Haploinsufficient Rock1+/− and Rock2+/− Mice Are Not Protected from Cardiac Inflammation and Postinflammatory Fibrosis in Experimental Autoimmune Myocarditis

 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Mice
2.2. EAM Induction
2.3. Cell Cultures
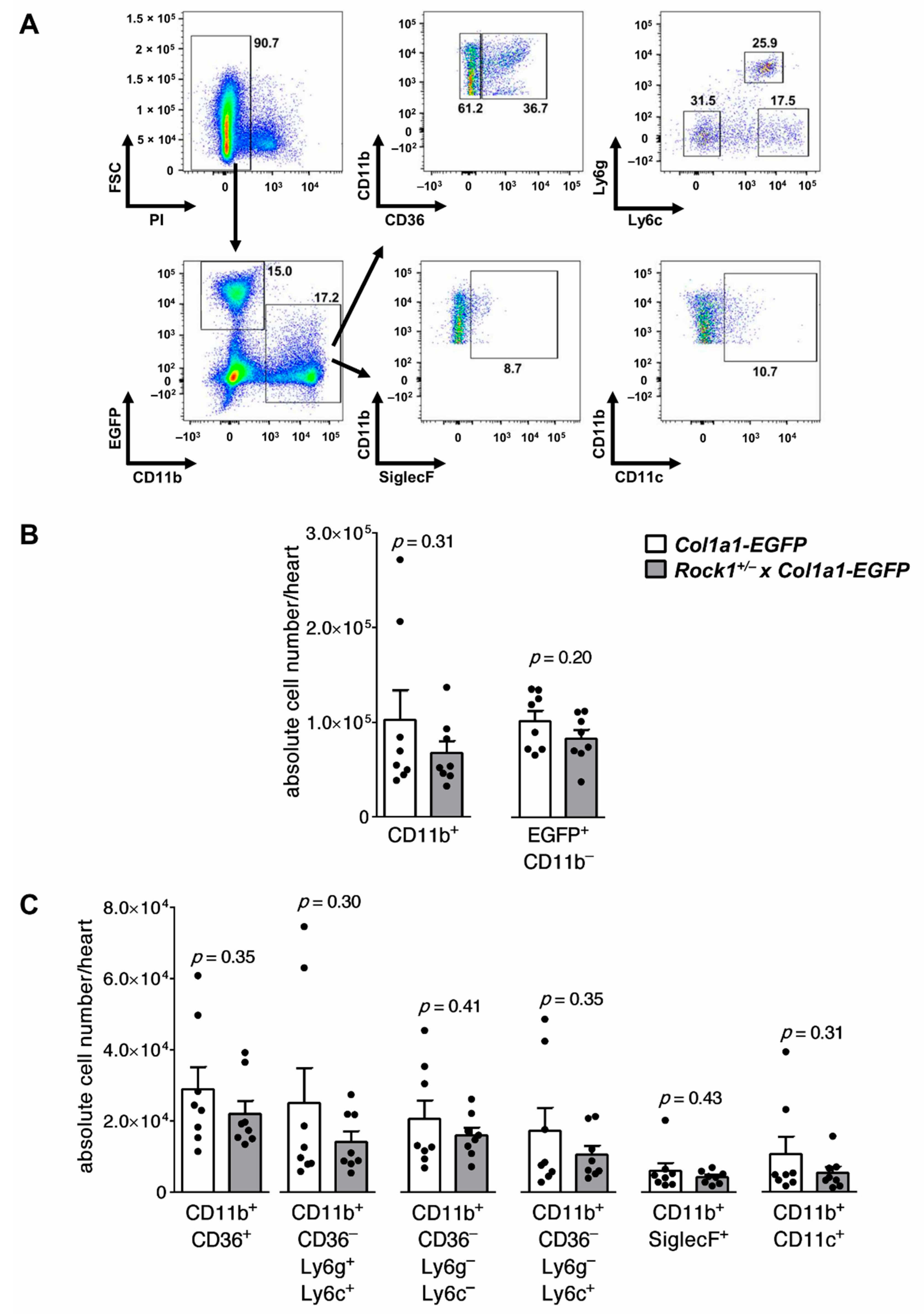
2.4. Flow Cytometry
2.5. Histology and Immunohistochemistry
2.6. Immunoblotting
2.7. Quantitative RT-PCR
2.8. Immunocytochemistry
2.9. Statistics
3. Results
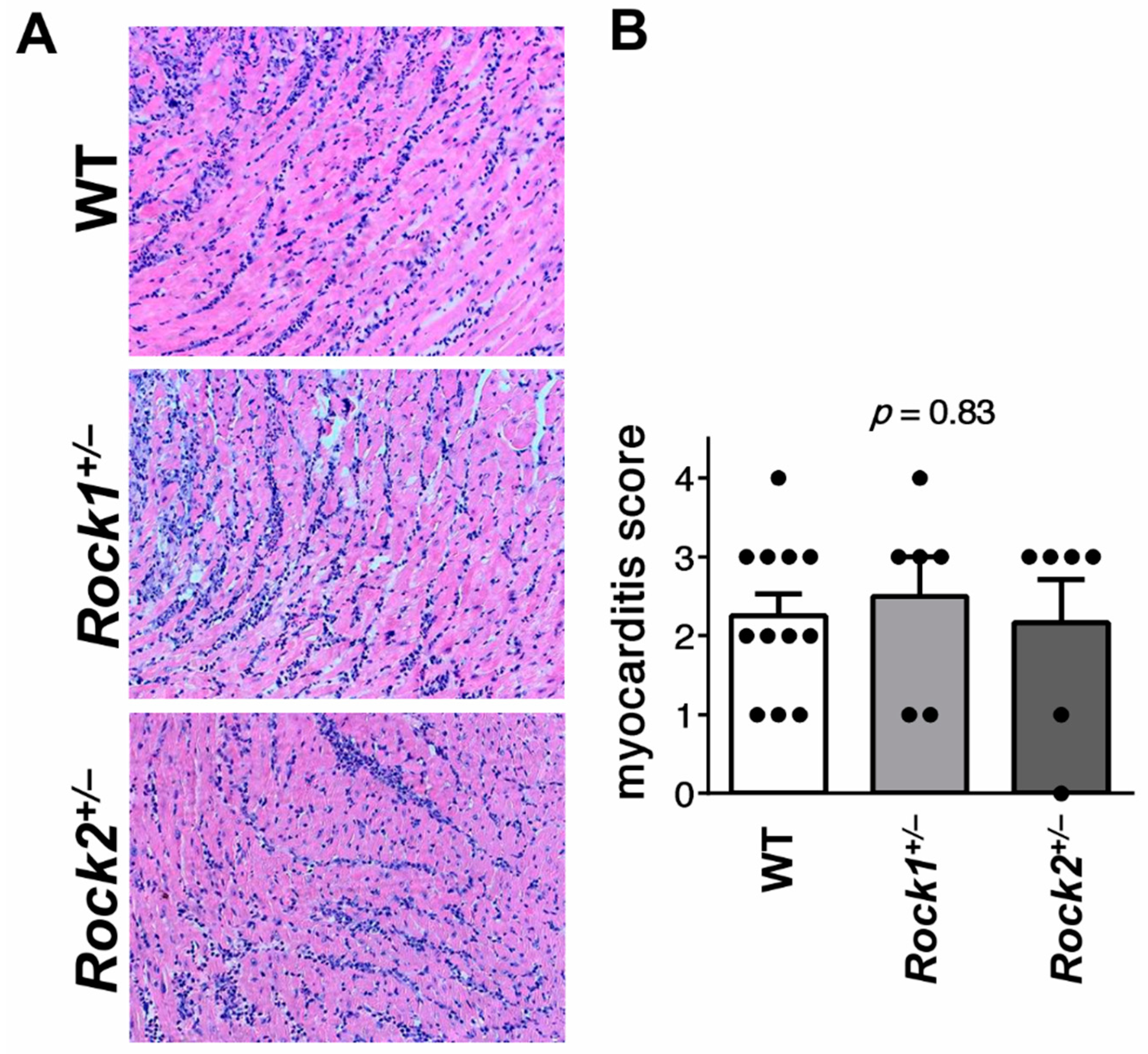
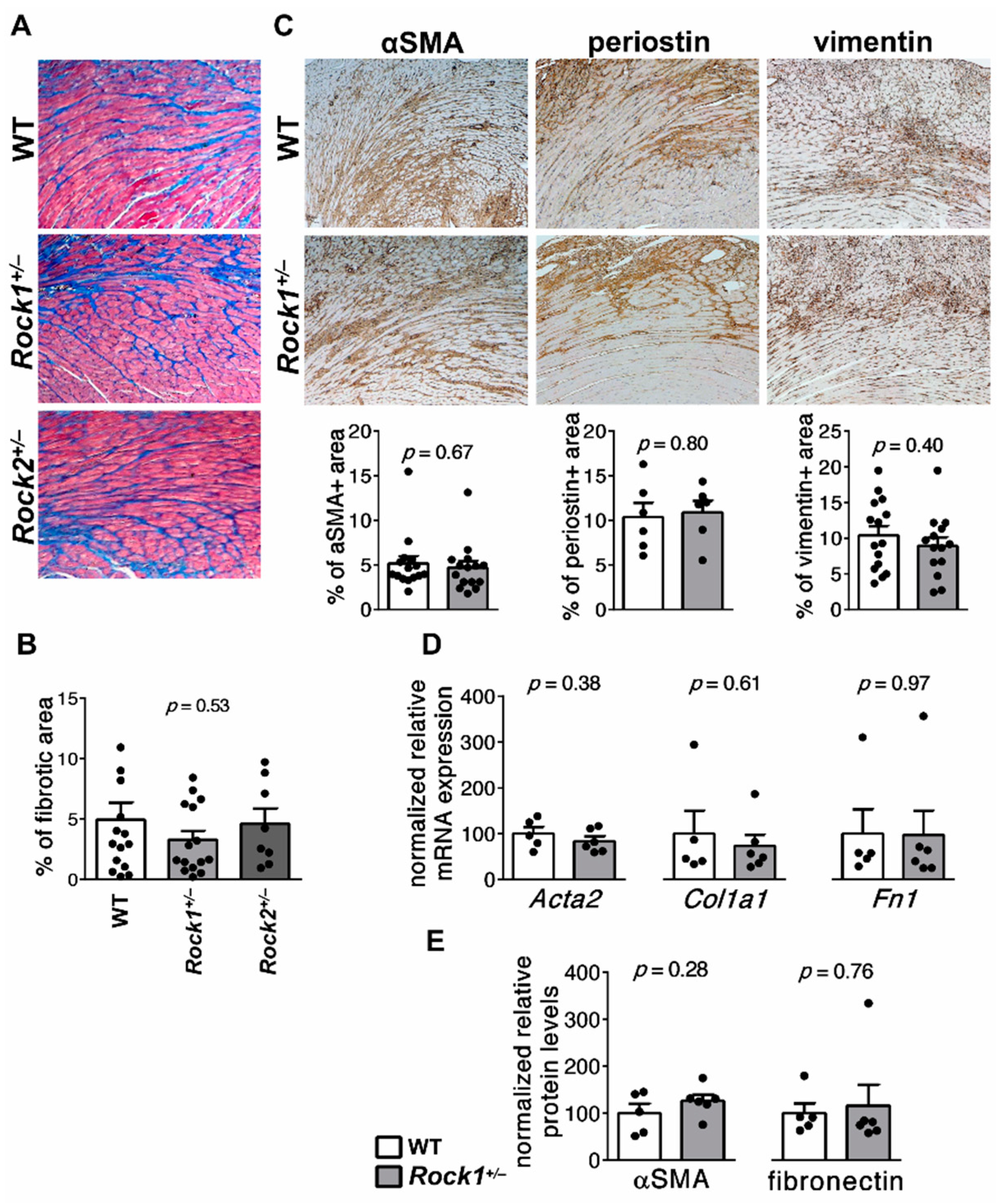
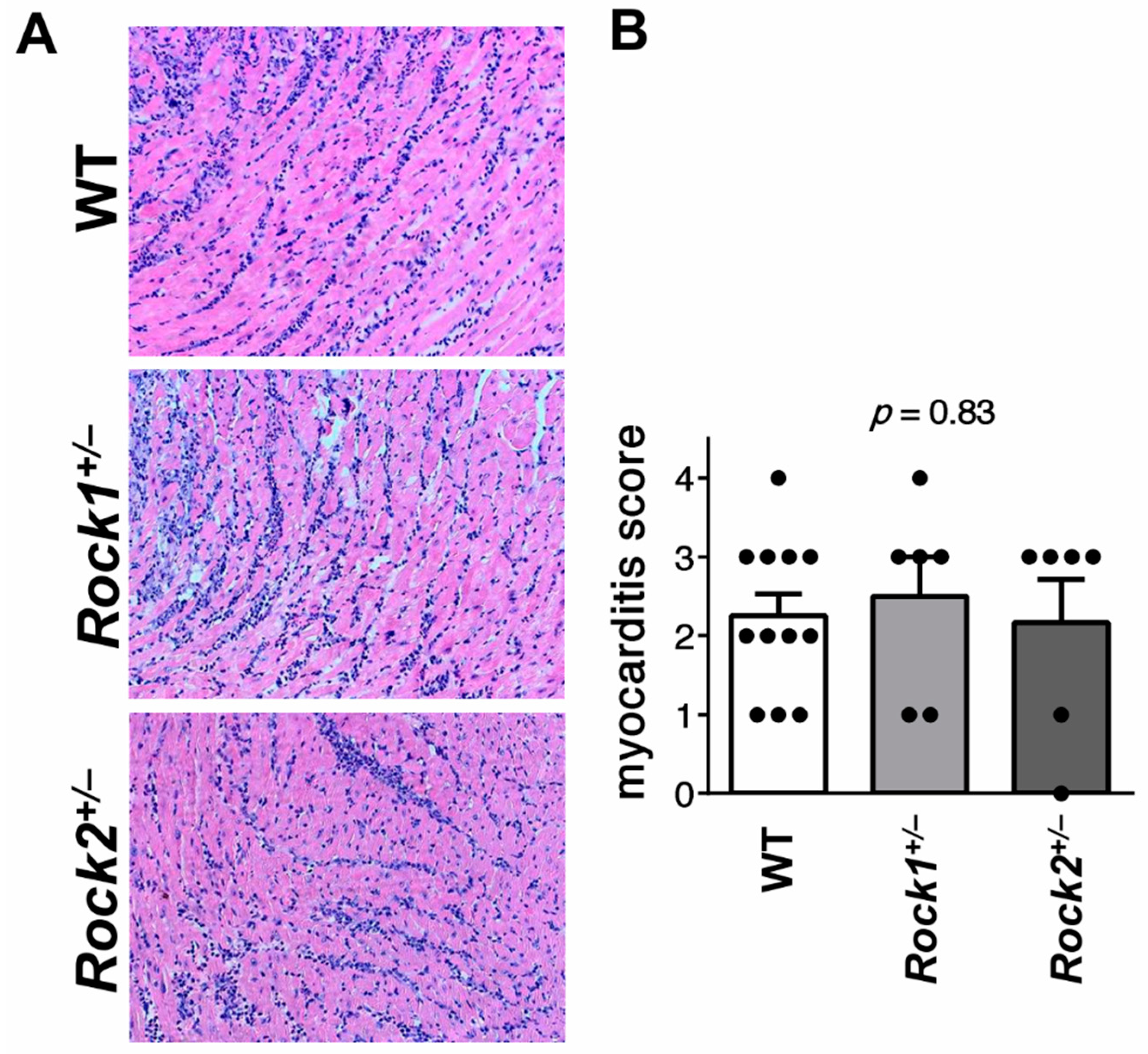
3.1. Rock1+/− and Rock2+/− Haploinsufficient Mice Develop Unaffected Myocarditis
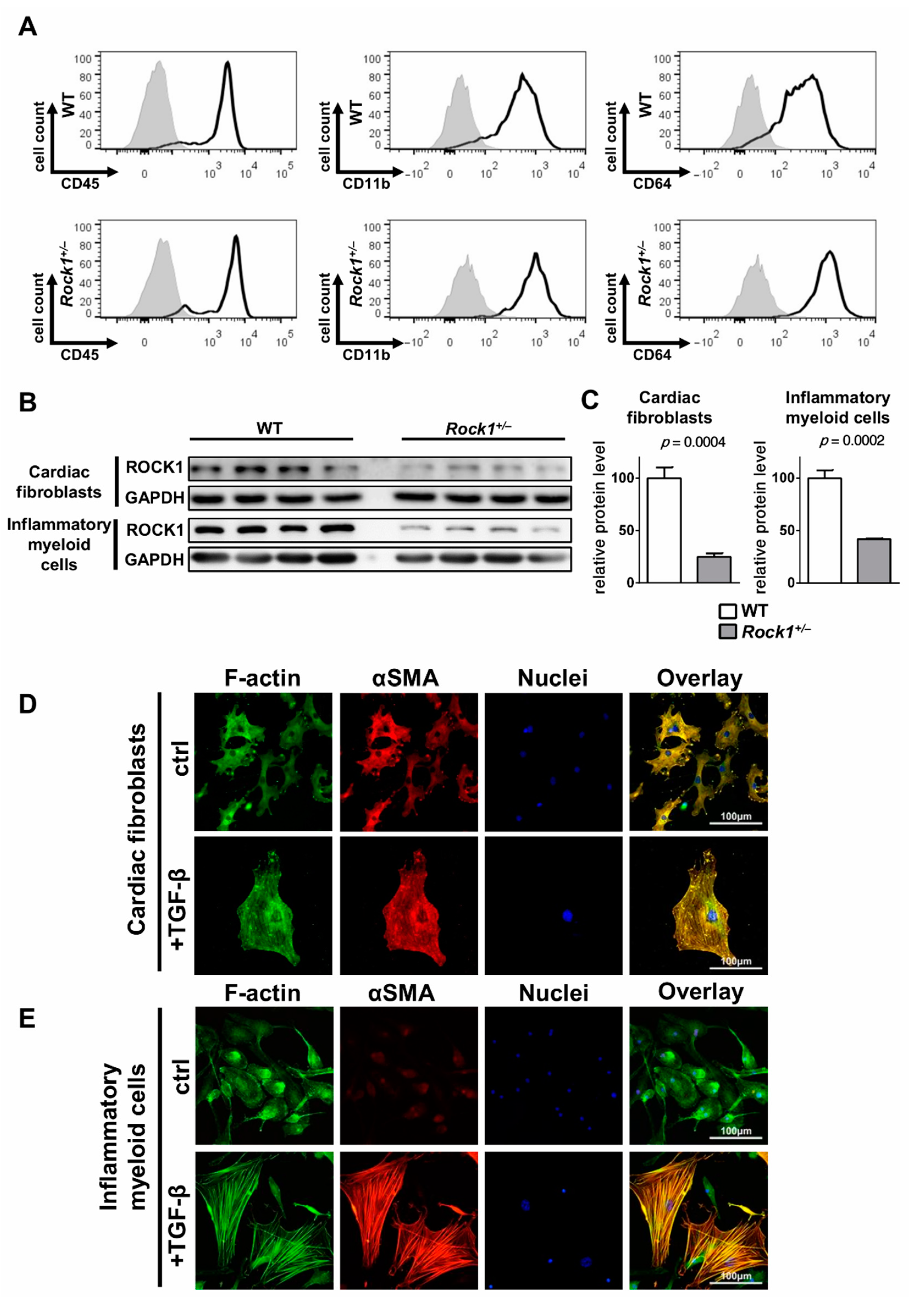
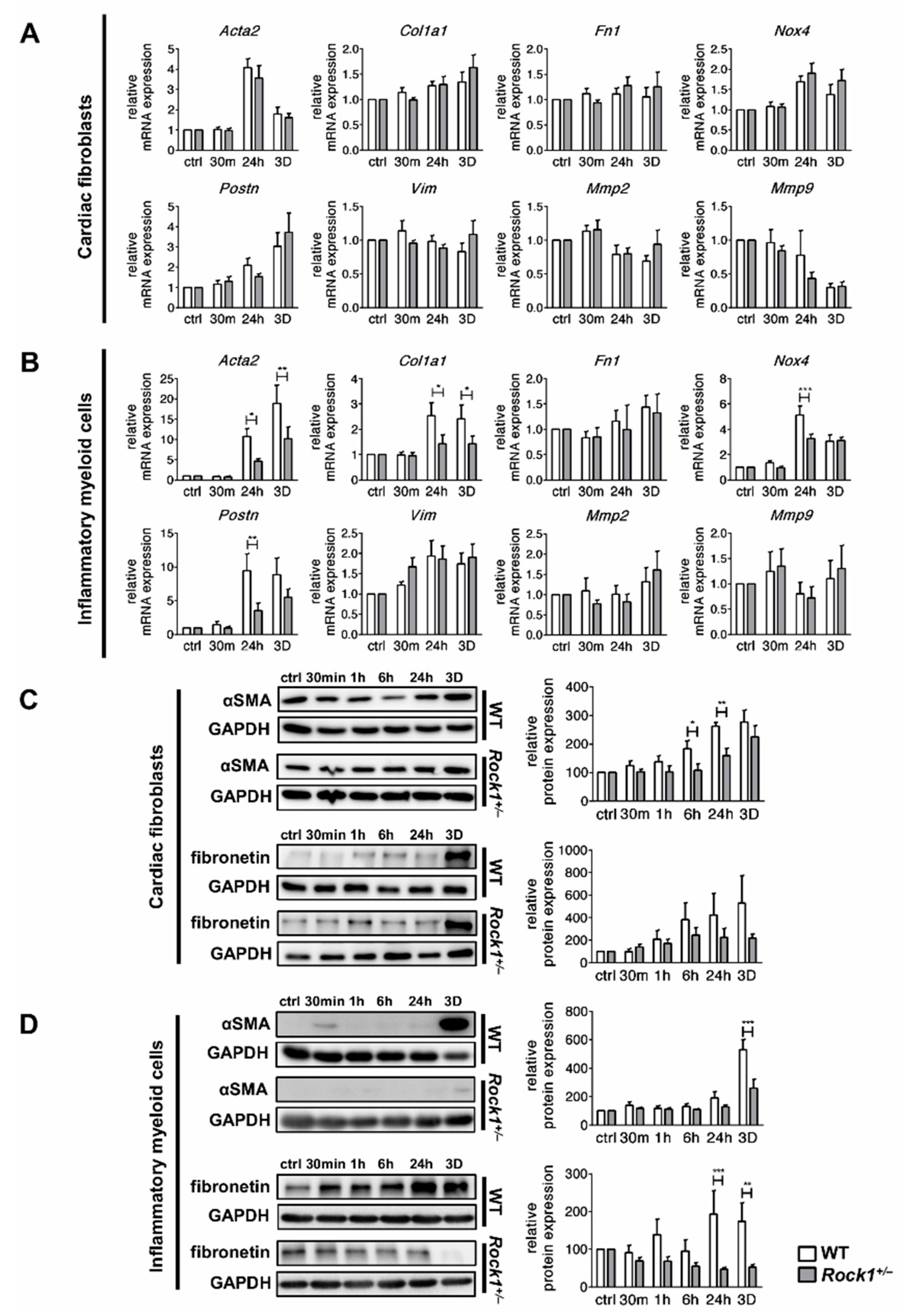
3.2. TGF-β Induces Profibrotic Changes in Cardiac Fibroblasts and in Inflammatory Myeloid Cells
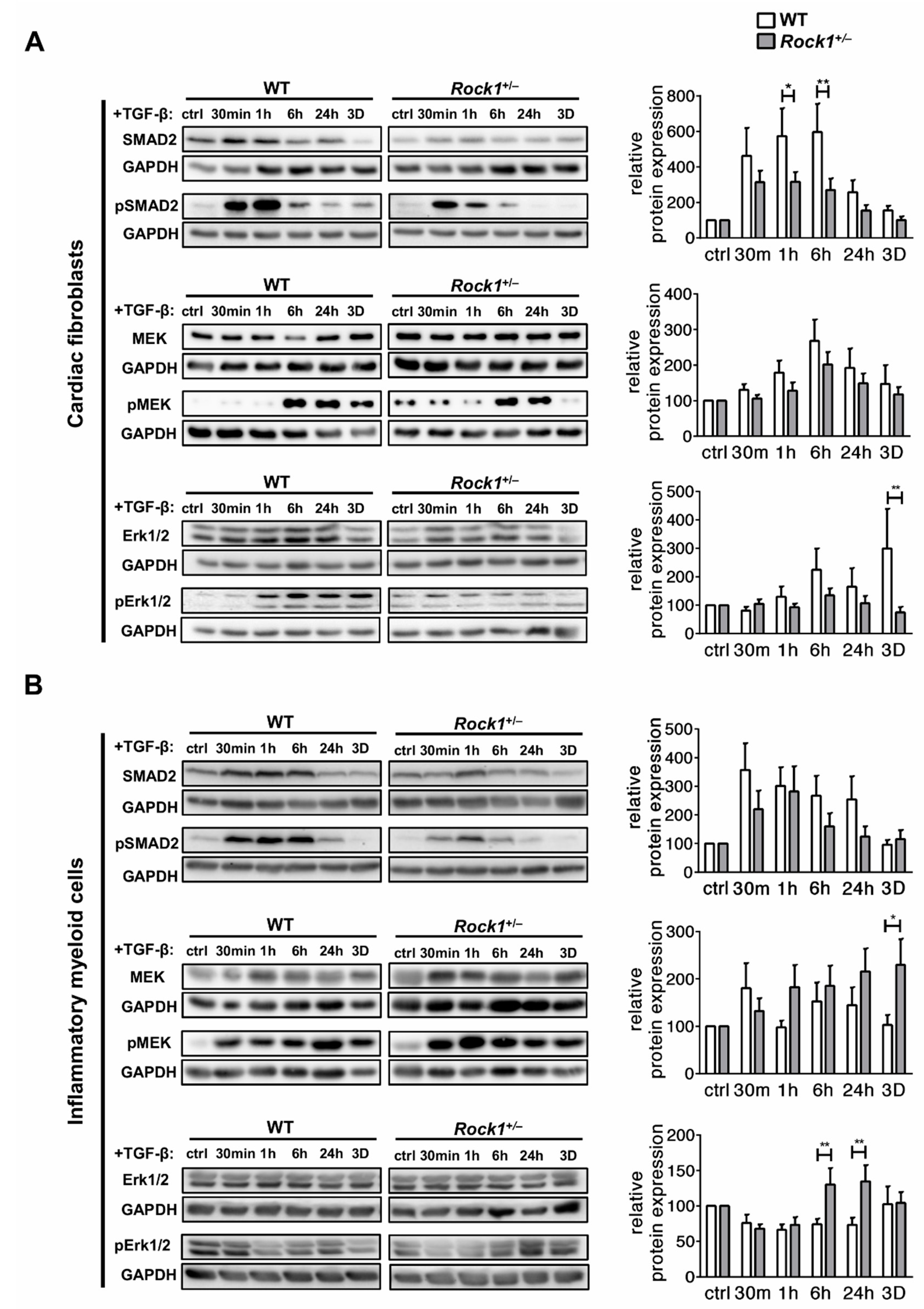
3.3. ROCK1 Differentially Regulates TGF-β Downstream Molecular Pathways in Cardiac Fibroblasts and in Inflammatory Myeloid Cells
3.4. ROCK1 Enhances Profibrotic TGF-β Response in Cardiac Fibroblasts and in Inflammatory Myeloid Cells
3.5. Rock1+/− and Rock2+/− Haploinsufficient Mice Are Not Protected from Cardiac Fibrosis in EAM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, L.; Zhao, Q.; Kong, W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 2018, 68–69, 490–506. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracamonte-Baran, W.; Čiháková, D. Cardiac Autoimmunity: Myocarditis. Adv. Exp. Med. Biol. 2017, 1003, 187–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambrosio, A.; Patti, G.; Manzoli, A.; Di Sciascio, G.; Sinagra, G.; Di Lenarda, A.; Silvestri, F. The fate of acute myocarditis between spontaneous improvement and evolution to dilated cardiomyopathy: A review. Heart 2001, 85, 499–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Błyszczuk, P. Myocarditis in Humans and in Experimental Animal Models. Front. Cardiovasc. Med. 2019, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Kania, G.; Blyszczuk, P.; Eriksson, U. Mechanisms of cardiac fibrosis in inflammatory heart disease. Trends Cardiovasc. Med. 2009, 19, 247–252. [Google Scholar] [CrossRef]
- Rosenkranz, S.; Flesch, M.; Amann, K.; Haeuseler, C.; Kilter, H.; Seeland, U.; Schlüter, K.D.; Böhm, M. Alterations of β-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-β1. Am. J. Physiol. Hear. Circ. Physiol. 2002, 283, 1253–1262. [Google Scholar] [CrossRef]
- Kania, G.; Blyszczuk, P.; Stein, S.; Valaperti, A.; Germano, D.; Dirnhofer, S.; Hunziker, L.; Matter, C.M.; Eriksson, U. Heart-infiltrating prominin-1+/CD133+ progenitor cells represent the cellular source of transforming growth factor β-mediated cardiac fibrosis in experimental autoimmune myocarditis. Circ. Res. 2009, 105, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.G.; Yuan, Y.P.; Wu, H.M.; Zhang, X.; Tang, Q.Z. Cardiac fibrosis: New insights into the pathogenesis. Int. J. Biol. Sci. 2018, 14, 1645–1657. [Google Scholar] [CrossRef] [Green Version]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.S.; Haak, A.J.; Khanna, D.; Neubig, R.R. Cellular mechanisms of tissue fibrosis. 8. Current and future drug targets in fibrosis: Focus on RHO GTPase-regulated gene transcription. Am. J. Physiol. Cell Physiol. 2014, 307, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The function of rho-associated kinases ROCK1 and ROCK2 in the pathogenesis of cardiovascular disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhang, Y.W.; Yang, Y.; Zhang, L.; Wei, L. ROCK1 plays an essential role in the transition from cardiac hypertrophy to failure in mice. J. Mol. Cell. Cardiol. 2010, 49, 819–828. [Google Scholar] [CrossRef] [Green Version]
- Haudek, S.B.; Gupta, D.; Dewald, O.; Schwartz, R.J.; Wei, L.; Trial, J.; Entman, M.L. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc. Res. 2009, 83, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Rikitake, Y.; Oyama, N.; Wang, C.Y.C.; Noma, K.; Satoh, M.; Kim, H.H.; Liao, J.K. Decreased perivascular fibrosis but not cardiac hypertrophy in ROCK1+/− haploinsufficient mice. Circulation 2005, 112, 2959–2965. [Google Scholar] [CrossRef] [Green Version]
- Hattori, T.; Shimokawa, H.; Higashi, M.; Hiroki, J.; Mukai, Y.; Tsutsui, H.; Kaibuchi, K.; Takeshita, A. Long-Term Inhibition of Rho-Kinase Suppresses Left Ventricular Remodeling after Myocardial Infarction in Mice. Circulation 2004, 109, 2234–2239. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xu, Y.; Li, X.; Guo, Y.; Liu, G. Inhibition of Rho-kinase ameliorates myocardial remodeling and fibrosis in pressure overload and myocardial infarction: Role of TGF-β1-TAK1. Toxicol. Lett. 2012, 211, 91–97. [Google Scholar] [CrossRef]
- Mera, C.; Godoy, I.; Ramírez, R.; Moya, J.; Ocaranza, M.P.; Jalil, J.E. Mechanisms of favorable effects of Rho kinase inhibition on myocardial remodeling and systolic function after experimental myocardial infarction in the rat. Ther. Adv. Cardiovasc. Dis. 2016, 10, 4–20. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Hieu, T.B.; Ma, F.; Yu, Y.; Cao, Z.; Wang, M.; Wu, W.; Mao, Y.; Rose, P.; Law, B.Y.K.; et al. ZYZ-168 alleviates cardiac fibrosis after myocardial infarction through inhibition of ERK1/2-dependent ROCK1 activation. Sci. Rep. 2017, 7, 43242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.M.; Bo, J.; Taffet, G.E.; Chang, J.; Shi, J.; Reddy, A.K.; Michael, L.H.; Schneider, M.D.; Entman, M.L.; Schwartz, R.J.; et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. FASEB J. 2006, 20, 916–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, M.; Shimokawa, H.; Hattori, T.; Hiroki, J.; Mukai, Y.; Morikawa, K.; Ichiki, T.; Takahashi, S.; Takeshita, A. Long-Term Inhibition of Rho-Kinase Suppresses Angiotensin II-Induced Cardiovascular Hypertrophy in Rats In Vivo: Effect on Endothelial NAD(P)H Oxidase System. Circ. Res. 2003, 93, 767–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, R.; Li, Y.; Noma, K.; Hiroi, Y.; Liu, P.Y.; Taniguchi, M.; Ito, M.; Liao, J.K. FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 2013, 27, 1439–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Li, Q.; Lin, X.; Ma, Y.; Yue, X.; Tao, Z.; Wang, F.; Mckeehan, W.L.; Wei, L.; Schwartz, R.J.; et al. Mechanism of fibrotic cardiomyopathy in mice expressing truncated Rho-associated coiled-coil protein kinase 1. FASEB J. 2012, 26, 2105–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Surma, M.; Yang, Y.; Wei, L. Disruption of both ROCK1 and ROCK2 genes in cardiomyocytes promotes autophagy and reduces cardiac fibrosis during aging. FASEB J. 2019, 33, 7348–7362. [Google Scholar] [CrossRef]
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2, 93187. [Google Scholar] [CrossRef]
- Stellato, M.; Czepiel, M.; Distler, O.; Błyszczuk, P.; Kania, G. Identification and Isolation of Cardiac Fibroblasts From the Adult Mouse Heart Using Two-Color Flow Cytometry. Front. Cardiovasc. Med. 2019, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- National Research Council. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011; ISBN 978-0-309-15400-0. [Google Scholar]
- Kania, G.; Blyszczuk, P.; Valaperti, A.; Dieterle, T.; Leimenstoll, B.; Dirnhofer, S.; Zulewski, H.; Eriksson, U. Prominin-1+/CD133+ bone marrow-derived heart-resident cells suppress experimental autoimmune myocarditis. Cardiovasc. Res. 2008, 80, 236–245. [Google Scholar] [CrossRef] [Green Version]
- Zarak-Crnkovic, M.; Kania, G.; Jaźwa-Kusior, A.; Czepiel, M.; Wijnen, W.J.; Czyż, J.; Müller-Edenborn, B.; Vdovenko, D.; Lindner, D.; Gil-Cruz, C.; et al. Heart non-specific effector CD4+ T cells protect from postinflammatory fibrosis and cardiac dysfunction in experimental autoimmune myocarditis. Basic Res. Cardiol. 2020, 115, 6. [Google Scholar] [CrossRef] [Green Version]
- Działo, E.; Rudnik, M.; Koning, R.I.; Czepiel, M.; Tkacz, K.; Baj-Krzyworzeka, M.; Distler, O.; Siedlar, M.; Kania, G.; Błyszczuk, P. WNT3a and WNT5a Transported by Exosomes Activate WNT Signaling Pathways in Human Cardiac Fibroblasts. Int. J. Mol. Sci. 2019, 20, 1436. [Google Scholar] [CrossRef] [Green Version]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Invest. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, T.; Liao, J.K. Rho kinases and cardiac remodeling. Circ. J. 2016, 80, 1491–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surma, M.; Wei, L.; Shi, J. Rho kinase as a therapeutic target in cardiovascular disease. Future Cardiol. 2011, 7, 657–671. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Hu, E.; Tao, L.; Boyce, R.; Mirabile, R.; Thudium, D.T.; Ma, X.L.; Willette, R.N.; Yue, T.L. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc. Res. 2004, 61, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, K.; Ueno, H.; Kagitani, S.; Takabayashi, D.; Takata, M.; Inoue, H. Fasudil attenuates myocardial fibrosis in association with inhibition of monocyte/macrophage infiltration in the heart of DOCA/salt hypertensive rats. J. Cardiovasc. Pharmacol. 2007, 50, 187–194. [Google Scholar] [CrossRef]
- Knipe, R.S.; Probst, C.K.; Lagares, D.; Franklin, A.; Spinney, J.J.; Brazee, P.L.; Grasberger, P.; Zhang, L.; Black, K.E.; Sakai, N.; et al. The rho kinase isoforms ROCK1 and ROCK2 each contribute to the development of experimental pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2018, 58, 471–481. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, F.; Huang, X.R.; Liu, F.; Chen, H.; Chung, A.C.K.; Shi, J.; Wei, L.; Lan, H.Y.; Fu, P. Amelioration of albuminuria in ROCK1 knockout mice with streptozotocin-induced diabetic kidney disease. Am. J. Nephrol. 2011, 34, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef]
- Fu, P.; Liu, F.; Su, S.; Wang, W.; Huang, X.R.; Entman, M.L.; Schwartz, R.J.; Wei, L.; Lan, H.Y. Signaling mechanism of renal fibrosis in unilateral ureteral obstructive kidney disease in ROCK1 knockout mice. J. Am. Soc. Nephrol. 2006, 17, 3105–3114. [Google Scholar] [CrossRef] [Green Version]
- Yu, O.M.; Brown, J.H. G Protein-Coupled Receptor and RhoA-Stimulated Transcriptional Responses: Links to Inflammation, Differentiation, and Cell Proliferation. Mol. Pharmacol. 2015, 88, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Ohtsu, H.; Suzuki, H.; Nakashima, H.; Dhobale, S.; Frank, G.D.; Motley, E.D.; Eguchi, S. Angiotensin II signal transduction through small GTP-binding proteins: Mechanism and significance in vascular smooth muscle cells. Hypertens. (Dallas, Tex. 1979) 2006, 48, 534–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blyszczuk, P.; Müller-Edenborn, B.; Valenta, T.; Osto, E.; Stellato, M.; Behnke, S.; Glatz, K.; Basler, K.; Lüscher, T.F.; Distler, O.; et al. Transforming growth factor-b-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart, J. 2017, 38, 1413–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.R.; Neumann, D.A.; Lafond-Walker, A.; Herskowitz, A.; Rose, N.R. Role of IL-1 and tumor necrosis factor in coxsackie virus-induced autoimmune myocarditis. J. Immunol. 1993, 151, 1682–1690. [Google Scholar] [PubMed]
- Liu, Y.; Zhu, H.; Su, Z.; Sun, C.; Yin, J.; Yuan, H.; Sandoghchian, S.; Jiao, Z.; Wang, S.; Xu, H. IL-17 contributes to cardiac fibrosis following experimental autoimmune myocarditis by a PKCβ/Erk1/2/NF-κB-dependent signaling pathway. Int. Immunol. 2012, 24, 605–612. [Google Scholar] [CrossRef]
- Chen, S.; Crawford, M.; Day, R.M.; Briones, V.R.; Leader, J.E.; Jose, P.A.; Lechleider, R.J. RhoA modulates Smad signaling during transforming growth factor-beta-induced smooth muscle differentiation. J. Biol. Chem. 2006, 281, 1765–1770. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Kimoto, K.; Imaizumi, M.; Nakatsuka, K. Inhibition of RhoA/Rho-kinase pathway suppresses the expression of type I collagen induced by TGF-β2 in human retinal pigment epithelial cells. Exp. Eye Res. 2007, 84, 464–472. [Google Scholar] [CrossRef]
- Manresa-Arraut, A.; Johansen, F.F.; Brakebusch, C.; Issazadeh-Navikas, S.; Hasseldam, H. RhoA Drives T-Cell Activation and Encephalitogenic Potential in an Animal Model of Multiple Sclerosis. Front. Immunol. 2018, 9, 1235. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Minohara, M.; Kikuchi, H.; Ishizu, T.; Tanaka, M.; Piao, H.; Osoegawa, M.; Ohyagi, Y.; Shimokawa, H.; Kira, J.-I. The selective Rho-kinase inhibitor Fasudil is protective and therapeutic in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2006, 180, 126–134. [Google Scholar] [CrossRef]
- Biswas, P.S.; Gupta, S.; Chang, E.; Song, L.; Stirzaker, R.A.; Liao, J.K.; Bhagat, G.; Pernis, A.B. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Invest. 2010, 120, 3280–3295. [Google Scholar] [CrossRef] [Green Version]
- Dai, K.; Wang, Y.; Tai, S.; Ni, H.; Lian, H.; Yu, Y.; Liao, W.; Zheng, C.; Chen, Q.; Kuver, A.; et al. Fasudil exerts a cardio-protective effect on mice with coxsackievirus B3-induced acute viral myocarditis. Cardiovasc. Ther. 2018, 36, 12477. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Liu, P.-Y.; Kasahara, D.I.; Williams, A.S.; Verbout, N.G.; Halayko, A.J.; Fedulov, A.; Shoji, T.; Williams, E.S.; Noma, K.; et al. Role of Rho kinase isoforms in murine allergic airway responses. Eur. Respir. J. 2011, 38, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Tönges, L. ROCKing regeneration: Rho kinase inhibition as molecular target for neurorestoration. Front. Mol. Neurosci. 2011, 4, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tkacz, K.; Rolski, F.; Czepiel, M.; Działo, E.; Siedlar, M.; Eriksson, U.; Kania, G.; Błyszczuk, P. Haploinsufficient Rock1+/− and Rock2+/− Mice Are Not Protected from Cardiac Inflammation and Postinflammatory Fibrosis in Experimental Autoimmune Myocarditis. Cells 2020, 9, 700. https://doi.org/10.3390/cells9030700
Tkacz K, Rolski F, Czepiel M, Działo E, Siedlar M, Eriksson U, Kania G, Błyszczuk P. Haploinsufficient Rock1+/− and Rock2+/− Mice Are Not Protected from Cardiac Inflammation and Postinflammatory Fibrosis in Experimental Autoimmune Myocarditis. Cells. 2020; 9(3):700. https://doi.org/10.3390/cells9030700
Chicago/Turabian StyleTkacz, Karolina, Filip Rolski, Marcin Czepiel, Edyta Działo, Maciej Siedlar, Urs Eriksson, Gabriela Kania, and Przemysław Błyszczuk. 2020. "Haploinsufficient Rock1+/− and Rock2+/− Mice Are Not Protected from Cardiac Inflammation and Postinflammatory Fibrosis in Experimental Autoimmune Myocarditis" Cells 9, no. 3: 700. https://doi.org/10.3390/cells9030700
APA StyleTkacz, K., Rolski, F., Czepiel, M., Działo, E., Siedlar, M., Eriksson, U., Kania, G., & Błyszczuk, P. (2020). Haploinsufficient Rock1+/− and Rock2+/− Mice Are Not Protected from Cardiac Inflammation and Postinflammatory Fibrosis in Experimental Autoimmune Myocarditis. Cells, 9(3), 700. https://doi.org/10.3390/cells9030700