol. Brain 2016, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, M.M.; Barth, S.; Greve, B.; Schumann, K.M.; Bartels, A.; Weissert, R. Identification of gene expression patterns crucially involved in experimental autoimmune encephalomyelitis and multiple sclerosis. Dis. Model. Mech. 2016, 9, 1211–1220. [Google Scholar] [CrossRef] [Green Version]
- Roboon, J.; Hattori, T.; Ishii, H.; Takarada-Iemata, M.; Le, T.M.; Shiraishi, Y.; Ozaki, N.; Yamamoto, Y.; Sugawara, A.; Okamoto, H.; et al. Deletion of CD38 Suppresses Glial Activation and Neuroinflammation in a Mouse Model of Demyelination. Front. Cell. Neurosci. 2019, 13, 258. [Google Scholar] [CrossRef]
- Chini, C.; Hogan, K.A.; Warner, G.M.; Tarragó, M.G.; Peclat, T.R.; Tchkonia, T.; Kirkland, J.L.; Chini, E. The NADase CD38 is induced by factors secreted from senescent cells providing a potential link between senescence and age-related cellular NAD+ decline. Biochem. Biophys. Res. Commun. 2019, 513, 486–493. [Google Scholar] [CrossRef]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [Green Version]
- Turnquist, C.; Horikawa, I.; Foran, E.; Major, E.O.; Vojtesek, B.; Lane, D.P.; Lu, X.; Harris, B.T.; Harris, C.C. p53 isoforms regulate astrocyte-mediated neuroprotection and neurodegeneration. Cell Death Differ. 2016, 23, 1515–1528. [Google Scholar] [CrossRef] [Green Version]
- Kritsilis, M.; Rizou, S.; Koutsoudaki, P.; Evangelou, K.; Gorgoulis, V.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Wiley, C.D.; Iyer, S.S.; Basisty, N.; et al. Aging-related inflammation driven by cellular senescence enhances NAD consumption via activation of CD38+ pro-inflammatory macrophages. bioRxiv 2019, 609438. [Google Scholar]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095.e10. [Google Scholar] [CrossRef] [Green Version]
- van de Donk, N.W.C.J.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Milo, R. Therapies for multiple sclerosis targeting B cells. Croat. Med. J. 2019, 60, 87–98. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerreiro, S.; Privat, A.-L.; Bressac, L.; Toulorge, D. CD38 in Neurodegeneration and Neuroinflammation. Cells 2020, 9, 471. https://doi.org/10.3390/cells9020471
Guerreiro S, Privat A-L, Bressac L, Toulorge D. CD38 in Neurodegeneration and Neuroinflammation. Cells. 2020; 9(2):471. https://doi.org/10.3390/cells9020471
Chicago/Turabian StyleGuerreiro, Serge, Anne-Laure Privat, Laurence Bressac, and Damien Toulorge. 2020. "CD38 in Neurodegeneration and Neuroinflammation" Cells 9, no. 2: 471. https://doi.org/10.3390/cells9020471