Non-Phosphorylatable PEA-15 Sensitises SKOV-3 Ovarian Cancer Cells to Cisplatin

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Lines and Cell Culture
2.3. Plasmid Transfection
2.4. Cell Fractionation
2.5. Western Blot
2.6. MTT Assay
2.7. cDNA Microarray Analysis
2.8. Correlation of Tumour Gene Expression Levels with the Survival of Cisplatin-Treated Patients in The Cancer Genome Atlas (TCGA)
2.9. Statistical Analysis
3. Results
3.1. PEA-15AA Sensitised SKOV-3 Cells to Cisplatin
3.2. PEA-15AA-Transfected SKOV-3 Cells Contain more Cytosolic p-ERK1/2 than PEA-15DD-Transfected Cells
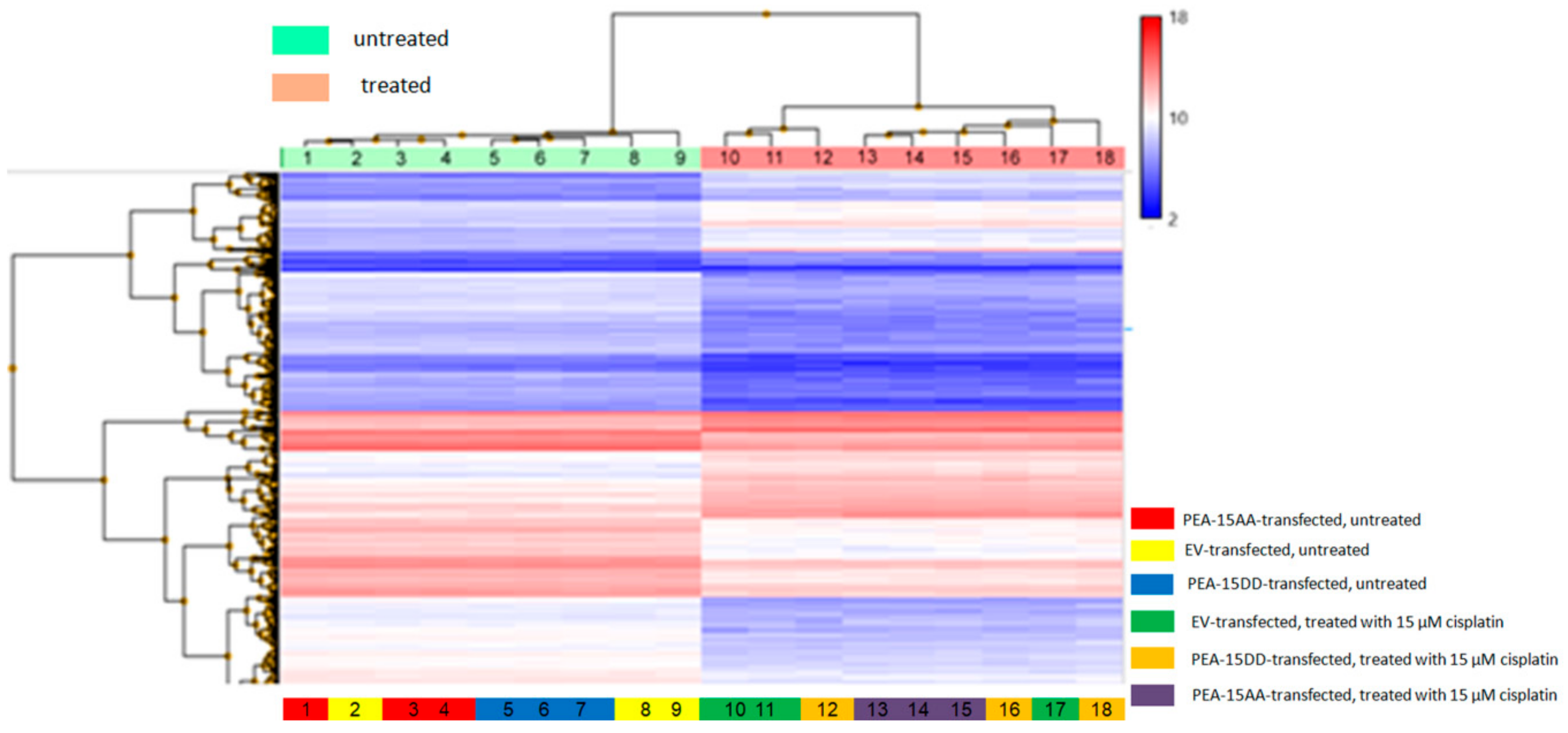
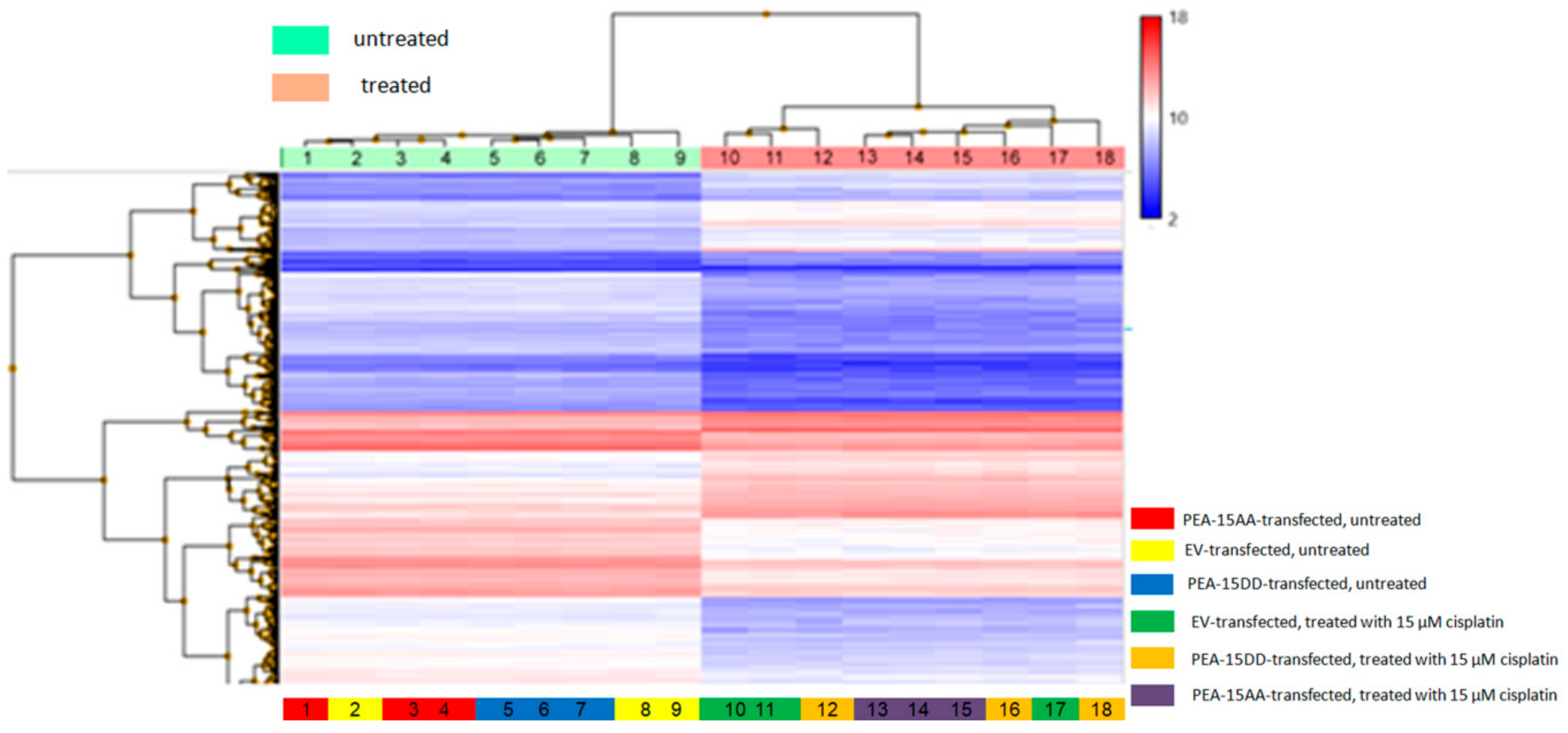
3.3. Differentially Expressed Genes in Transfected Cells
3.4. Correlation of Genes Differentially Expressed in the Comparisons Untreated SKOV-3-AA vs. SKOV-3-EV and SKOV-3-AA vs. SKOV-3-DD Cells with the Survival of Cisplatin-Treated Patients in The Cancer Genome Atlas (TCGA)
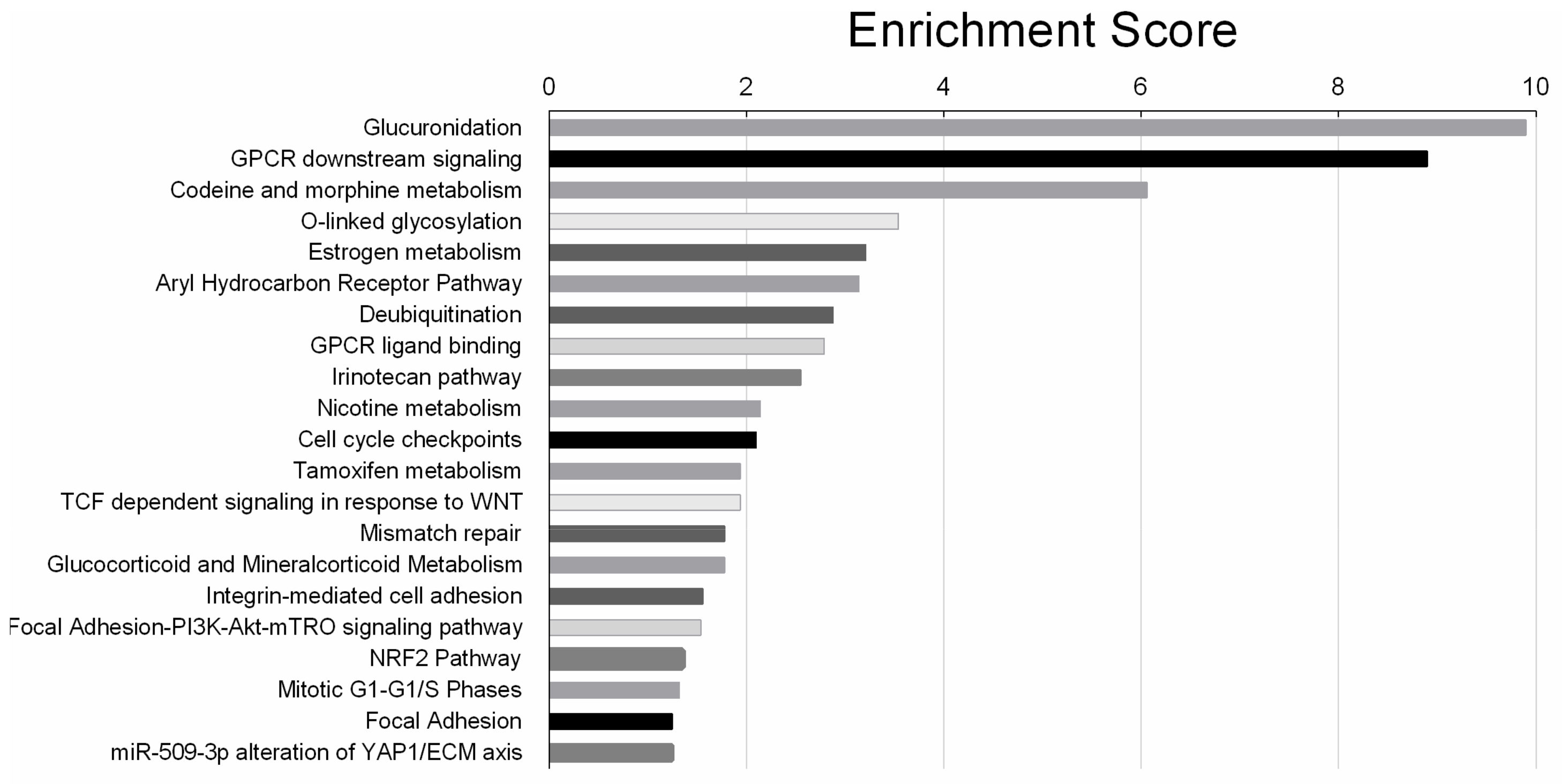
3.5. Pathway Analysis for the Genes Exclusively Regulated in SKOV-3-AA Cells Following Cisplatin Treatment
3.6. Evaluation of the Responsible Genes Within the Affected Pathways
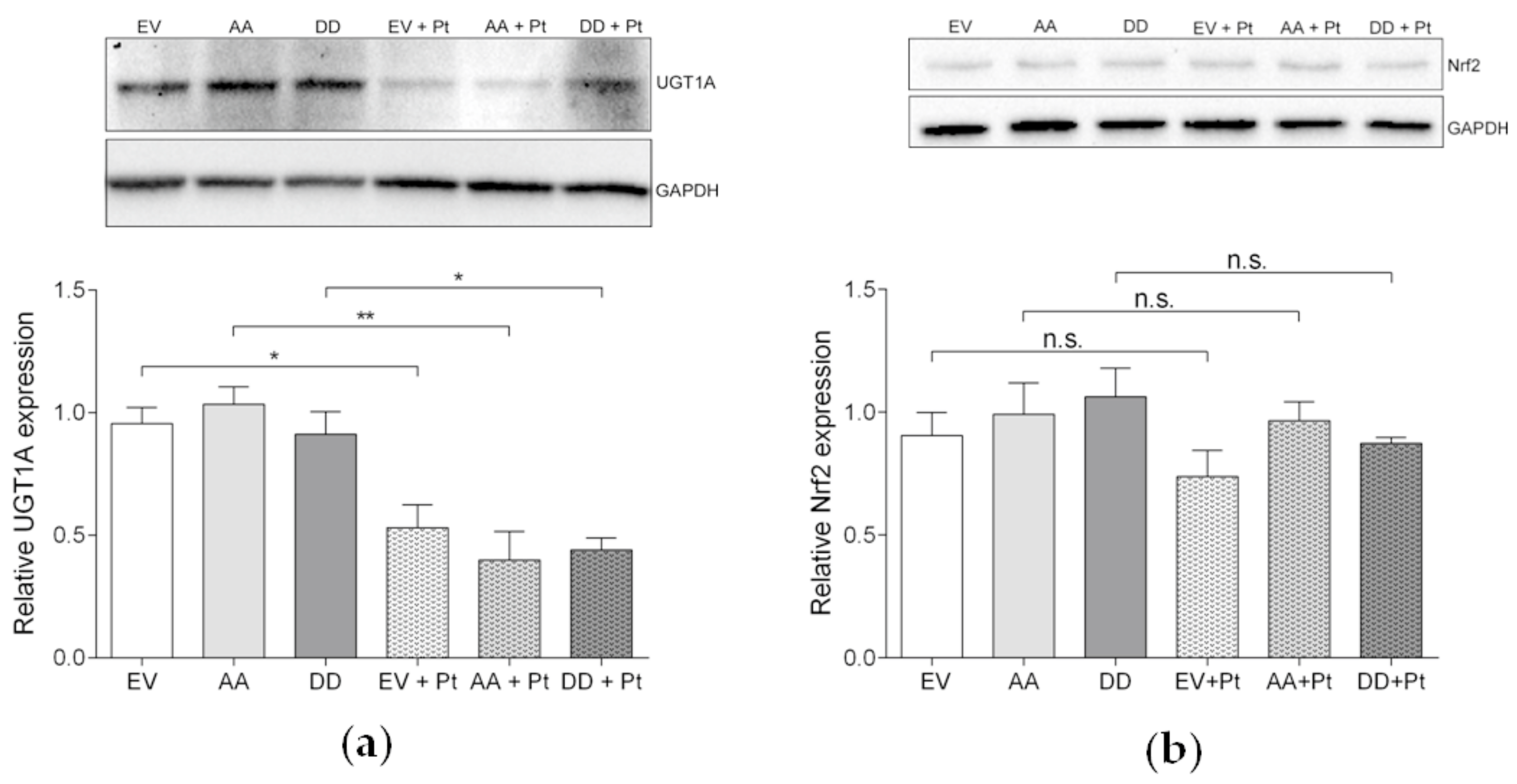
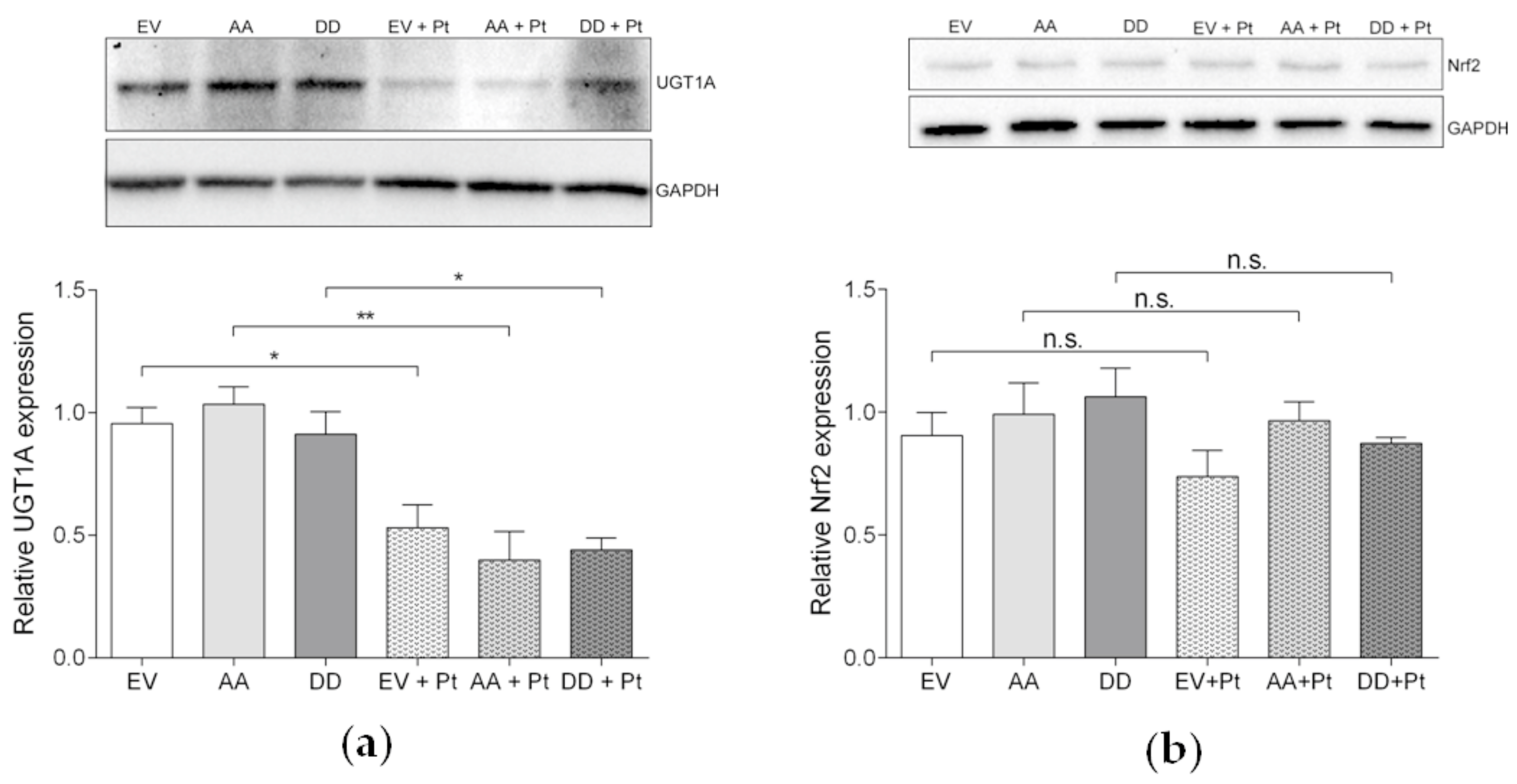
3.6.1. UGT1A and Nrf2 Pathway
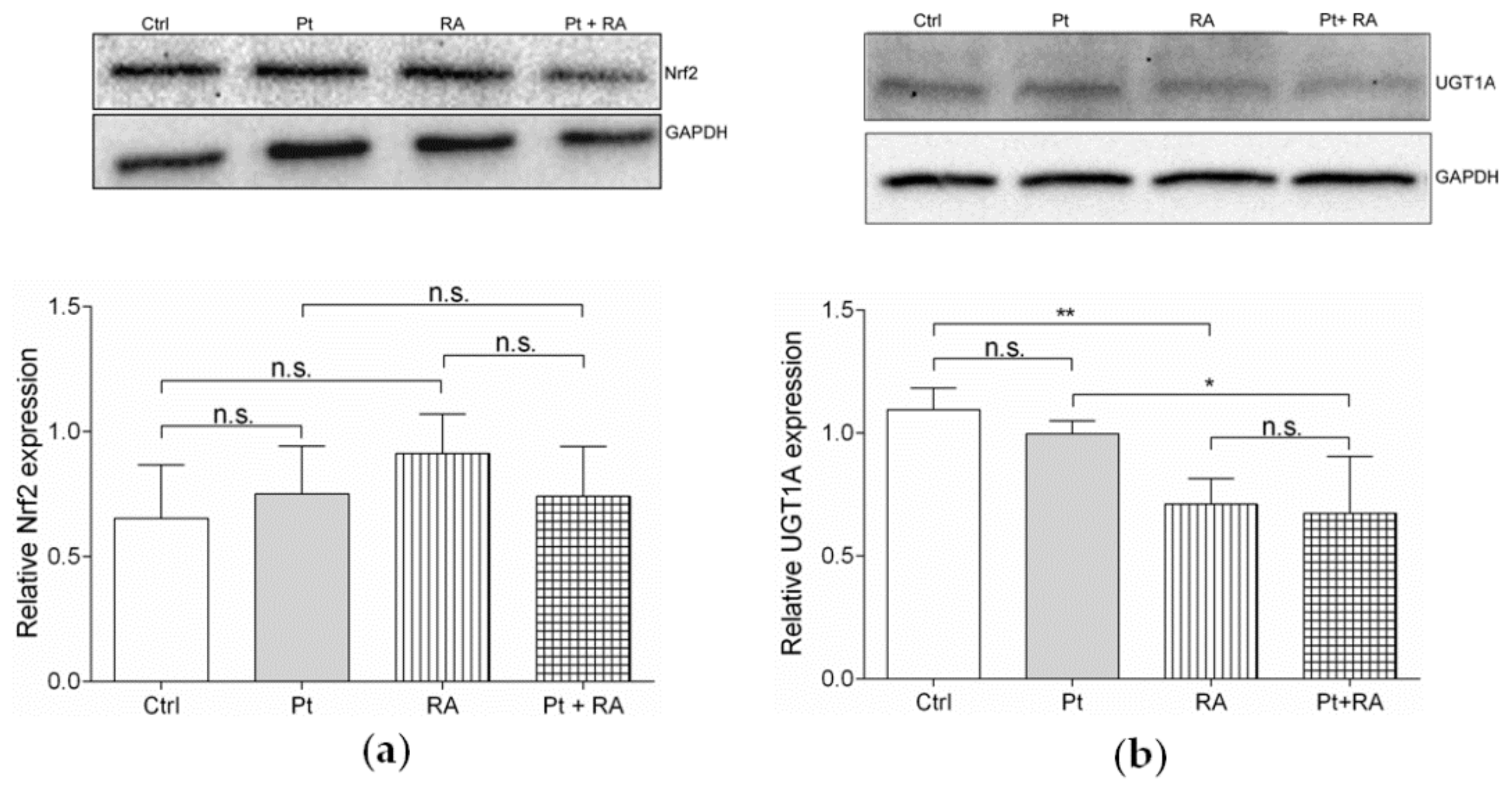
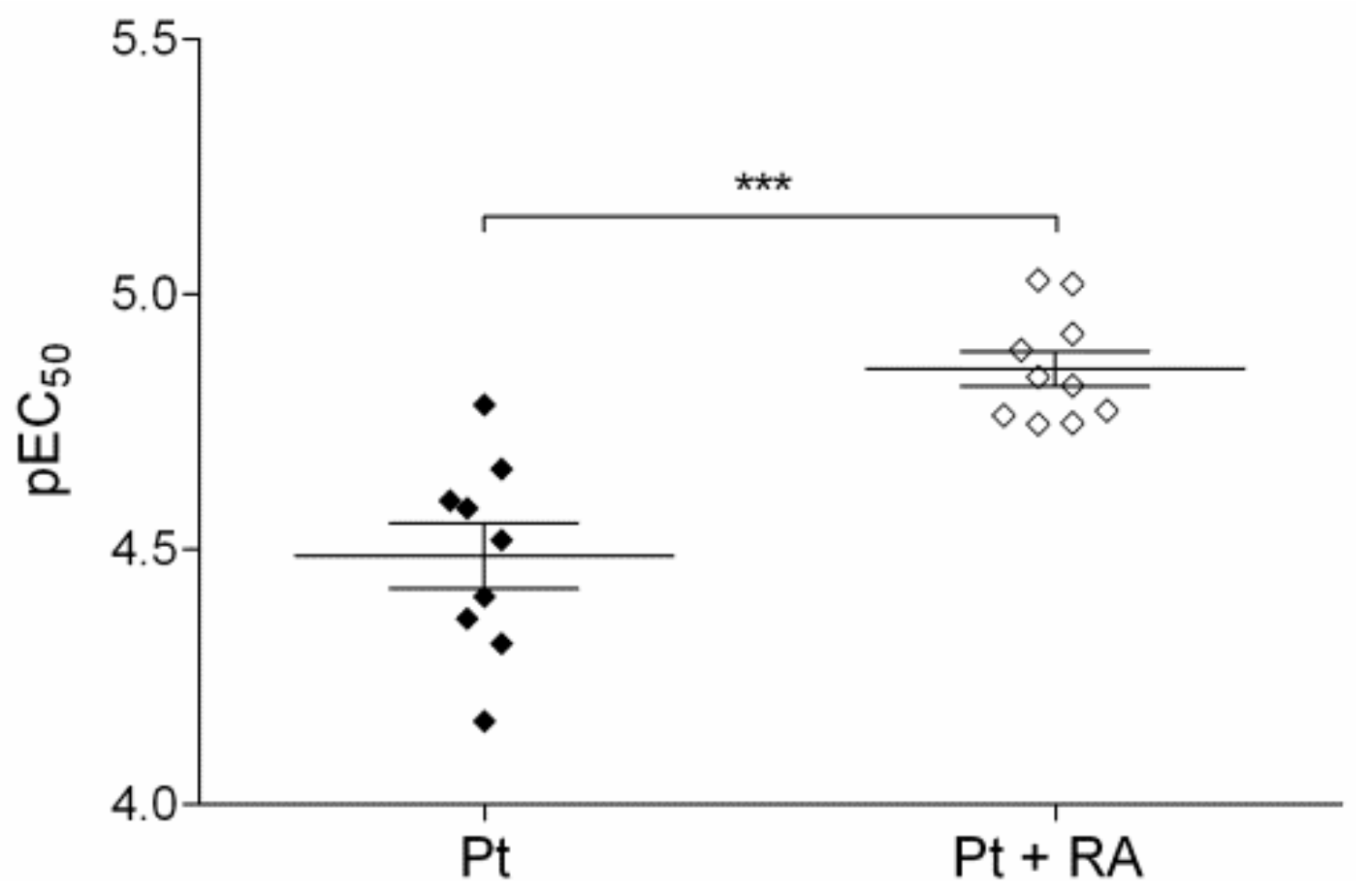
3.6.2. Retinoic Acid, an Inhibitor of Nrf2/ARE Pathway, Increases Cisplatin Sensitivity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharm. 2016, 77, 1103–1124. [Google Scholar] [CrossRef] [PubMed]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Gille, H.; Sharrocks, A.D.; Shaw, P.E. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature 1992, 358, 414–417. [Google Scholar] [CrossRef] [PubMed]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radziejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Boulton, T.G.; Yancopoulos, G.D.; Gregory, J.S.; Slaughter, C.; Moomaw, C.; Hsu, J.; Cobb, M.H. An insulin-stimulated protein kinase similar to yeast kinases involved in cell cycle control. Science 1990, 249, 64–67. [Google Scholar] [CrossRef]
- Fukuda, M.; Gotoh, Y.; Nishida, E. Interaction of MAP kinase with MAP kinase kinase: Its possible role in the control of nucleocytoplasmic transport of MAP kinase. EMBO J. 1997, 16, 1901–1908. [Google Scholar] [CrossRef] [Green Version]
- Reszka, A.A.; Seger, R.; Diltz, C.D.; Krebs, E.G.; Fischer, E.H. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc. Natl. Acad. Sci. USA 1995, 92, 8881–8885. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, M.; Mathers, J.; Dickinson, R.J.; Mandl, M.; Keyse, S.M. Both nuclear cytoplasmic shuttling of the dual specificity phosphatase MKP-3 and its ability to anchor MAP kinase in the cytoplasm are mediated by a conserved nuclear export signal. J. Biol. Chem. 2004, 279, 41882–41891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, M.; Fukuda, M.; Nishida, E. Two co-existing mechanisms for nuclear import of MAP kinase: Passive diffusion of a monomer and active transport of a dimer. EMBO J. 1999, 18, 5347–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehurst, A.W.; Wilsbacher, J.L.; You, Y.; Luby-Phelps, K.; Moore, M.S.; Cobb, M.H. ERK2 enters the nucleus by a carrier-independent mechanism. Proc. Natl. Acad. Sci. USA 2002, 99, 7496–7501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubayashi, Y.; Fukuda, M.; Nishida, E. Evidence for existence of a nuclear pore complex-mediated, cytosol-independent pathway of nuclear translocation of ERK MAP kinase in permeabilized cells. J. Biol. Chem. 2001, 276, 41755–41760. [Google Scholar] [CrossRef] [Green Version]
- Lenormand, P.; Sardet, C.; Pages, G.; L’Allemain, G.; Brunet, A.; Pouyssegur, J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not of their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 1993, 122, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.; Sarnecki, C.; Blenis, J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol. Cell Biol. 1992, 12, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Yuan, X.; Frodin, M.; Grummt, I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol. Cell 2003, 11, 405–413. [Google Scholar] [CrossRef]
- Marais, R.; Wynne, J.; Treisman, R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell 1993, 73, 381–393. [Google Scholar] [CrossRef]
- Danziger, N.; Yokoyama, M.; Jay, T.; Cordier, J.; Glowinski, J.; Chneiweiss, H. Cellular expression, developmental regulation, and phylogenic conservation of PEA-15, the astrocytic major phosphoprotein and protein kinase C substrate. J. Neurochem. 1995, 64, 1016–1025. [Google Scholar] [CrossRef]
- Estelles, A.; Yokoyama, M.; Nothias, F.; Vincent, J.D.; Glowinski, J.; Vernier, P. The major astrocytic phosphoprotein PEA-15 is encoded by two mRNAs conserved on their full length in mouse and human. J. Biol. Chem. 1996, 271, 14800–14806. [Google Scholar] [CrossRef] [Green Version]
- Ramos, J.W.; Kojima, T.K.; Hughes, P.E.; Fenczik, C.A.; Ginsberg, M.H. The death effector domain of PEA-15 is involved in its regulation of integrin activation. J. Biol. Chem. 1998, 273, 33897–33900. [Google Scholar] [CrossRef] [Green Version]
- Fiory, F.; Formisano, P.; Perruolo, G.; Beguinot, F. PED/PEA-15, a multifunctional protein controlling cell survival and glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E592–E601. [Google Scholar] [CrossRef] [Green Version]
- Hao, C.; Beguinot, F.; Condorelli, G.; Trencia, A.; Van Meir, E.G.; Yong, V.W.; Parney, I.F.; Roa, W.H.; Petruk, K.C. Induction and intracellular regulation of tumor necrosis factor related apoptosis-inducing ligand (TRAIL) mediated apoptosis in human malignant glioma cells. Cancer Res. 2001, 61, 1162–1170. [Google Scholar]
- Glading, A.; Koziol, J.A.; Krueger, J.; Ginsberg, M.H. PEA-15 inhibits tumor cell invasion by binding to extracellular signal-regulated kinase 1/2. Cancer Res. 2007, 67, 1536–1544. [Google Scholar] [CrossRef] [Green Version]
- Formisano, P.; Perruolo, G.; Libertini, S.; Santopietro, S.; Troncone, G.; Raciti, G.A.; Oriente, F.; Portella, G.; Miele, C.; Beguinot, F. Raised expression of the antiapoptotic protein ped/pea-15 increases susceptibility to chemically induced skin tumor development. Oncogene 2005, 24, 7012–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulzmaier, F.; Opoku-Ansah, J.; Ramos, J.W. Phosphorylation is the switch that turns PEA-15 from tumor suppressor to tumor promoter. Small GTPases 2012, 3, 173–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, H.; Danziger, N.; Cordier, J.; Glowinski, J.; Chneiweiss, H. Characterization of PEA-15, a major substrate for protein kinase C in astrocytes. J. Biol. Chem. 1993, 268, 5911–5920. [Google Scholar] [PubMed]
- Kubes, M.; Cordier, J.; Glowinski, J.; Girault, J.A.; Chneiweiss, H. Endothelin induces a calcium-dependent phosphorylation of PEA-15 in intact astrocytes: Identification of Ser104 and Ser116 phosphorylated, respectively, by protein kinase C and calcium/calmodulin kinase II in vitro. J. Neurochem. 1998, 71, 1307–1314. [Google Scholar] [CrossRef] [Green Version]
- Callaway, K.; Abramczyk, O.; Martin, L.; Dalby, K.N. The anti-apoptotic protein PEA-15 is a tight binding inhibitor of ERK1 and ERK2, which blocks docking interactions at the D-recruitment site. Biochemistry 2007, 46, 9187–9198. [Google Scholar] [CrossRef]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Camonis, J.; Ginsberg, M.H.; et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Bartholomeusz, C.; Itamochi, H.; Nitt, M.; Saya, H.; Ginsberg, M.H.; Ueno, N.T. Antitumor effect of E1A in ovarian cancer by cytoplasmic sequestration of activated ERK by PEA-15. Oncogene 2006, 25, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomeusz, C.; Rosen, D.; Wei, C.; Kazansky, A.; Yamasaki, F.; Takahashi, T.; Itamochi, H.; Kondo, S.; Liu, J.; Ueno, N.T. PEA-15 induces autophagy in human ovarian cancer cells and is associated with prolonged overall survival. Cancer Res. 2008, 68, 9302–9310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Bartholomeusz, C.; Krishnamurthy, S.; Liu, P.; Saso, H.; LaFortune, T.A.; Hortobagyi, G.N.; Ueno, N.T. PEA-15 unphosphorylated at both serine 104 and serine 116 inhibits ovarian cancer cell tumorigenicity and progression through blocking β-catenin. Oncogenesis 2012, 1, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Bartholomeusz, C.; Ahmed, A.A.; Kazansky, A.; Diao, L.; Baggerly, K.A.; Hortobagyi, G.N.; Ueno, N.T. Bisphosphorylated PEA-15 sensitizes ovarian cancer cells to Paclitaxel by impairing the microtubule-destabilizing effect of SCLIP. Mol. Cancer 2013, 12, 1099–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, P.K.; Krohn, R.I.; Hermanson, G.T.; Mallia, A.K.; Gartner, F.H.; Provenzano, M.D.; Fujimoto, E.K.; Goeke, N.M.; Olson, B.J.; Klenk, D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985, 150, 76–85. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human tumor cell lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. Cancer Genome Atlas Research Network. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Li, Q.; Du, Y.; Yun, J.; Xie, Y.; DeBerardinis, R.J.; Xiao, G. Genomic regression analysis of coordinated expression. Nat. Commun. 2017, 8, 2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 Axis in Cancer Cell Growth and Chemoresistance. Oxid. Med. Cell Longev. 2016, 1958174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atencia, R.; Garcia-Sanz, M.; Unda, F.; Arechaga, J. Apoptosis during retinoic acid-induced differentiation of F9 embryonal carcinoma cells. Exp. Cell Res. 1994, 214, 663–667. [Google Scholar] [CrossRef]
- Funke, V.; Lehmann-Koch, J.; Bickeböller, M.; Benner, A.; Tagscherer, K.E.; Grund, K.; Pfeifer, M.; Herpel, E.; Schirmacher, P.; Chang-Claude, J.; et al. The PEA-15/PED protein regulates cellular survival and invasiveness in colorectal carcinomas. Cancer Lett. 2013, 335, 431–440. [Google Scholar] [CrossRef]
- Eckert, A.; Böck, B.C.; Tagscherer, K.E.; Haas, T.L.; Grund, K.; Sykora, J.; Herold-Mende, C.; Ehemann, V.; Hollstein, M.; Chneiweiss, H.; et al. The PEA-15/PED protein protects glioblastoma cells from glucose deprivation induced apoptosis via the ERK/MAP kinase pathway. Oncogene 2008, 27, 1155–1166. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Verdier-Pinard, P.; Fernandez-Fuentes, N.; Burd, B.; Angeletti, R.; Fiser, A.; Horwitz, S.B.; Orr, G.A. Insights into the mechanism of microtubule stabilization by Taxol. Proc. Natl. Acad. Sci. USA 2006, 103, 10166–10173. [Google Scholar] [CrossRef] [Green Version]
- Gregg, J.; Fraizer, G. Transcriptional Regulation of EGR1 by EGF and the ERK Signaling Pathway in Prostate Cancer Cells. Genes Cancer 2011, 2, 900–909. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Sutariya, V.; Bishayee, A.; Bhatia, D. Sequential activation of Elk-1/Egr-1/GADD45α by arsenic. Oncotarget 2014, 5, 3862–3870. [Google Scholar] [CrossRef]
- Calogero, A.; Porcellini, A.; Lombari, V.; Fabbiano, C.; Arcella, A.; Miscusi, M.; Ponti, D.; Ragona, G. Sensitivity to cisplatin in primary cell lines derived from human glioma correlates with levels of EGR-1 expression. Cancer Cell Int. 2011, 11, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Addario, M.; Arora, P.D.; McCulloch, C.A. Role of p38 in stress activation of Sp1. Genes 2006, 379, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Milanini-Mongiat, J.; Pouysségur, J.; Pagès, G. Identification of two Sp1 phosphorylation sites for p42/p44 mitogen-activated protein kinases: Their implication in vascular endothelial growth factor gene transcription. J. Biol. Chem. 2002, 277, 20631–20639. [Google Scholar] [CrossRef] [Green Version]
- Yue, J.; Lan, S.; Yuan, C.; Shen, Z. Prognostic values of filamin-A status for topoisomerase II poison chemotherapy. Int. J. Biol. Sci. 2012, 8, 442–450. [Google Scholar] [CrossRef]
- Jin, Y.Z.; Pei, C.Z.; Wen, L.Y. FLNA is a predictor of chemoresistance and poor survival in cervical cancer. Biomark. Med. 2016, 10, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Savoy, R.M.; Ghosh, P.M. The dual role of filamin A in cancer: Can’t live with (too much of) it, can’t live without it. Endocr. Relat. Cancer 2013, 20, R341–R356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999, 286, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Tang, H.; Liu, P.; Kong, Y.; Wu, M.; Xiao, X.; Yang, L.; Gao, J.; Wei, W.; Lee, J.; et al. Development of PEA-15 using a potent non-viral vector for therapeutic application in breast cancer. Cancer Lett. 2015, 356, 374–381. [Google Scholar] [CrossRef] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment Condition 1 | Treatment Condition 2 | Number of Genes |
|---|---|---|
| SKOV-3-EV, untreated | SKOV-3- AA, untreated | 3 |
| SKOV-3-EV, untreated | SKOV-3-DD, untreated | 18 |
| SKOV-3-AA, untreated | SKOV-3- DD, untreated | 10 |
| SKOV-3-EV, untreated | SKOV-3-EV, 15 µM cisplatin, 24 h | 4430 |
| SKOV-3-AA, untreated | SKOV-3-AA, 15 µM cisplatin, 24 h | 4197 |
| SKOV-3-DD, untreated | SKOV-3-DD, 15 µM cisplatin, 24 h | 4110 |
| Gene | Fold Change | Hazard Ratio 1 | p-Value | FDR (thr. 0.2) 2 | FDR (thr. 0.05) |
|---|---|---|---|---|---|
| EGR1 | −2.22 | 0.7068 | 0.00322 ** | 0.034099 | 0.008525 |
| NAV3 | −2.18 | 0.7062 | 0.00317 ** | 0.033948 | 0.008487 |
| GPRC5C | 2.14 | 1.4745 | 0.000971 ** | 0.023936 | 0.005984 |
| Gene | Fold change | Hazard ratio 1 | p-value | FDR (thr. 0.2) 2 | FDR (thr. 0.05) |
| TSTD3 | 2.16 | 0.7545 | 0.0162 * | 0.053933 | 0.013483 |
| ELFN2 | 2.09 | 0.9036 | 0.385 | 0.139261 | 0.034815 |
| LOC100287225 | −2.37 | n.a. 3 | n.a. | n.a. | n.a. |
| TIPARP | −2.19 | 0.7505 | 0.015 * | 0.052694 | 0.013174 |
| PRKG1 | −2.00 | 0.7532 | 0.0154 * | 0.053205 | 0.013301 |
| ND6 | −2.07 | 0.8492 | 0.163 | 0.10641 | 0.026602 |
| RBM26 | −2.42 | 0.8951 | 0.342 | 0.133674 | 0.033418 |
| FLNA | −2.86 | 0.7523 | 0.0152 * | 0.052997 | 0.013249 |
| MAVS | −2.24 | 0.6656 | 0.000569 ** | 0.020231 | 0.005058 |
| GTSF1L | −2.07 | n.a. | n.a. | n.a. | n.a. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilruba, S.; Grondana, A.; Schiedel, A.C.; Ueno, N.T.; Bartholomeusz, C.; Cinatl Jr, J.; McLaughlin, K.-M.; Wass, M.N.; Michaelis, M.; Kalayda, G.V. Non-Phosphorylatable PEA-15 Sensitises SKOV-3 Ovarian Cancer Cells to Cisplatin. Cells 2020, 9, 515. https://doi.org/10.3390/cells9020515
Dilruba S, Grondana A, Schiedel AC, Ueno NT, Bartholomeusz C, Cinatl Jr J, McLaughlin K-M, Wass MN, Michaelis M, Kalayda GV. Non-Phosphorylatable PEA-15 Sensitises SKOV-3 Ovarian Cancer Cells to Cisplatin. Cells. 2020; 9(2):515. https://doi.org/10.3390/cells9020515
Chicago/Turabian StyleDilruba, Shahana, Alessia Grondana, Anke C. Schiedel, Naoto T. Ueno, Chandra Bartholomeusz, Jindrich Cinatl Jr, Katie-May McLaughlin, Mark N. Wass, Martin Michaelis, and Ganna V. Kalayda. 2020. "Non-Phosphorylatable PEA-15 Sensitises SKOV-3 Ovarian Cancer Cells to Cisplatin" Cells 9, no. 2: 515. https://doi.org/10.3390/cells9020515
APA StyleDilruba, S., Grondana, A., Schiedel, A. C., Ueno, N. T., Bartholomeusz, C., Cinatl Jr, J., McLaughlin, K.-M., Wass, M. N., Michaelis, M., & Kalayda, G. V. (2020). Non-Phosphorylatable PEA-15 Sensitises SKOV-3 Ovarian Cancer Cells to Cisplatin. Cells, 9(2), 515. https://doi.org/10.3390/cells9020515