Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc


Abstract
:1. Introduction
1.1. Myc
1.2. Myc in Cancer
1.3. Myc as a Therapeutic Target
- The three Myc family members, MYC, MYCN, and MYCL, are partially-redundant transcription factors, so that Myc-inhibitory strategies, ideally, should target them all to obtain the most efficient therapeutic impact.
- Myc is an intrinsically disordered, non-enzymatic protein; hence, conventional small molecules that target highly conserved, fixed three-dimensional structures like ATP-binding pockets in kinases cannot be discovered or designed.
- Myc exerts its function in the nucleus of the cell, an elusive compartment to conventional therapeutics.
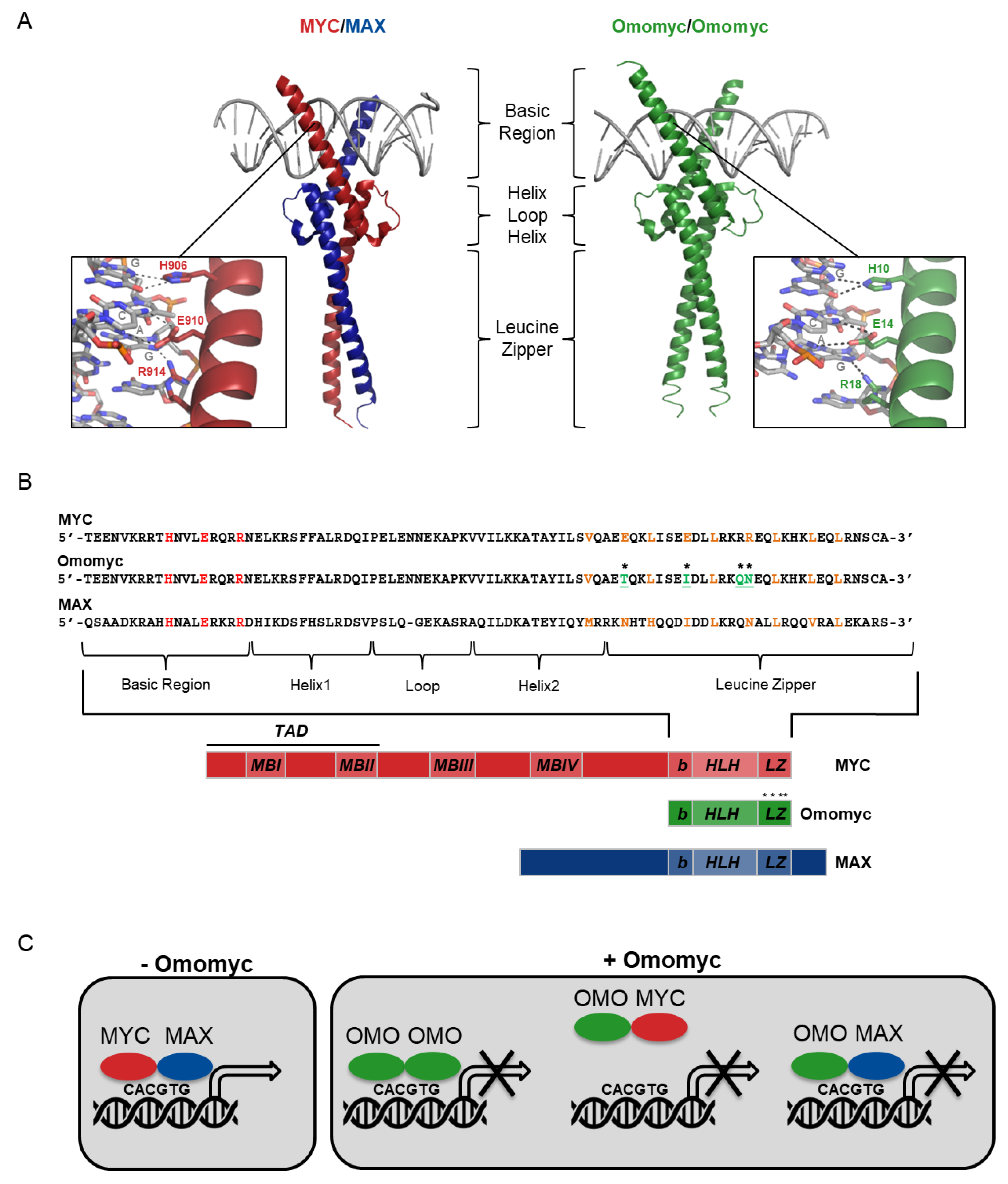
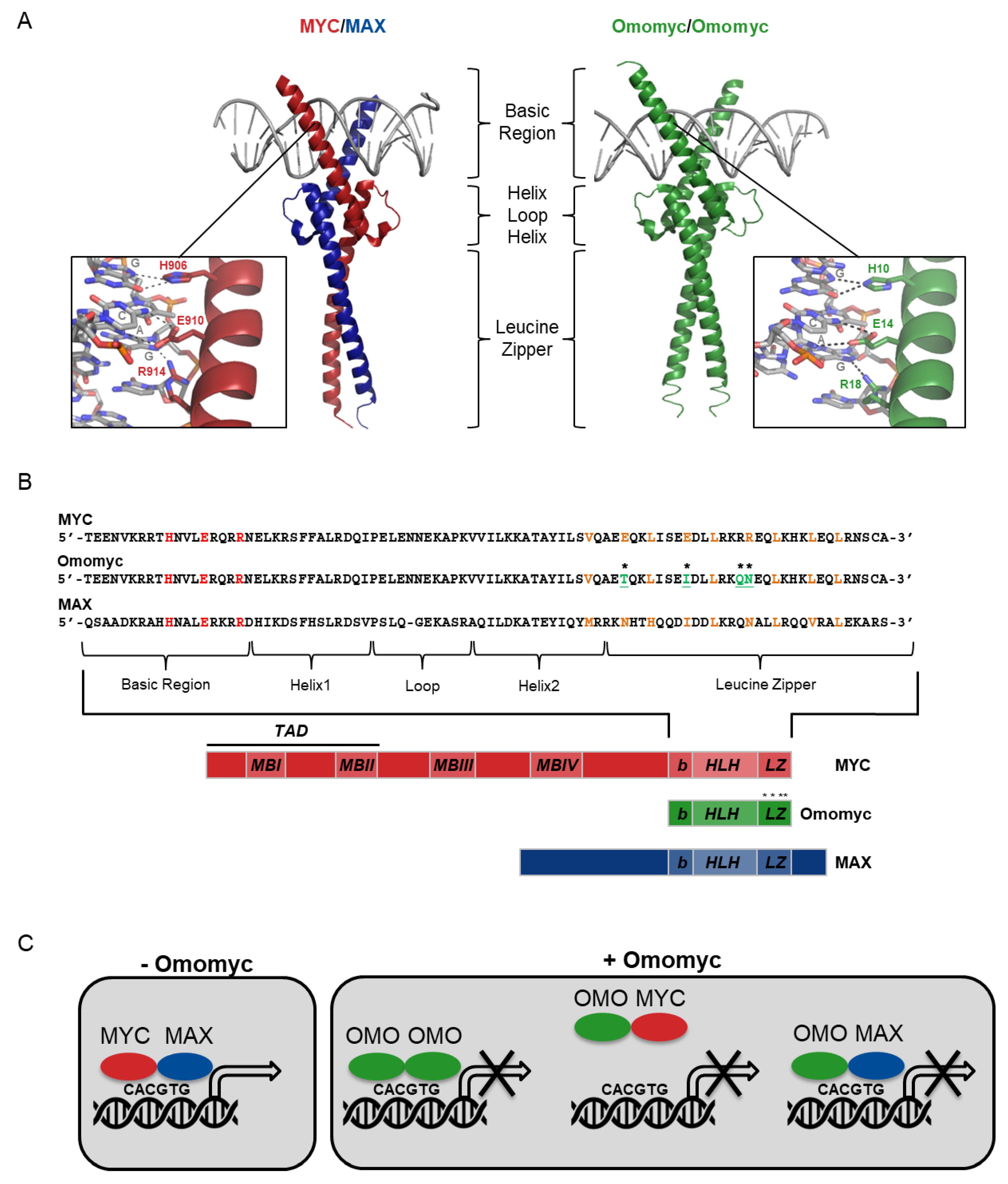
2. Omomyc Design and Characterization
2.1. Omomyc as a Myc Dominant Negative
2.2. Omomyc’s Role in Myc-induced Transactivation and Transrepression
2.3. Omomyc and Epigenetic Markers
2.4. Omomyc and Stemness
3. Omomyc as a Proof of Concept that Myc Inhibition is a Viable Therapeutic Option
3.1. Omomyc Efficacy In Vitro
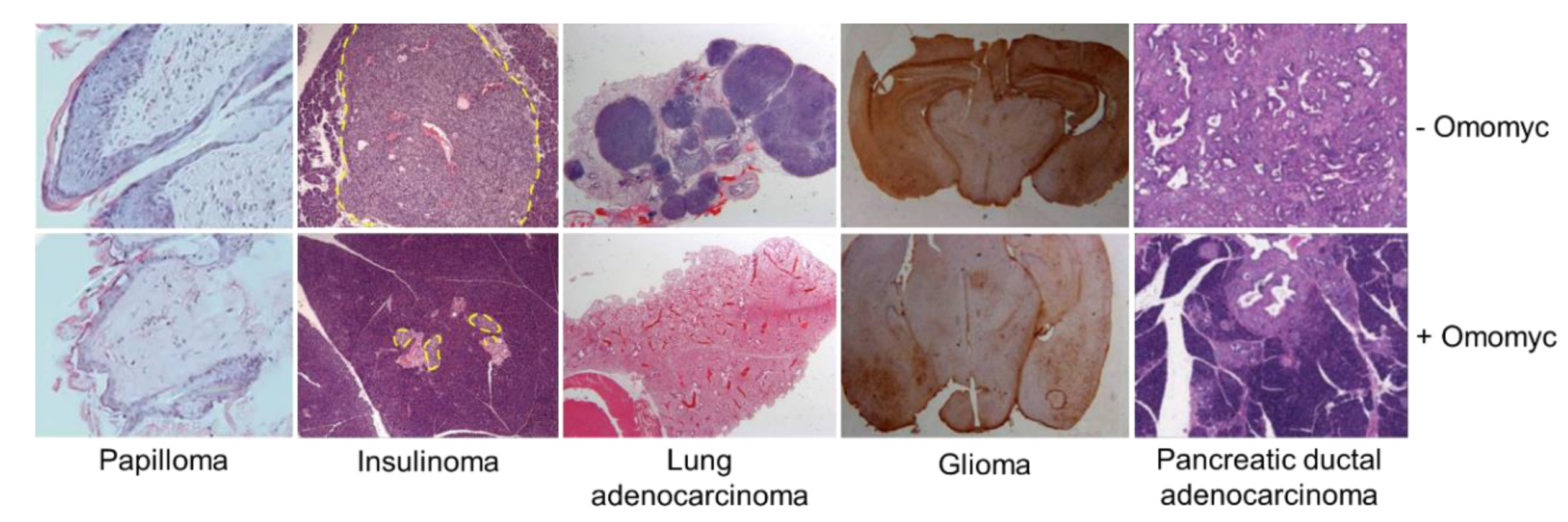
3.2. Omomyc Efficacy and Side Effects In Vivo
4. From Proof of Concept to Pharmacological Approach
4.1. Recombinant Omomyc is a Cell-penetrating Peptide
4.2. The Omomyc Mini-Protein Behaves as its Transgenically-expressed Counterpart in Cancer Cells
4.3. Intranasal or Intravenous Administration of Omomyc is Safe and Efficacious In Vivo
5. Ongoing Research and Future Directions
5.1. Omomyc Fusion with “Phylomers”
5.2. Inclusion Bodies
5.3. Variants of Omomyc
5.4. Other bHLHLZ Mini-Proteins
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tansey, W.P. Mammalian MYC Proteins and Cancer. New J. Sci. 2014, 2014, 27. [Google Scholar] [CrossRef] [Green Version]
- Malynn, B.A.; de Alboran, I.M.; O’Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [PubMed]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vita, M.; Henriksson, M. The Myc oncoprotein as a therapeutic target for human cancer. Semin. Cancer Biol. 2006, 16, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. Enigmatic MYC Conducts an Unfolding Systems Biology Symphony. Genes Cancer 2010, 1, 526–531. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Patel, J.H.; Loboda, A.P.; Showe, M.K.; Showe, L.C.; McMahon, S.B. Analysis of genomic targets reveals complex functions of MYC. Nat. Rev. Cancer 2004, 4, 562–568. [Google Scholar] [CrossRef]
- Knoepfler, P.S.; Zhang, X.Y.; Cheng, P.F.; Gafken, P.R.; McMahon, S.B.; Eisenman, R.N. Myc influences global chromatin structure. EMBO J. 2006, 25, 2723–2734. [Google Scholar] [CrossRef]
- Cotterman, R.; Jin, V.X.; Krig, S.R.; Lemen, J.M.; Wey, A.; Farnham, P.J.; Knoepfler, P.S. N-Myc regulates a widespread euchromatic program in the human genome partially independent of its role as a classical transcription factor. Cancer Res. 2008, 68, 9654–9662. [Google Scholar] [CrossRef] [Green Version]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [Green Version]
- Bedard, M.; Maltais, L.; Montagne, M.; Lavigne, P. Miz-1 and Max compete to engage c-Myc: Implication for the mechanism of inhibition of c-Myc transcriptional activity by Miz-1. Proteins 2017, 85, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.K.; Burley, S.K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell 2003, 112, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Jung, L.A.; Gebhardt, A.; Koelmel, W.; Ade, C.P.; Walz, S.; Kuper, J.; von Eyss, B.; Letschert, S.; Redel, C.; d’Artista, L.; et al. OmoMYC blunts promoter invasion by oncogenic MYC to inhibit gene expression characteristic of MYC-dependent tumors. Oncogene 2017, 36, 1911–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLano, W. The PyMOL Molecular Graphics System; DeLano Scientific LLC: San Carlos, CA, USA, 2002. [Google Scholar]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.R.; Ayer, D.E. Interactions between Myc and MondoA transcription factors in metabolism and tumourigenesis. Br. J. Cancer 2015, 113, 1529–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masso-Valles, D.; Beaulieu, M.E.; Soucek, L. MYC, MYCL and MYCN as therapeutic targets in lung cancer. Expert Opin. Ther. Targets 2020. [Google Scholar] [CrossRef]
- Levens, D. You Don’t Muck with MYC. Genes Cancer 2010, 1, 547–554. [Google Scholar] [CrossRef]
- Vervoorts, J.; Luscher-Firzlaff, J.; Luscher, B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006, 281, 34725–34729. [Google Scholar] [CrossRef] [Green Version]
- Zanet, J.; Pibre, S.; Jacquet, C.; Ramirez, A.; de Alboran, I.M.; Gandarillas, A. Endogenous Myc controls mammalian epidermal cell size, hyperproliferation, endoreplication and stem cell amplification. J. Cell Sci. 2005, 118, 1693–1704. [Google Scholar] [CrossRef] [Green Version]
- McMahon, S.B. MYC and the control of apoptosis. Cold Spring Harb. Perspect. Med. 2014, 4, a014407. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Qi, Y.; Hann, S.R. The ARF tumor suppressor: Keeping Myc on a leash. Cell Cycle 2005, 4, 249–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phesse, T.J.; Myant, K.B.; Cole, A.M.; Ridgway, R.A.; Pearson, H.; Muncan, V.; van den Brink, G.R.; Vousden, K.H.; Sears, R.; Vassilev, L.T.; et al. Endogenous c-Myc is essential for p53-induced apoptosis in response to DNA damage in vivo. Cell Death Differ. 2014, 21, 956–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boone, D.N.; Qi, Y.; Li, Z.; Hann, S.R. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 632–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, J.H.; McMahon, S.B. BCL2 is a downstream effector of MIZ-1 essential for blocking c-MYC-induced apoptosis. J. Biol. Chem. 2007, 282, 5–13. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.H.; McMahon, S.B. Targeting of Miz-1 is essential for Myc-mediated apoptosis. J. Biol. Chem. 2006, 281, 3283–3289. [Google Scholar] [CrossRef]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Boxer, L.M.; Dang, C.V. Translocations involving c-myc and c-myc function. Oncogene 2001, 20, 5595–5610. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Wang, J.; Ou, C.; Zhang, Y.; Ma, L.; Weng, W.; Pan, Q.; Sun, F. Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochem. Biophys. Res. Commun. 2013, 439, 167–172. [Google Scholar] [CrossRef]
- Whitfield, J.R.; Beaulieu, M.E.; Soucek, L. Strategies to Inhibit Myc and Their Clinical Applicability. Front. Cell Dev. Biol. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeid, R.; Lawlor, M.A.; Poon, E.; Reyes, J.M.; Fulciniti, M.; Lopez, M.A.; Scott, T.G.; Nabet, B.; Erb, M.A.; Winter, G.E.; et al. Enhancer invasion shapes MYCN-dependent transcriptional amplification in neuroblastoma. Nat. Genet. 2018, 50, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.M.; Edwards, M.C.; Meyers, Z.R.; Talbot, C.C., Jr.; Hao, H.; Blum, D.; Iorns, E.; Tsui, R.; Denis, A.; Perfito, N.; et al. Replication Study: Transcriptional amplification in tumor cells with elevated c-Myc. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. c-Myc target genes involved in cell growth, apoptosis, and metabolism. Mol. Cell. Biol. 1999, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.L.; Wang, P.; Lu, M.Z.; Zhang, S.D.; Zheng, L. c-Myc maintains the self-renewal and chemoresistance properties of colon cancer stem cells. Oncol. Lett. 2019, 17, 4487–4493. [Google Scholar] [CrossRef] [Green Version]
- Poli, V.; Fagnocchi, L.; Fasciani, A.; Cherubini, A.; Mazzoleni, S.; Ferrillo, S.; Miluzio, A.; Gaudioso, G.; Vaira, V.; Turdo, A.; et al. MYC-driven epigenetic reprogramming favors the onset of tumorigenesis by inducing a stem cell-like state. Nat. Commun. 2018, 9, 1024. [Google Scholar] [CrossRef] [Green Version]
- Baudino, T.A.; McKay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [Green Version]
- Brandvold, K.A.; Neiman, P.; Ruddell, A. Angiogenesis is an early event in the generation of myc-induced lymphomas. Oncogene 2000, 19, 2780–2785. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Cai, B.; Ding, M.; Su, Z.; Liu, Y.; Shen, L. c-Myc Overexpression Promotes Oral Cancer Cell Proliferation and Migration by Enhancing Glutaminase and Glutamine Synthetase Activity. Am. J. Med. Sci. 2019, 358, 235–242. [Google Scholar] [CrossRef]
- Wolfer, A.; Ramaswamy, S. MYC and metastasis. Cancer Res. 2011, 71, 2034–2037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casacuberta-Serra, S.; Soucek, L. Myc and Ras, the Bonnie and Clyde of immune evasion. Transl. Cancer Res. 2018, 7, S457–S459. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, N.; Leyland-Jones, B.; De, P. MYC-xing it up with PIK3CA mutation and resistance to PI3K inhibitors: Summit of two giants in breast cancers. Am. J. Cancer Res. 2015, 5, 1–19. [Google Scholar] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawai, S.; Shimono, A.; Wakamatsu, Y.; Palmes, C.; Hanaoka, K.; Kondoh, H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development 1993, 117, 1445–1455. [Google Scholar]
- Hatton, K.S.; Mahon, K.; Chin, L.; Chiu, F.C.; Lee, H.W.; Peng, D.; Morgenbesser, S.D.; Horner, J.; DePinho, R.A. Expression and activity of L-Myc in normal mouse development. Mol. Cell. Biol. 1996, 16, 1794–1804. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Huang, J.; Tian, Y.; Chen, Y.; Yang, Y.; Zhang, X.; Zhang, F.; Xue, L. Myc suppresses tumor invasion and cell migration by inhibiting JNK signaling. Oncogene 2017, 36, 3159–3167. [Google Scholar] [CrossRef]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P.; et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V.; Reddy, E.P.; Shokat, K.M.; Soucek, L. Drugging the ‘undruggable’ cancer targets. Nat. Rev. Cancer 2017, 17, 502–508. [Google Scholar] [CrossRef]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentino, F.P.; Tokgun, E.; Sole-Sanchez, S.; Giampaolo, S.; Tokgun, O.; Jauset, T.; Kohno, T.; Perucho, M.; Soucek, L.; Yokota, J. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget 2016, 7, 31014–31028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tato, F.; Nasi, S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002, 62, 3507–3510. [Google Scholar]
- Soucek, L.; Nasi, S.; Evan, G.I. Omomyc expression in skin prevents Myc-induced papillomatosis. Cell Death Differ. 2004, 11, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Conzen, S.D.; Gottlob, K.; Kandel, E.S.; Khanduri, P.; Wagner, A.J.; O’Leary, M.; Hay, N. Induction of cell cycle progression and acceleration of apoptosis are two separable functions of c-Myc: Transrepression correlates with acceleration of apoptosis. Mol. Cell. Biol. 2000, 20, 6008–6018. [Google Scholar] [CrossRef] [Green Version]
- Greasley, P.J.; Bonnard, C.; Amati, B. Myc induces the nucleolin and BN51 genes: Possible implications in ribosome biogenesis. Nucleic Acids Res. 2000, 28, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Cetinkaya, C.; Munoz-Alonso, M.J.; von der Lehr, N.; Bahram, F.; Beuger, V.; Eilers, M.; Leon, J.; Larsson, L.G. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003, 22, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef]
- Von Eyss, B.; Eilers, M. Addicted to Myc—But why? Genes Dev. 2011, 25, 895–897. [Google Scholar] [CrossRef] [Green Version]
- Luscher, B.; Larsson, L.G. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: Function and regulation. Oncogene 1999, 18, 2955–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallant, P.; Steiger, D. Myc’s secret life without Max. Cell Cycle 2009, 8, 3848–3853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wert, M.; Kennedy, S.; Palfrey, H.C.; Hay, N. Myc drives apoptosis in PC12 cells in the absence of Max. Oncogene 2001, 20, 3746–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mongiardi, M.P.; Savino, M.; Bartoli, L.; Beji, S.; Nanni, S.; Scagnoli, F.; Falchetti, M.L.; Favia, A.; Farsetti, A.; Levi, A.; et al. Myc and Omomyc functionally associate with the Protein Arginine Methyltransferase 5 (PRMT5) in glioblastoma cells. Sci. Rep. 2015, 5, 15494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galardi, S.; Savino, M.; Scagnoli, F.; Pellegatta, S.; Pisati, F.; Zambelli, F.; Illi, B.; Annibali, D.; Beji, S.; Orecchini, E.; et al. Resetting cancer stem cell regulatory nodes upon MYC inhibition. EMBO Rep. 2016, 17, 1872–1889. [Google Scholar] [CrossRef] [Green Version]
- Annibali, D.; Whitfield, J.R.; Favuzzi, E.; Jauset, T.; Serrano, E.; Cuartas, I.; Redondo-Campos, S.; Folch, G.; Gonzalez-Junca, A.; Sodir, N.M.; et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014, 5, 4632. [Google Scholar] [CrossRef]
- Alimova, I.; Pierce, A.; Danis, E.; Donson, A.; Birks, D.K.; Griesinger, A.; Foreman, N.K.; Santi, M.; Soucek, L.; Venkataraman, S.; et al. Inhibition of MYC attenuates tumor cell self-renewal and promotes senescence in SMARCB1-deficient Group 2 atypical teratoid rhabdoid tumors to suppress tumor growth in vivo. Int. J. Cancer 2019, 144, 1983–1995. [Google Scholar] [CrossRef]
- Flores, I.; Murphy, D.J.; Swigart, L.B.; Knies, U.; Evan, G.I. Defining the temporal requirements for Myc in the progression and maintenance of skin neoplasia. Oncogene 2004, 23, 5923–5930. [Google Scholar] [CrossRef] [Green Version]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Masso-Valles, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Sodir, N.M.; Kortlever, R.M.; Barthet, V.J.A.; Campos, T.; Pellegrinet, L.; Kupczak, S.; Anastasiou, P.; Brown Swigart, L.; Soucek, L.; Arends, M.J.; et al. Myc instructs and maintains pancreatic adenocarcinoma phenotype. Cancer Discov. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, A.; Maclaren, O.J.; Fletcher, A.G.; Muraro, D.; Kreuzaler, P.A.; Byrne, H.M.; Maini, P.K.; Watson, A.J.; Pin, C. Cell proliferation within small intestinal crypts is the principal driving force for cell migration on villi. FASEB J. 2017, 31, 636–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- von Eyss, B.; Jaenicke, L.A.; Kortlever, R.M.; Royla, N.; Wiese, K.E.; Letschert, S.; McDuffus, L.A.; Sauer, M.; Rosenwald, A.; Evan, G.I.; et al. A MYC-Driven Change in Mitochondrial Dynamics Limits YAP/TAZ Function in Mammary Epithelial Cells and Breast Cancer. Cancer Cell 2015, 28, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagne, M.; Beaudoin, N.; Fortin, D.; Lavoie, C.L.; Klinck, R.; Lavigne, P. The Max b-HLH-LZ can transduce into cells and inhibit c-Myc transcriptional activities. PLoS ONE 2012, 7, e32172. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Beaulieu, M.E.; Jauset, T.; Masso-Valles, D.; Martinez-Martin, S.; Rahl, P.; Maltais, L.; Zacarias-Fluck, M.F.; Casacuberta-Serra, S.; Serrano Del Pozo, E.; Fiore, C.; et al. Intrinsic cell-penetrating activity propels Omomyc from proof of concept to viable anti-MYC therapy. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Colin, M.; Delporte, C.; Janky, R.; Lechon, A.S.; Renard, G.; Van Antwerpen, P.; Maltese, W.A.; Mathieu, V. Dysregulation of Macropinocytosis Processes in Glioblastomas May Be Exploited to Increase Intracellular Anti-Cancer Drug Levels: The Example of Temozolomide. Cancers 2019, 11, 411. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, H.; Bonner-Weir, S.; Wei, F.Y.; Matsushita, M.; Matsumoto, S. BETA2/NeuroD protein can be transduced into cells due to an arginine- and lysine-rich sequence. Diabetes 2005, 54, 2859–2866. [Google Scholar] [CrossRef] [Green Version]
- Demma, M.J.; Mapelli, C.; Sun, A.; Bodea, S.; Ruprecht, B.; Javaid, S.; Wiswell, D.; Muise, E.; Chen, S.; Zelina, J.; et al. Omomyc Reveals New Mechanisms To Inhibit the MYC Oncogene. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, S.; Prochownik, E.V. Small-molecule inhibitors of the Myc oncoprotein. Biochim. Biophys. Acta 2015, 1849, 525–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Sorolla, A.; Cunningham, P.T.; Bogdawa, H.M.; Beck, S.; Golden, E.; Dewhurst, R.E.; Florez, L.; Cruickshank, M.N.; Hoffmann, K.; et al. Tumor penetrating peptides inhibiting MYC as a potent targeted therapeutic strategy for triple-negative breast cancers. Oncogene 2019, 38, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Milech, N.; Juraja, S.M.; Cunningham, P.T.; Stone, S.R.; Francis, R.W.; Anastasas, M.; Hall, C.M.; Heinrich, T.; Bogdawa, H.M.; et al. A platform for discovery of functional cell-penetrating peptides for efficient multi-cargo intracellular delivery. Sci. Rep. 2018, 8, 12538. [Google Scholar] [CrossRef] [PubMed]
- Pesarrodona, M.; Jauset, T.; Diaz-Riascos, Z.V.; Sanchez-Chardi, A.; Beaulieu, M.E.; Seras-Franzoso, J.; Sanchez-Garcia, L.; Balta-Foix, R.; Mancilla, S.; Fernandez, Y.; et al. Targeting Antitumoral Proteins to Breast Cancer by Local Administration of Functional Inclusion Bodies. Adv. Sci. 2019, 6, 1900849. [Google Scholar] [CrossRef] [PubMed]
- Calo-Lapido, R.; Penas, C.; Jimenez-Balsa, A.; Vazquez, M.E.; Mascarenas, J.L. A chemical approach for the synthesis of the DNA-binding domain of the oncoprotein MYC. Org. Biomol. Chem. 2019, 17, 6748–6752. [Google Scholar] [CrossRef]
- Brown, Z.Z.; Mapelli, C.; Farasat, I.; Shoultz, A.V.; Johnson, S.A.; Orvieto, F.; Santoprete, A.; Bianchi, E.; McCracken, A.B.; Chen, K.; et al. Multiple Synthetic Routes to the Mini-Protein Omomyc and Coiled-Coil Domain Truncations. J. Org. Chem. 2019. [Google Scholar] [CrossRef]
- Demma, M.J.; Hohn, M.J.; Sun, A.; Mapelli, C.; Hall, B.; Walji, A.; O’Neil, J. Inhibition of Myc Transcriptional Activity by a Mini Protein Based Upon Mxd1. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- De Marco, A.; Ferrer-Miralles, N.; Garcia-Fruitos, E.; Mitraki, A.; Peternel, S.; Rinas, U.; Trujillo-Roldan, M.A.; Valdez-Cruz, N.A.; Vazquez, E.; Villaverde, A. Bacterial inclusion bodies are industrially exploitable amyloids. FEMS Microbiol. Rev. 2019, 43, 53–72. [Google Scholar] [CrossRef]
- Hoffmann, D.; Ebrahimi, M.; Gerlach, D.; Salzig, D.; Czermak, P. Reassessment of inclusion body-based production as a versatile opportunity for difficult-to-express recombinant proteins. Crit. Rev. Biotechnol. 2018, 38, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhao, S.; Karnad, A.; Freeman, J.W. The biology and role of CD44 in cancer progression: Therapeutic implications. J. Hematol. Oncol. 2018, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Type of Variant | Main Differences with Omomyc | Efficacy in vitro | Efficacy in vivo | Efficacy and Lack of Toxicity after Systemic Administration |
|---|---|---|---|---|
| Omomyc [79] | √ | √ | √ | |
| FPPa-Omomyc [85,86] | Efficacious at lower concentrations | √ | √ | Not reported |
| Omomyc-FNI/II/IV-H6 inclusion bodies [87] | Slow release Targeted to CD44+ cells | √ | Some | Not reported |
| [AQ]Omomyc(SH) [88] | Enhanced cell penetration | Not reported | Not reported | Not reported |
| Shorter Omomyc derivatives with enhanced DNA binding activity [89] | Much shorter than Omomyc Not able to dimerize with Myc or MAX | √ | Not reported | Not reported |
| Mad (not derived from Omomyc) [90] | Binds to MAX but not Myc Protected from ubiquitination More potent than Omomyc in vitro | √ | Not reported | Not reported |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massó-Vallés, D.; Soucek, L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells 2020, 9, 883. https://doi.org/10.3390/cells9040883
Massó-Vallés D, Soucek L. Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells. 2020; 9(4):883. https://doi.org/10.3390/cells9040883
Chicago/Turabian StyleMassó-Vallés, Daniel, and Laura Soucek. 2020. "Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc" Cells 9, no. 4: 883. https://doi.org/10.3390/cells9040883
APA StyleMassó-Vallés, D., & Soucek, L. (2020). Blocking Myc to Treat Cancer: Reflecting on Two Decades of Omomyc. Cells, 9(4), 883. https://doi.org/10.3390/cells9040883