Hepatocyte Injury and Hepatic Stem Cell Niche in the Progression of Non-Alcoholic Steatohepatitis




Abstract
:1. Introduction
2. Hepatocyte Damage in NAFLD
2.1. Hepatocytes in Physiological Turnover and Regeneration
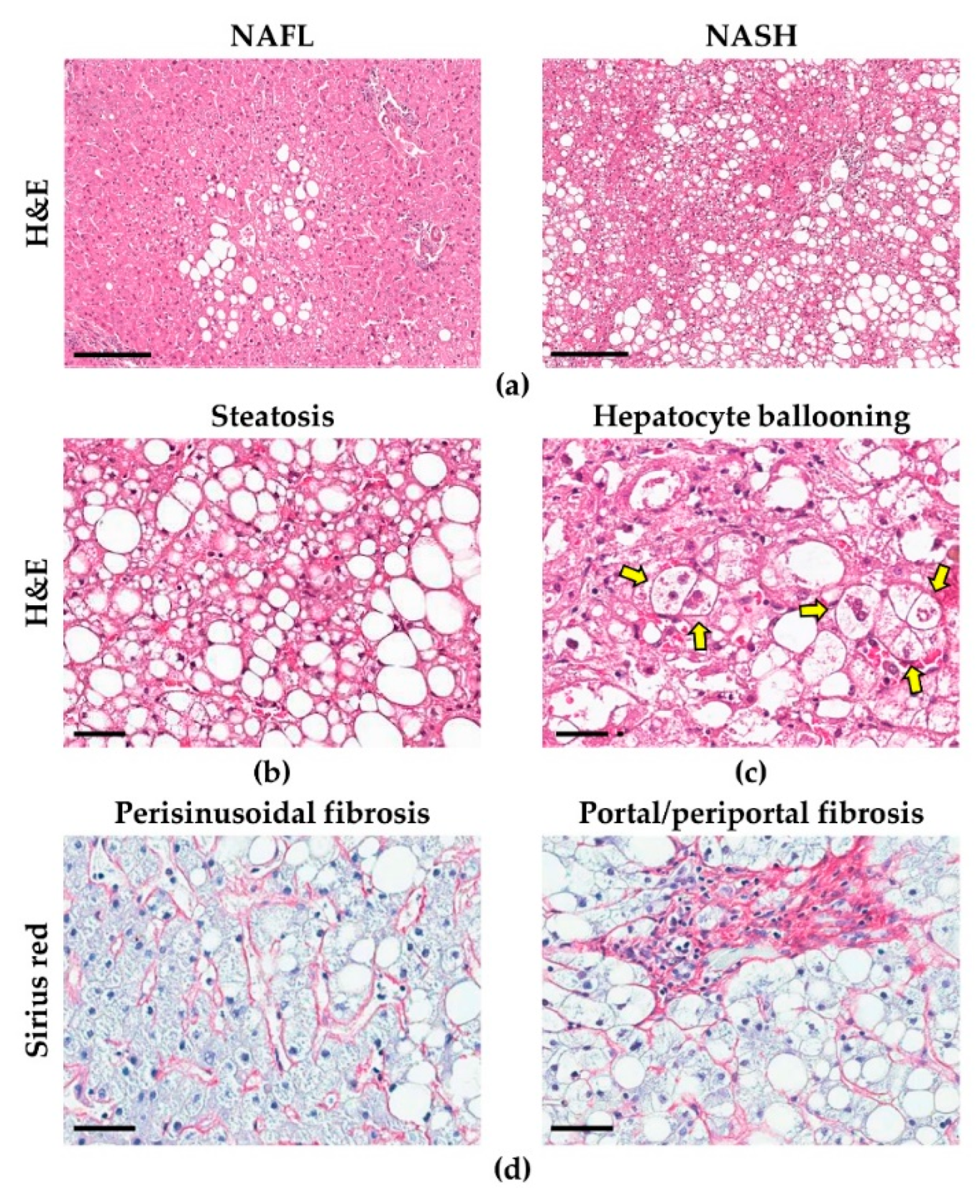
2.2. Morphological Alterations in Hepatocytes
2.3. Lipotoxicity in Hepatocytes
2.4. Endoplasmic Reticulum Stress and Mitochondrial Dysfunction in NAFLD
2.5. Hepatocyte Autophagy and Apoptosis in NAFLD
3. Hepatic Stem/progenitor Cells (HpSCs)
3.1. HpSCs are Involved in the Liver Regenerative Response
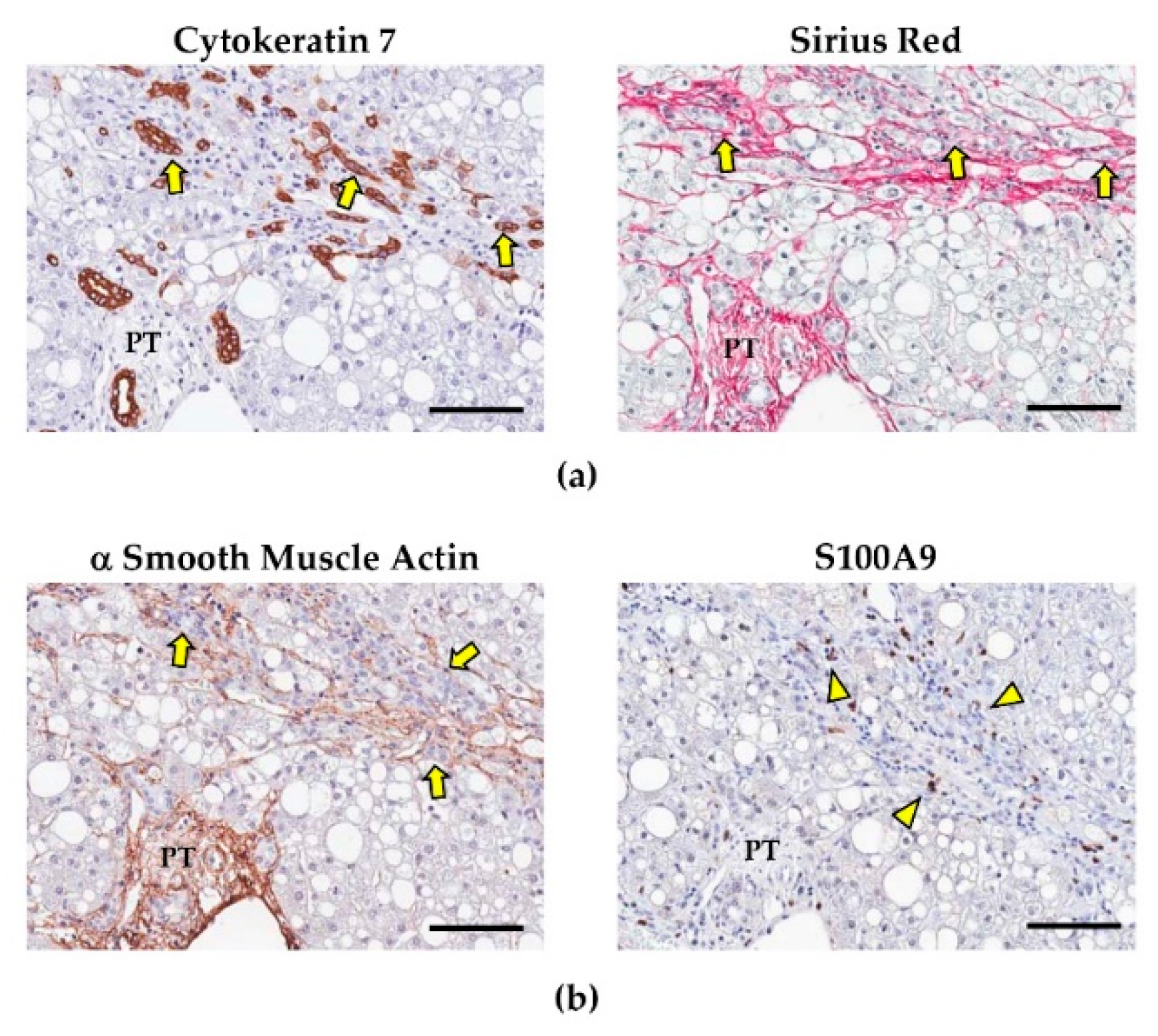
3.2. HpSCs and Their Niche
3.3. HpSCs and Their Involvement in NAFLD Progression
4. Non-Parenchymal Cells: Supporting the HpSC Response in NAFLD
4.1. Hepatic Stellate Cells and Portal Myofibroblasts: Fibrogenetic Pathways in NAFLD
4.2. Liver Macrophages and Their Role in Influencing Fibrogenesis and HpSC Response
4.3. Re-Shaping HpSC Niche as a Therapeutic Approach in NAFLD Patients
5. Interaction of Liver Cellular Compartments with Extra-Hepatic Organs
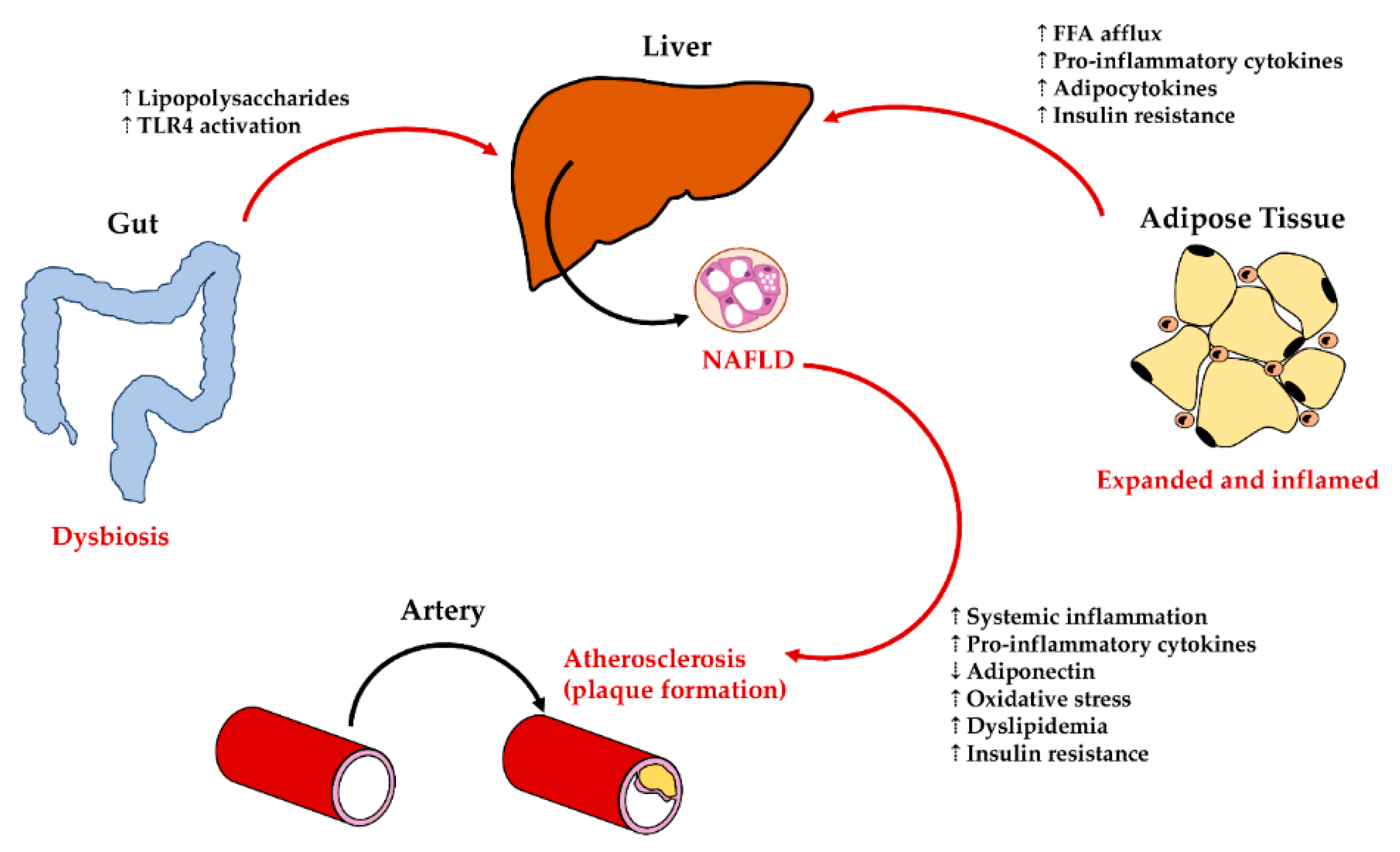
5.1. Liver—Adipose Tissue Axis: Influences on Liver Cells in NAFLD
5.2. Liver—Gut Axis: Influences on Liver Damage in NAFLD
5.3. Liver—Cardiovascular System Interplay in NAFLD
6. Conclusions
Funding
Conflicts of Interest
References
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Stefan, N.; Haring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.P.; Dhindsa, D.S.; Lee, S.K.; Sandesara, P.B.; Chalasani, N.P.; Sperling, L.S. Nonalcoholic fatty liver disease and the heart: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2019, 73, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.P.; Tang, G.Y.; Nobili, V.; Armstrong, M.J. Evaluations of lifestyle, dietary, and pharmacologic treatments for pediatric nonalcoholic fatty liver disease: A systematic review. Clin. Gastroenterol. Hepatol. 2018. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Alisi, A.; Newton, K.P.; Schwimmer, J.B. Comparison of the phenotype and approach to pediatric vs. adult patients with nonalcoholic fatty liver disease. Gastroenterology 2016, 150, 1798–1810. [Google Scholar] [CrossRef] [Green Version]
- Skinner, A.C.; Skelton, J.A. Prevalence and trends in obesity and severe obesity among children in the United States, 1999–2012. JAMA Pediatr. 2014, 168, 561–566. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Nascimento, E.M.; Gajera, C.R.; Chen, L.; Neuhofer, P.; Garbuzov, A.; Wang, S.; Artandi, S.E. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 2018, 556, 244–248. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, L.; Fish, M.; Logan, C.Y.; Nusse, R. Self-renewing diploid Axin2(+) cells fuel homeostatic renewal of the liver. Nature 2015, 524, 180–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, C.H.; Hsu, S.H.; Guo, F.; Tan, C.T.; Yu, V.C.; Visvader, J.E.; Chow, P.K.H.; Fu, N.Y. Lgr5(+) pericentral hepatocytes are self-maintained in normal liver regeneration and susceptible to hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 19530–19540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Pikiolek, M.; Orsini, V.; Bergling, S.; Holwerda, S.; Morelli, L.; Hoppe, P.S.; Planas-Paz, L.; Yang, Y.; Ruffner, H.; et al. AXIN2(+) pericentral hepatocytes have limited contributions to liver homeostasis and regeneration. Cell Stem Cell 2020, 26, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Planas-Paz, L.; Orsini, V.; Boulter, L.; Calabrese, D.; Pikiolek, M.; Nigsch, F.; Xie, Y.; Roma, G.; Donovan, A.; Marti, P.; et al. The RSPO-LGR4/5-ZNRF3/RNF43 module controls liver zonation and size. Nat. Cell Biol. 2016, 18, 467–479. [Google Scholar] [CrossRef]
- Font-Burgada, J.; Shalapour, S.; Ramaswamy, S.; Hsueh, B.; Rossell, D.; Umemura, A.; Taniguchi, K.; Nakagawa, H.; Valasek, M.A.; Ye, L.; et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015, 162, 766–779. [Google Scholar] [CrossRef] [Green Version]
- Pu, W.; Zhang, H.; Huang, X.; Tian, X.; He, L.; Wang, Y.; Zhang, L.; Liu, Q.; Li, Y.; Li, Y.; et al. Mfsd2a+ hepatocytes repopulate the liver during injury and regeneration. Nat. Commun. 2016, 7, 13369. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Jimenez, R.J.; Sharma, K.; Luu, H.Y.; Hsu, B.Y.; Ravindranathan, A.; Stohr, B.A.; Willenbring, H. Broad distribution of hepatocyte proliferation in liver homeostasis and regeneration. Cell Stem Cell 2020, 26, 27–33. [Google Scholar] [CrossRef]
- Schaub, J.R.; Malato, Y.; Gormond, C.; Willenbring, H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 2014, 8, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Kleiner, D.E.; Makhlouf, H.R. Histology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in adults and children. Clin. Liver Dis. 2016, 20, 293–312. [Google Scholar] [CrossRef] [Green Version]
- Hall, Z.; Bond, N.J.; Ashmore, T.; Sanders, F.; Ament, Z.; Wang, X.; Murray, A.J.; Bellafante, E.; Virtue, S.; Vidal-Puig, A.; et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1165–1180. [Google Scholar] [CrossRef]
- Kucukoglu, O.; Guldiken, N.; Chen, Y.; Usachov, V.; El-Heliebi, A.; Haybaeck, J.; Denk, H.; Trautwein, C.; Strnad, P. High-fat diet triggers Mallory-Denk body formation through misfolding and crosslinking of excess keratin 8. Hepatology 2014, 60, 169–178. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Gattolliat, C.H.; Asselah, T. Mitochondrial dysfunction and signaling in chronic liver diseases. Gastroenterology 2018, 155, 629–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive lipid species and metabolic pathways in progression and resolution of nonalcoholic steatohepatitis. Gastroenterology 2018, 155, 282–302.e288. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER stress drives lipogenesis and steatohepatitis via caspase-2 activation of S1P. Cell 2018, 175, 133–145.e115. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Cao, Z.; Lai, X.; Shi, Y.; Zhou, N. Niacin ameliorates hepatic steatosis by inhibiting de novo lipogenesis via a GPR109A-mediated PKC-ERK1/2-AMPK signaling pathway in C57BL/6 mice fed a high-fat diet. J. Nutr. 2019. [Google Scholar] [CrossRef]
- Zhang, M.; Tang, Y.; Tang, E.; Lu, W. MicroRNA-103 represses hepatic de novo lipogenesis and alleviates NAFLD via targeting FASN and SCD1. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Elhafiz, M.; Zhao, G.; Ismail, M.; Xu, D.; Das, D.; Fan, S.; Cheng, N.; Yousef, B.A.; Jiang, Z.; Zhang, L. Imbalanced insulin substrate-1 and insulin substrate-2 signaling trigger hepatic steatosis in vitamin D deficient rats: 8-methoxypsoralen, a vitamin D receptor ligand with a promising anti-steatotic action. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 158657. [Google Scholar] [CrossRef] [PubMed]
- Siculella, L.; Giannotti, L.; Testini, M.; Gnoni, G.V.; Damiano, F. In steatotic cells, ATP-citrate lyase mRNA is efficiently translated through a cap-independent mechanism, contributing to the stimulation of de novo lipogenesis. Int. J. Mol. Sci. 2020, 21, 1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.E.; Fisher, E.A. Lipoprotein metabolism, dyslipidemia, and nonalcoholic fatty liver disease. Semin. Liver Dis. 2013, 33, 380–388. [Google Scholar] [PubMed] [Green Version]
- Canivet, C.M.; Bonnafous, S.; Rousseau, D.; Leclere, P.S.; Lacas-Gervais, S.; Patouraux, S.; Sans, A.; Luci, C.; Bailly-Maitre, B.; Iannelli, A.; et al. Hepatic FNDC5 is a potential local protective factor against Non-Alcoholic Fatty Liver. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165705. [Google Scholar] [CrossRef] [PubMed]
- Win, S.; Than, T.A.; Le, B.H.; Garcia-Ruiz, C.; Fernandez-Checa, J.C.; Kaplowitz, N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 2015, 62, 1367–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xu, W.; Zhai, T.; You, J.; Chen, Y. Silibinin ameliorates hepatic lipid accumulation and oxidative stress in mice with non-alcoholic steatohepatitis by regulating CFLAR-JNK pathway. Acta Pharm. Sin. B 2019, 9, 745–757. [Google Scholar] [CrossRef]
- Kojima, S.; Kuo, T.F.; Tatsukawa, H. Regulation of transglutaminase-mediated hepatic cell death in alcoholic steatohepatitis and non-alcoholic steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27, 52–57. [Google Scholar] [CrossRef]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Biol. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Pastori, D.; Baratta, F.; Overi, D.; Labbadia, G.; Polimeni, L.; Di Costanzo, A.; Pannitteri, G.; Carnevale, R.; Del Ben, M.; et al. PNPLA3 variant and portal/periportal histological pattern in patients with biopsy-proven non-alcoholic fatty liver disease: A possible role for oxidative stress. Sci. Rep. 2017, 7, 15756. [Google Scholar] [CrossRef]
- Bruschi, F.V.; Tardelli, M.; Herac, M.; Claudel, T.; Trauner, M. Metabolic regulation of hepatic PNPLA3 expression and severity of liver fibrosis in patients with NASH. Liver Int. Off. J. Int. Assoc. Study Liver 2020. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Lin, W.; Liu, Y.; Guo, H.; Wang, L.; Yang, L.; Li, L.; Li, D.; Tang, R. Chronic Microcystin-LR exposure induces abnormal lipid metabolism via endoplasmic reticulum stress in male zebrafish. Toxins (Basel) 2020, 12, 107. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Zhu, M.; Liu, X.; Chen, X.; Yuan, Y.; Li, L.; Liu, J.; Lu, Y.; Cheng, J.; Chen, Y. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis. Nutr. Metab. (Lond.) 2020, 17, 11. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.; Uddin, M.J.; Pak, E.S.; Kang, H.; Jin, E.J.; Jo, S.; Kang, D.; Lee, H.; Ha, H. The impaired redox balance in peroxisomes of catalase knockout mice accelerates nonalcoholic fatty liver disease through endoplasmic reticulum stress. Free Radic. Biol. Med. 2020, 148, 22–32. [Google Scholar] [CrossRef]
- Xiao, T.; Liang, X.; Liu, H.; Zhang, F.; Meng, W.; Hu, F. Mitochondrial stress protein HSP60 regulates ER stress-induced hepatic lipogenesis. J. Mol. Endocrinol. 2020, 64, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hanelt, I.; Hummer, G.; Ernst, R. Activation of the unfolded protein response by lipid bilayer stress. Mol. Cell 2017, 67, 673–684.e678. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Wang, S.; Malhotra, J.; Hassler, J.R.; Back, S.H.; Wang, G.; Chang, L.; Xu, W.; Miao, H.; Leonardi, R.; et al. The unfolded protein response transducer IRE1alpha prevents ER stress-induced hepatic steatosis. EMBO J. 2011, 30, 1357–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepulveda, D.; Rojas-Rivera, D.; Rodriguez, D.A.; Groenendyk, J.; Kohler, A.; Lebeaupin, C.; Ito, S.; Urra, H.; Carreras-Sureda, A.; Hazari, Y.; et al. Interactome screening identifies the ER luminal chaperone Hsp47 as a regulator of the unfolded protein response transducer IRE1alpha. Mol. Cell 2018, 69, 238–252.e237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.K.; Lee, J.Y.; Jang, Y.M.; Kwon, Y.H. Involvement of endoplasmic reticulum stress in palmitate-induced apoptosis in HepG2 cells. Toxicol. Res. 2008, 24, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Xiong, X.; Wang, X.; Lu, Y.; Wang, E.; Zhang, Z.; Yang, J.; Zhang, H.; Li, X. Hepatic steatosis exacerbated by endoplasmic reticulum stress-mediated downregulation of FXR in aging mice. J. Hepatol. 2014, 60, 847–854. [Google Scholar] [CrossRef]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.P.; Liu, X.J.; Xie, L.; Shen, X.Z.; Wu, J. Impaired mitophagy triggers NLRP3 inflammasome activation during the progression from nonalcoholic fatty liver to nonalcoholic steatohepatitis. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 749–763. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Kotiadis, V.N.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 2014, 1840, 1254–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunny, N.E.; Bril, F.; Cusi, K. Mitochondrial adaptation in nonalcoholic fatty liver disease: Novel mechanisms and treatment strategies. Trends Endocrinol. Metab. 2017, 28, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Banerjee, A.; Yoo, S.H.; Jang, S.; Gonzalez, F.J.; Song, B.J. Critical role of cytochrome P450 2E1 (CYP2E1) in the development of high fat-induced non-alcoholic steatohepatitis. J. Hepatol. 2012, 57, 860–866. [Google Scholar] [CrossRef] [Green Version]
- Boland, M.L.; Oldham, S.; Boland, B.B.; Will, S.; Lapointe, J.M.; Guionaud, S.; Rhodes, C.J.; Trevaskis, J.L. Nonalcoholic steatohepatitis severity is defined by a failure in compensatory antioxidant capacity in the setting of mitochondrial dysfunction. World J. Gastroenterol. WJG 2018, 24, 1748–1765. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Bockowska, S.B.; Lebensztejn, D.M. Pediatric non-alcoholic steatohepatitis: the first report on the ultrastructure of hepatocyte mitochondria. World J. Gastroenterol. WJG 2014, 20, 4335–4340. [Google Scholar] [CrossRef]
- Satapati, S.; Sunny, N.E.; Kucejova, B.; Fu, X.; He, T.T.; Mendez-Lucas, A.; Shelton, J.M.; Perales, J.C.; Browning, J.D.; Burgess, S.C. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J. Lipid Res. 2012, 53, 1080–1092. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.D.; Meers, G.M.; Morris, E.M.; Linden, M.A.; Cunningham, R.P.; Ibdah, J.A.; Thyfault, J.P.; Laughlin, M.H.; Rector, R.S. eNOS deletion impairs mitochondrial quality control and exacerbates Western diet-induced NASH. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E605–E616. [Google Scholar] [CrossRef]
- Hinke, S.A.; Martens, G.A.; Cai, Y.; Finsi, J.; Heimberg, H.; Pipeleers, D.; Van de Casteele, M. Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br. J. Pharmacol. 2007, 150, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Ikura, Y.; Ohsawa, M.; Suekane, T.; Fukushima, H.; Itabe, H.; Jomura, H.; Nishiguchi, S.; Inoue, T.; Naruko, T.; Ehara, S.; et al. Localization of oxidized phosphatidylcholine in nonalcoholic fatty liver disease: Impact on disease progression. Hepatology 2006, 43, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Noureddin, M.; Yates, K.P.; Vaughn, I.A.; Neuschwander-Tetri, B.A.; Sanyal, A.J.; McCullough, A.; Merriman, R.; Hameed, B.; Doo, E.; Kleiner, D.E.; et al. Clinical and histological determinants of nonalcoholic steatohepatitis and advanced fibrosis in elderly patients. Hepatology 2013, 58, 1644–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, T.; Murata, D.; Adachi, Y.; Itoh, K.; Kameoka, S.; Igarashi, A.; Kato, T.; Araki, Y.; Huganir, R.L.; Dawson, T.M.; et al. Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 2018, 28, 588–604.e585. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Park, J.S.; Roh, Y.S. Molecular insights into the role of mitochondria in non-alcoholic fatty liver disease. Arch. Pharm. Res. 2019, 42, 935–946. [Google Scholar] [CrossRef]
- Zeng, X.; Yang, J.; Hu, O.; Huang, J.; Ran, L.; Chen, M.; Zhang, Y.; Zhou, X.; Zhu, J.; Zhang, Q.; et al. Dihydromyricetin ameliorates nonalcoholic fatty liver disease by improving mitochondrial respiratory capacity and redox homeostasis through modulation of SIRT3 signaling. Antioxid. Redox Signal. 2019, 30, 163–183. [Google Scholar] [CrossRef]
- Baker, P.R., 2nd; Friedman, J.E. Mitochondrial role in the neonatal predisposition to developing nonalcoholic fatty liver disease. J. Clin. Investig. 2018, 128, 3692–3703. [Google Scholar] [CrossRef]
- Elsheikh, A.; Lavergne, S.N.; Castrejon, J.L.; Farrell, J.; Wang, H.; Sathish, J.; Pichler, W.J.; Park, B.K.; Naisbitt, D.J. Drug antigenicity, immunogenicity, and costimulatory signaling: evidence for formation of a functional antigen through immune cell metabolism. J. Immunol. 2010, 185, 6448–6460. [Google Scholar] [CrossRef]
- Marques, P.E.; Oliveira, A.G.; Pereira, R.V.; David, B.A.; Gomides, L.F.; Saraiva, A.M.; Pires, D.A.; Novaes, J.T.; Patricio, D.O.; Cisalpino, D.; et al. Hepatic DNA deposition drives drug-induced liver injury and inflammation in mice. Hepatology 2015, 61, 348–360. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bonten, E.J.; Annunziata, I.; d’Azzo, A. Lysosomal multienzyme complex: Pros and cons of working together. Cell. Mol. Life Sci. 2014, 71, 2017–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Tajeddine, N.; Vitale, I.; Criollo, A.; Vicencio, J.M.; Hickman, J.A.; Geneste, O.; Kroemer, G. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle 2007, 6, 2263–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.M.; Ding, W.X.; Gao, W. Autophagy in the liver. Hepatology 2008, 47, 1773–1785. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Weller, S.G.; Drizyte-Miller, K.; Chen, J.; Krueger, E.W.; Mehall, B.; Casey, C.A.; Cao, H.; McNiven, M.A. Maturation of lipophagic organelles in hepatocytes is dependent upon a Rab10-dynamin-2 complex. Hepatology 2019. [Google Scholar] [CrossRef]
- Manco, R.; Clerbaux, L.A.; Verhulst, S.; Bou Nader, M.; Sempoux, C.; Ambroise, J.; Bearzatto, B.; Gala, J.L.; Horsmans, Y.; van Grunsven, L.; et al. Reactive cholangiocytes differentiate into proliferative hepatocytes with efficient DNA repair in mice with chronic liver injury. J. Hepatol. 2019, 70, 1180–1191. [Google Scholar] [CrossRef]
- Takahashi, S.S.; Sou, Y.S.; Saito, T.; Kuma, A.; Yabe, T.; Sugiura, Y.; Lee, H.C.; Suematsu, M.; Yokomizo, T.; Koike, M.; et al. Loss of autophagy impairs physiological steatosis by accumulation of NCoR1. Life Sci Alliance 2020, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.E.; Singh, B.K.; Hsu, M.C.; Huang, C.; Yen, P.M.; Wu, L.S.; Jong, D.S.; Chiu, C.H. Increasing dietary medium-chain fatty acid ratio mitigates high-fat diet-induced non-alcoholic steatohepatitis by regulating autophagy. Sci. Rep. 2017, 7, 13999. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010, 24, 3052–3065. [Google Scholar] [CrossRef] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Inhibitory effect of intracellular lipid load on macroautophagy. Autophagy 2010, 6, 825–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Wang, J.; Jiang, W.; Shi, C.; Wang, X.; Huang, Y.; Hu, C. Caveolin-1 alleviates lipid accumulation in NAFLD associated with promoting autophagy by inhibiting the Akt/mTOR pathway. Eur. J. Pharmacol. 2020, 871, 172910. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.K.; Kwon, S.W.; Han, D.H.; Lee, Y.H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Harada, M. Autophagy is involved in the elimination of intracellular inclusions, Mallory-Denk bodies, in hepatocytes. Med. Mol. Morphol. 2010, 43, 13–18. [Google Scholar] [CrossRef]
- Harada, M.; Hanada, S.; Toivola, D.M.; Ghori, N.; Omary, M.B. Autophagy activation by rapamycin eliminates mouse Mallory-Denk bodies and blocks their proteasome inhibitor-mediated formation. Hepatology 2008, 47, 2026–2035. [Google Scholar] [CrossRef]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepat. Med. 2016, 8, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Singh, R.; Xiang, Y.; Czaja, M.J. Macroautophagy and chaperone-mediated autophagy are required for hepatocyte resistance to oxidant stress. Hepatology 2010, 52, 266–277. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.C.; Cullen, S.P.; Martin, S.J. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- McArthur, K.; Kile, B.T. Apoptotic caspases: Multiple or mistaken identities? Trends Cell Biol. 2018, 28, 475–493. [Google Scholar] [CrossRef]
- Hatting, M.; Zhao, G.; Schumacher, F.; Sellge, G.; Al Masaoudi, M.; Gabetaler, N.; Boekschoten, M.; Muller, M.; Liedtke, C.; Cubero, F.J.; et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology 2013, 57, 2189–2201. [Google Scholar] [CrossRef]
- Li, C.P.; Li, J.H.; He, S.Y.; Li, P.; Zhong, X.L. Roles of Fas/Fasl, Bcl-2/Bax, and Caspase-8 in rat nonalcoholic fatty liver disease pathogenesis. Genet. Mol. Res. 2014, 13, 3991–3999. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, P.X.; Zhao, L.P.; Zhang, X.; Ji, Y.X.; Zhang, X.J.; Fang, C.; Lu, Y.X.; Yang, X.; Gao, M.M.; et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat. Med. 2018, 24, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.; Lu, J.; Huang, K.; Han, Y.; Chen, J.; Yang, Y.; Liu, B. PPAR delta inhibition protects against palmitic acid-LPS induced lipidosis and injury in cultured hepatocyte L02 cell. Int. J. Med. Sci. 2019, 16, 1593–1603. [Google Scholar] [CrossRef]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. WJG 2018, 24, 2661–2672. [Google Scholar] [CrossRef]
- Ribeiro, P.S.; Cortez-Pinto, H.; Sola, S.; Castro, R.E.; Ramalho, R.M.; Baptista, A.; Moura, M.C.; Camilo, M.E.; Rodrigues, C.M. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am. J. Gastroenterol. 2004, 99, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Alkhouri, N.; Alisi, A.; Okwu, V.; Matloob, A.; Ferrari, F.; Crudele, A.; De Vito, R.; Lopez, R.; Feldstein, A.E.; Nobili, V. Circulating soluble fas and fas ligand levels are elevated in children with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2015, 60, 2353–2359. [Google Scholar] [CrossRef]
- Ferreira, D.M.; Castro, R.E.; Machado, M.V.; Evangelista, T.; Silvestre, A.; Costa, A.; Coutinho, J.; Carepa, F.; Cortez-Pinto, H.; Rodrigues, C.M. Apoptosis and insulin resistance in liver and peripheral tissues of morbidly obese patients is associated with different stages of non-alcoholic fatty liver disease. Diabetologia 2011, 54, 1788–1798. [Google Scholar] [CrossRef]
- Hsieh, S.; Leaderer, B.P.; Feldstein, A.E.; Santoro, N.; McKay, L.A.; Caprio, S.; McConnell, R. Traffic-related air pollution associations with cytokeratin-18, a marker of hepatocellular apoptosis, in an overweight and obese paediatric population. Pediatr. Obes. 2018, 13, 342–347. [Google Scholar] [CrossRef]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; De Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar] [CrossRef]
- Guy, C.D.; Suzuki, A.; Zdanowicz, M.; Abdelmalek, M.F.; Burchette, J.; Unalp, A.; Diehl, A.M.; NAFLD CRN. Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012, 55, 1711–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakisaka, K.; Cazanave, S.C.; Werneburg, N.W.; Razumilava, N.; Mertens, J.C.; Bronk, S.F.; Gores, G.J. A hedgehog survival pathway in ‘undead’ lipotoxic hepatocytes. J. Hepatol. 2012, 57, 844–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.V.; Michelotti, G.A.; Pereira Tde, A.; Boursier, J.; Kruger, L.; Swiderska-Syn, M.; Karaca, G.; Xie, G.; Guy, C.D.; Bohinc, B.; et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase-2 deficient mice with non-alcoholic steatohepatitis. Gut 2015, 64, 1148–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; In Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef]
- Mirea, A.M.; Stienstra, R.; Kanneganti, T.D.; Tack, C.J.; Chavakis, T.; Toonen, E.J.M.; Joosten, L.A.B. Mice deficient in the IL-1beta activation genes Prtn3, elane, and Casp1 are protected against the development of obesity-induced NAFLD. Inflammation 2020. [Google Scholar] [CrossRef] [Green Version]
- Roskams, T.A.; Theise, N.D.; Balabaud, C.; Bhagat, G.; Bhathal, P.S.; Bioulac-Sage, P.; Brunt, E.M.; Crawford, J.M.; Crosby, H.A.; Desmet, V.; et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology 2004, 39, 1739–1745. [Google Scholar] [CrossRef]
- Schmelzer, E.; Zhang, L.; Bruce, A.; Wauthier, E.; Ludlow, J.; Yao, H.L.; Moss, N.; Melhem, A.; McClelland, R.; Turner, W.; et al. Human hepatic stem cells from fetal and postnatal donors. J. Exp. Med. 2007, 204, 1973–1987. [Google Scholar] [CrossRef] [Green Version]
- Libbrecht, L.; Desmet, V.; Van Damme, B.; Roskams, T. The immunohistochemical phenotype of dysplastic foci in human liver: Correlation with putative progenitor cells. J. Hepatol. 2000, 33, 76–84. [Google Scholar] [CrossRef]
- Spee, B.; Carpino, G.; Schotanus, B.A.; Katoonizadeh, A.; Vander Borght, S.; Gaudio, E.; Roskams, T. Characterisation of the liver progenitor cell niche in liver diseases: Potential involvement of Wnt and Notch signalling. Gut 2010, 59, 247–257. [Google Scholar] [CrossRef]
- Overi, D.; Carpino, G.; Cardinale, V.; Franchitto, A.; Safarikia, S.; Onori, P.; Alvaro, D.; Gaudio, E. Contribution of resident stem cells to liver and biliary tree regeneration in human diseases. Int. J. Mol. Sci. 2018, 19, 2917. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.J.; Clouston, A.D.; Forbes, S.J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 2014, 146, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Olivero, F.; Carpino, G.; Overi, D.; Rosa, L.; Lepanto, M.S.; Cutone, A.; Franchitto, A.; Alpini, G.; Onori, P.; et al. Role of lactoferrin and its receptors on biliary epithelium. Biometals 2018, 31, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Alpini, G.; McGill, J.M.; Larusso, N.F. The pathobiology of biliary epithelia. Hepatology 2002, 35, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Gaudio, E.; Carpino, G.; Cardinale, V.; Franchitto, A.; Onori, P.; Alvaro, D. New insights into liver stem cells. Dig. Liver Dis. 2009, 41, 455–462. [Google Scholar] [CrossRef]
- Espanol-Suner, R.; Carpentier, R.; Van Hul, N.; Legry, V.; Achouri, Y.; Cordi, S.; Jacquemin, P.; Lemaigre, F.; Leclercq, I.A. Liver progenitor cells yield functional hepatocytes in response to chronic liver injury in mice. Gastroenterology 2012, 143, 1564–1575.e1567. [Google Scholar] [CrossRef]
- Malato, Y.; Naqvi, S.; Schurmann, N.; Ng, R.; Wang, B.; Zape, J.; Kay, M.A.; Grimm, D.; Willenbring, H. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J. Clin. Investig. 2011, 121, 4850–4860. [Google Scholar] [CrossRef]
- Rodrigo-Torres, D.; Affo, S.; Coll, M.; Morales-Ibanez, O.; Millan, C.; Blaya, D.; Alvarez-Guaita, A.; Rentero, C.; Lozano, J.J.; Maestro, M.A.; et al. The biliary epithelium gives rise to liver progenitor cells. Hepatology 2014, 60, 1367–1377. [Google Scholar] [CrossRef] [Green Version]
- Raven, A.; Lu, W.Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef]
- Russell, J.O.; Lu, W.Y.; Okabe, H.; Abrams, M.; Oertel, M.; Poddar, M.; Singh, S.; Forbes, S.J.; Monga, S.P. Hepatocyte-specific beta-catenin deletion during severe liver injury provokes cholangiocytes to differentiate into hepatocytes. Hepatology 2019, 69, 742–759. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.Y.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, X.; Li, W.; Feng, R.X.; Li, L.; Yi, G.R.; Zhang, X.N.; Yin, C.; Yu, H.Y.; Zhang, J.P.; et al. Chronic liver injury induces conversion of biliary epithelial cells into hepatocytes. Cell Stem Cell 2018, 23, 114–122.e113. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Cardinale, V.; Folseraas, T.; Overi, D.; Floreani, A.; Franchitto, A.; Onori, P.; Cazzagon, N.; Berloco, P.B.; Karlsen, T.H.; et al. Hepatic stem/progenitor cell activation differs between primary sclerosing and primary biliary cholangitis. Am. J. Pathol. 2018, 188, 627–639. [Google Scholar] [CrossRef] [Green Version]
- Lanzoni, G.; Cardinale, V.; Carpino, G. The hepatic, biliary, and pancreatic network of stem/progenitor cell niches in humans: A new reference frame for disease and regeneration. Hepatology 2016, 64, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Boulter, L.; Lu, W.Y.; Forbes, S.J. Differentiation of progenitors in the liver: A matter of local choice. J. Clin. Investig. 2013, 123, 1867–1873. [Google Scholar] [CrossRef] [Green Version]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; Van Rooijen, N.; et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med. 2012, 18, 572–579. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Chen, C.C.; Alpini, G.; Lau, L.F. CCN1 induces hepatic ductular reaction through integrin alphavbeta(5)-mediated activation of NF-kappaB. J. Clin. Investig. 2015, 125, 1886–1900. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Nobili, V.; Renzi, A.; De Stefanis, C.; Stronati, L.; Franchitto, A.; Alisi, A.; Onori, P.; De Vito, R.; Alpini, G.; et al. Macrophage Activation in Pediatric Nonalcoholic Fatty Liver Disease (NAFLD) correlates with hepatic progenitor cell response via Wnt3a pathway. PLoS ONE 2016, 11, e0157246. [Google Scholar] [CrossRef]
- Bird, T.G.; Lu, W.Y.; Boulter, L.; Gordon-Keylock, S.; Ridgway, R.A.; Williams, M.J.; Taube, J.; Thomas, J.A.; Wojtacha, D.; Gambardella, A.; et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated TWEAK signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 6542–6547. [Google Scholar] [CrossRef] [Green Version]
- Jakubowski, A.; Ambrose, C.; Parr, M.; Lincecum, J.M.; Wang, M.Z.; Zheng, T.S.; Browning, B.; Michaelson, J.S.; Baetscher, M.; Wang, B.; et al. TWEAK induces liver progenitor cell proliferation. J. Clin. Investig. 2005, 115, 2330–2340. [Google Scholar] [CrossRef] [Green Version]
- Grzelak, C.A.; Martelotto, L.G.; Sigglekow, N.D.; Patkunanathan, B.; Ajami, K.; Calabro, S.R.; Dwyer, B.J.; Tirnitz-Parker, J.E.; Watkins, D.N.; Warner, F.J.; et al. The intrahepatic signalling niche of hedgehog is defined by primary cilia positive cells during chronic liver injury. J. Hepatol. 2014, 60, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lopategi, A.; Ge, X.; Lu, Y.; Kitamura, N.; Urtasun, R.; Leung, T.M.; Fiel, M.I.; Nieto, N. Osteopontin induces ductular reaction contributing to liver fibrosis. Gut 2014, 63, 1805–1818. [Google Scholar] [CrossRef]
- Coombes, J.D.; Swiderska-Syn, M.; Dolle, L.; Reid, D.; Eksteen, B.; Claridge, L.; Briones-Orta, M.A.; Shetty, S.; Oo, Y.H.; Riva, A.; et al. Osteopontin neutralisation abrogates the liver progenitor cell response and fibrogenesis in mice. Gut 2015, 64, 1120–1131. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, L.; Chen, R.; Zhou, X.; Fan, X.; Liang, Y.; Jia, R.; Wang, H.; Liu, G.; Guo, Y.; et al. Osteopontin promotes hepatic progenitor cell expansion and tumorigenicity via activation of beta-catenin in mice. Stem Cells 2015. [Google Scholar] [CrossRef]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Wood, M.J.; Gadd, V.L.; Powell, L.W.; Ramm, G.A.; Clouston, A.D. Ductular reaction in hereditary hemochromatosis: The link between hepatocyte senescence and fibrosis progression. Hepatology 2014, 59, 848–857. [Google Scholar] [CrossRef]
- Nobili, V.; Alisi, A.; Cutrera, R.; Carpino, G.; De Stefanis, C.; D’Oria, V.; De Vito, R.; Cucchiara, S.; Gaudio, E.; Musso, G. Altered gut-liver axis and hepatic adiponectin expression in OSAS: novel mediators of liver injury in paediatric non-alcoholic fatty liver. Thorax 2015, 70, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Zhu, N.L.; Asahina, K.; Mann, D.A.; Mann, J. Epigenetic cell fate regulation of hepatic stellate cells. Hepatol. Res. 2011, 41, 675–682. [Google Scholar] [CrossRef]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Carotti, S.; Morini, S.; Corradini, S.G.; Burza, M.A.; Molinaro, A.; Carpino, G.; Merli, M.; De Santis, A.; Muda, A.O.; Rossi, M.; et al. Glial fibrillary acidic protein as an early marker of hepatic stellate cell activation in chronic and posttransplant recurrent hepatitis C. Liver Transplant. 2008, 14, 806–814. [Google Scholar] [CrossRef]
- Carpino, G.; Morini, S.; Ginanni Corradini, S.; Franchitto, A.; Merli, M.; Siciliano, M.; Gentili, F.; Onetti Muda, A.; Berloco, P.; Rossi, M.; et al. Alpha-SMA expression in hepatic stellate cells and quantitative analysis of hepatic fibrosis in cirrhosis and in recurrent chronic hepatitis after liver transplantation. Dig. Liver Dis. 2005, 37, 349–356. [Google Scholar] [CrossRef]
- Martinez-Hernandez, A.; Amenta, P.S. The hepatic extracellular matrix. I. Components and distribution in normal liver. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 1–11. [Google Scholar] [CrossRef]
- Klaas, M.; Kangur, T.; Viil, J.; Maemets-Allas, K.; Minajeva, A.; Vadi, K.; Antsov, M.; Lapidus, N.; Jarvekulg, M.; Jaks, V. The alterations in the extracellular matrix composition guide the repair of damaged liver tissue. Sci. Rep. 2016, 6, 27398. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Ronnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Daniels, S.J.; Holm Nielsen, S.; Bager, C.; Rasmussen, D.G.K.; Loomba, R.; Surabattula, R.; Villesen, I.F.; Luo, Y.; Shevell, D.; et al. Collagen biology and non-invasive biomarkers of liver fibrosis. Liver Int. Off. J. Int. Assoc. Study Liver 2020. [Google Scholar] [CrossRef] [Green Version]
- Decaris, M.L.; Li, K.W.; Emson, C.L.; Gatmaitan, M.; Liu, S.; Wang, Y.; Nyangau, E.; Colangelo, M.; Angel, T.E.; Beysen, C.; et al. Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood. Hepatology 2017, 65, 78–88. [Google Scholar] [CrossRef]
- Del Ben, M.; Overi, D.; Polimeni, L.; Carpino, G.; Labbadia, G.; Baratta, F.; Pastori, D.; Noce, V.; Gaudio, E.; Angelico, F.; et al. Overexpression of the vitronectin V10 subunit in patients with nonalcoholic steatohepatitis: Implications for noninvasive diagnosis of NASH. Int. J. Mol. Sci. 2018, 19, 603. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Bracht, T.; Schweinsberg, V.; Trippler, M.; Kohl, M.; Ahrens, M.; Padden, J.; Naboulsi, W.; Barkovits, K.; Megger, D.A.; Eisenacher, M.; et al. Analysis of disease-associated protein expression using quantitative proteomics-fibulin-5 is expressed in association with hepatic fibrosis. J. Proteome Res. 2015, 14, 2278–2286. [Google Scholar] [CrossRef]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heeboll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oro, D.; Thrane, S.W.; et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P. Reversibility of liver fibrosis. Fibrogenes. Tissue Repair 2012, 5, S26. [Google Scholar] [CrossRef] [Green Version]
- Munsterman, I.D.; Kendall, T.J.; Khelil, N.; Popa, M.; Lomme, R.; Drenth, J.P.H.; Tjwa, E. Extracellular matrix components indicate remodelling activity in different fibrosis stages of human non-alcoholic fatty liver disease. Histopathology 2018, 73, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Sircana, A.; Paschetta, E.; Saba, F.; Molinaro, F.; Musso, G. Recent insight into the role of fibrosis in nonalcoholic steatohepatitis-related hepatocellular carcinoma. Int. J. Mol. Sci. 2019, 20, 1745. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Carpino, G.; De Peppo, F.; Caccamo, R.; Mosca, A.; Romito, I.; Overi, D.; Franchitto, A.; Onori, P.; Alisi, A.; et al. Laparoscopic sleeve gastrectomy improves nonalcoholic fatty liver disease-related liver damage in adolescents by reshaping cellular interactions and hepatic adipocytokine production. J. Pediatr. 2018, 194, 100–108.e103. [Google Scholar] [CrossRef]
- Harrison, S.A.; Bashir, M.R.; Guy, C.D.; Zhou, R.; Moylan, C.A.; Frias, J.P.; Alkhouri, N.; Bansal, M.B.; Baum, S.; Neuschwander-Tetri, B.A.; et al. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2019, 394, 2012–2024. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Della Corte, C.; Carpino, G.; De Vito, R.; De Stefanis, C.; Alisi, A.; Cianfarani, S.; Overi, D.; Mosca, A.; Stronati, L.; Cucchiara, S.; et al. Docosahexanoic acid plus Vitamin D treatment improves features of NAFLD in children with serum Vitamin D deficiency: Results from a single centre trial. PLoS ONE 2016, 11, e0168216. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.J.; Nedergaard, A.F.; Sun, S.; Veidal, S.S.; Larsen, L.; Zheng, Q.; Suetta, C.; Henriksen, K.; Christiansen, C.; Karsdal, M.A.; et al. The neo-epitope specific PRO-C3 ELISA measures true formation of type III collagen associated with liver and muscle parameters. Am. J. Transl. Res. 2013, 5, 303–315. [Google Scholar]
- Nielsen, M.J.; Veidal, S.S.; Karsdal, M.A.; Orsnes-Leeming, D.J.; Vainer, B.; Gardner, S.D.; Hamatake, R.; Goodman, Z.D.; Schuppan, D.; Patel, K. Plasma Pro-C3 (N-terminal type III collagen propeptide) predicts fibrosis progression in patients with chronic hepatitis C. Liver Int. Off. J. Int. Assoc. Study Liver 2015, 35, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Boyle, M.; Tiniakos, D.; Schattenberg, J.M.; Ratziu, V.; Bugianessi, E.; Petta, S.; Oliveira, C.P.; Govaere, O.; Younes, R.; McPherson, S.; et al. Performance of the PRO-C3 collagen neo-epitope biomarker in non-alcoholic fatty liver disease. JHEP Rep. 2019, 1, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Janech, M.G.; Sobolesky, P.M.; Bland, A.M.; Samsuddin, S.; Alazawi, W.; Syn, W.K. Proteomic screening of plasma identifies potential noninvasive biomarkers associated with significant/advanced fibrosis in patients with nonalcoholic fatty liver disease. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoien, R.; Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; O’Brien, P.E.; Tilg, H.; et al. Heterogeneity of fibrosis patterns in non-alcoholic fatty liver disease supports the presence of multiple fibrogenic pathways. Liver Int. 2013, 33, 624–632. [Google Scholar] [CrossRef]
- Clouston, A.D.; Powell, E.E.; Walsh, M.J.; Richardson, M.M.; Demetris, A.J.; Jonsson, J.R. Fibrosis correlates with a ductular reaction in hepatitis C: roles of impaired replication, progenitor cells and steatosis. Hepatology 2005, 41, 809–818. [Google Scholar] [CrossRef]
- Eckert, C.; Klein, N.; Kornek, M.; Lukacs-Kornek, V. The complex myeloid network of the liver with diverse functional capacity at steady state and in inflammation. Front. Immunol. 2015, 6, 179. [Google Scholar] [CrossRef] [Green Version]
- Breous, E.; Somanathan, S.; Vandenberghe, L.H.; Wilson, J.M. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 2009, 50, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Kuniyasu, Y.; Marfani, S.M.; Inayat, I.B.; Sheikh, S.Z.; Mehal, W.Z. Kupffer cells required for high affinity peptide-induced deletion, not retention, of activated CD8+ T cells by mouse liver. Hepatology 2004, 39, 1017–1027. [Google Scholar] [CrossRef]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Lanthier, N.; Molendi-Coste, O.; Cani, P.D.; van Rooijen, N.; Horsmans, Y.; Leclercq, I.A. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J. 2011, 25, 4301–4311. [Google Scholar] [CrossRef] [PubMed]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.L.; Gaudin, F.; Naveau, S.; Prevot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kazankov, K.; Jorgensen, S.M.D.; Thomsen, K.L.; Moller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased liver localization of lipopolysaccharides in human and experimental non-alcoholic fatty liver disease. Hepatology 2019. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and non-alcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Yang, L.; van Rooijen, N.; Ohnishi, H.; Seki, E. Hepatic recruitment of macrophages promotes nonalcoholic steatohepatitis through CCR2. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1310–G1321. [Google Scholar] [CrossRef] [Green Version]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.M.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010, 59, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Carpino, G.; Renzi, A.; Onori, P.; Gaudio, E. Role of hepatic progenitor cells in nonalcoholic fatty liver disease development: Cellular cross-talks and molecular networks. Int. J. Mol. Sci. 2013, 14, 20112–20130. [Google Scholar] [CrossRef] [Green Version]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Sunami, Y.; Leithauser, F.; Gul, S.; Fiedler, K.; Guldiken, N.; Espenlaub, S.; Holzmann, K.H.; Hipp, N.; Sindrilaru, A.; Luedde, T.; et al. Hepatic activation of IKK/NFkappaB signaling induces liver fibrosis via macrophage-mediated chronic inflammation. Hepatology 2012, 56, 1117–1128. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Carpino, G.; Oliveira, F.L.; Panera, N.; Nobili, V.; Gaudio, E. The role of tissue macrophage-mediated inflammation on NAFLD pathogenesis and its clinical implications. Med. Inflamm. 2017, 2017, 8162421. [Google Scholar] [CrossRef] [PubMed]
- Van Hul, N.; Lanthier, N.; Espanol Suner, R.; Abarca Quinones, J.; van Rooijen, N.; Leclercq, I. Kupffer cells influence parenchymal invasion and phenotypic orientation, but not the proliferation, of liver progenitor cells in a murine model of liver injury. Am. J. Pathol. 2011, 179, 1839–1850. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [Green Version]
- Bertolani, C.; Sancho-Bru, P.; Failli, P.; Bataller, R.; Aleffi, S.; DeFranco, R.; Mazzinghi, B.; Romagnani, P.; Milani, S.; Gines, P.; et al. Resistin as an intrahepatic cytokine: Overexpression during chronic injury and induction of proinflammatory actions in hepatic stellate cells. Am. J. Pathol. 2006, 169, 2042–2053. [Google Scholar] [CrossRef] [Green Version]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef]
- Beier, J.I.; Guo, L.; von Montfort, C.; Kaiser, J.P.; Joshi-Barve, S.; Arteel, G.E. New role of resistin in lipopolysaccharide-induced liver damage in mice. J. Pharmacol. Exp. Ther. 2008, 325, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Carpino, G.; Alisi, A.; De Vito, R.; Franchitto, A.; Alpini, G.; Onori, P.; Gaudio, E. Role of docosahexaenoic acid treatment in improving liver histology in pediatric nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e88005. [Google Scholar] [CrossRef]
- Ali, A.T.; Hochfeld, W.E.; Myburgh, R.; Pepper, M.S. Adipocyte and adipogenesis. Eur. J. Cell Biol. 2013, 92, 229–236. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef]
- Cinti, S. The adipose organ: Morphological perspectives of adipose tissues. Proc. Nutr. Soc. 2001, 60, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayeser, T.; Basak, M.; Arslan, K.; Sayan, I. Investigating the correlation of the number of diagnostic criteria to serum adiponectin, leptin, resistin, TNF-alpha, EGFR levels and abdominal adipose tissue. Diabetes Metab. Syndr. 2016, 10, S165–S169. [Google Scholar] [CrossRef] [PubMed]
- Verboven, K.; Wouters, K.; Gaens, K.; Hansen, D.; Bijnen, M.; Wetzels, S.; Stehouwer, C.D.; Goossens, G.H.; Schalkwijk, C.G.; Blaak, E.E.; et al. Abdominal subcutaneous and visceral adipocyte size, lipolysis and inflammation relate to insulin resistance in male obese humans. Sci. Rep. 2018, 8, 4677. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Tordjman, J.; Clement, K.; Scherer, P.E. Fibrosis and adipose tissue dysfunction. Cell Metab. 2013, 18, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, R.W.; Allayee, H.; Inserra, A.; Fruhwirth, R.; Alisi, A.; Devito, R.; Carey, M.E.; Sinatra, F.; Goran, M.I.; Nobili, V. Macrophages and fibrosis in adipose tissue are linked to liver damage and metabolic risk in obese children. Obesity (Silver Spring) 2014, 22, 1512–1519. [Google Scholar] [CrossRef] [Green Version]
- Manco, M.; Bottazzo, G.; DeVito, R.; Marcellini, M.; Mingrone, G.; Nobili, V. Nonalcoholic fatty liver disease in children. J. Am. Coll. Nutr. 2008, 27, 667–676. [Google Scholar] [CrossRef]
- Wilson-Perez, H.E.; Chambers, A.P.; Ryan, K.K.; Li, B.; Sandoval, D.A.; Stoffers, D.; Drucker, D.J.; Perez-Tilve, D.; Seeley, R.J. Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like Peptide 1 receptor deficiency. Diabetes 2013, 62, 2380–2385. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Gianotti, T.F.; Rosselli, M.S.; Burgueno, A.L.; Castano, G.O.; Pirola, C.J. Liver transcriptional profile of atherosclerosis-related genes in human nonalcoholic fatty liver disease. Atherosclerosis 2011, 218, 378–385. [Google Scholar] [CrossRef]
- Cancello, R.; Tordjman, J.; Poitou, C.; Guilhem, G.; Bouillot, J.L.; Hugol, D.; Coussieu, C.; Basdevant, A.; Bar Hen, A.; Bedossa, P.; et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 2006, 55, 1554–1561. [Google Scholar] [CrossRef] [Green Version]
- Tordjman, J.; Divoux, A.; Prifti, E.; Poitou, C.; Pelloux, V.; Hugol, D.; Basdevant, A.; Bouillot, J.L.; Chevallier, J.M.; Bedossa, P.; et al. Structural and inflammatory heterogeneity in subcutaneous adipose tissue: Relation with liver histopathology in morbid obesity. J. Hepatol. 2012, 56, 1152–1158. [Google Scholar] [CrossRef]
- Tordjman, J.; Poitou, C.; Hugol, D.; Bouillot, J.L.; Basdevant, A.; Bedossa, P.; Guerre-Millo, M.; Clement, K. Association between omental adipose tissue macrophages and liver histopathology in morbid obesity: Influence of glycemic status. J. Hepatol. 2009, 51, 354–362. [Google Scholar] [CrossRef] [PubMed]
- D’Incao, R.B.; Tovo, C.V.; Mattevi, V.S.; Borges, D.O.; Ulbrich, J.M.; Coral, G.P.; Ramos, M.J.; Meinhardt, N.G. Adipokine levels versus hepatic histopathology in bariatric surgery patients. Obes. Surg. 2017, 27, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, R.N.; Karwoski, C.B. Endotoxin-mediated hepatic lipid accumulation during parenteral nutrition in rats. J. Am. Coll. Nutr. 2002, 21, 351–356. [Google Scholar] [CrossRef]
- Ferolla, S.M.; Armiliato, G.N.; Couto, C.A.; Ferrari, T.C. Probiotics as a complementary therapeutic approach in nonalcoholic fatty liver disease. World J. Hepatol. 2015, 7, 559–565. [Google Scholar] [CrossRef]
- Mimura, Y.; Sakisaka, S.; Harada, M.; Sata, M.; Tanikawa, K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology 1995, 109, 1969–1976. [Google Scholar] [CrossRef]
- Bikhazi, A.B.; Jurjus, A.R.; Kamal, M.T.; Al-Housseini, A.M.; Saab, R.N.; Jaroudi, W.A.; Bitar, K.M. Kinetics of lipopolysaccharide clearance by Kupffer and parenchyma cells in perfused rat liver. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2001, 129, 339–348. [Google Scholar] [CrossRef]
- Carnevale, R.; Nocella, C.; Petrozza, V.; Cammisotto, V.; Pacini, L.; Sorrentino, V.; Martinelli, O.; Irace, L.; Sciarretta, S.; Frati, G.; et al. Localization of lipopolysaccharide from Escherichia Coli into human atherosclerotic plaque. Sci. Rep. 2018, 8, 3598. [Google Scholar] [CrossRef]
- Mahfood Haddad, T.; Hamdeh, S.; Kanmanthareddy, A.; Alla, V.M. Nonalcoholic fatty liver disease and the risk of clinical cardiovascular events: A systematic review and meta-analysis. Diabetes Metab. Syndr. 2017, 11, S209–S216. [Google Scholar] [CrossRef]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Wu, F.; Ding, Y.; Hou, J.; Bi, J.; Zhang, Z. Association of non-alcoholic fatty liver disease with major adverse cardiovascular events: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 33386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francque, S.M.; van der Graaff, D.; Kwanten, W.J. Non-alcoholic fatty liver disease and cardiovascular risk: Pathophysiological mechanisms and implications. J. Hepatol. 2016, 65, 425–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725.e716. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NON-ALCOHOLIC FATTY LIVER | NON-ALCOHOLIC STEATOHEPATITIS |
|---|---|
|
|
| NON-ALCOHOLIC FATTY LIVER | NON-ALCOHOLIC STEATOHEPATITIS | |
|---|---|---|
| Hepatic stem/progenitor cells |
|
|
| Hepatic stellate cells & portal myofibroblast pool |
|
|
| Liver macrophage pool | Lobular macrophages
| Lobular macrophages
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Overi, D.; Carpino, G.; Franchitto, A.; Onori, P.; Gaudio, E. Hepatocyte Injury and Hepatic Stem Cell Niche in the Progression of Non-Alcoholic Steatohepatitis. Cells 2020, 9, 590. https://doi.org/10.3390/cells9030590
Overi D, Carpino G, Franchitto A, Onori P, Gaudio E. Hepatocyte Injury and Hepatic Stem Cell Niche in the Progression of Non-Alcoholic Steatohepatitis. Cells. 2020; 9(3):590. https://doi.org/10.3390/cells9030590
Chicago/Turabian StyleOveri, Diletta, Guido Carpino, Antonio Franchitto, Paolo Onori, and Eugenio Gaudio. 2020. "Hepatocyte Injury and Hepatic Stem Cell Niche in the Progression of Non-Alcoholic Steatohepatitis" Cells 9, no. 3: 590. https://doi.org/10.3390/cells9030590