Integrative Analysis of Methylome and Transcriptome Reveals the Regulatory Mechanisms of Hair Follicle Morphogenesis in Cashmere Goat

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
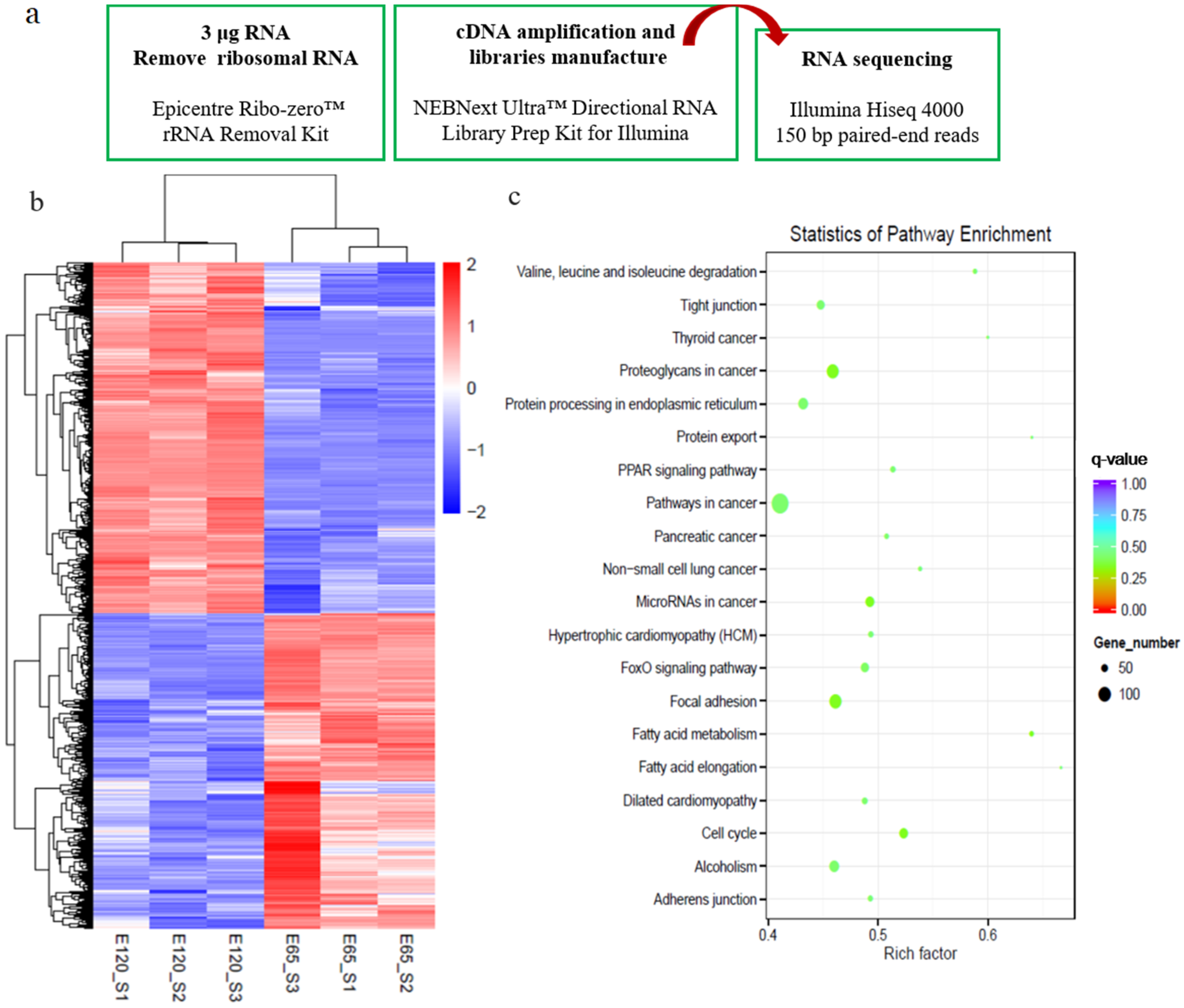
2.2. Transcriptome Sequencing and Bioinformatics Analysis
2.3. Quantitative Real-Time PCR (qRT-PCR)
2.4. Histology and Immunohistochemistry (IHC)
2.5. DNA Extraction, WGBS Library Construction, and Sequencing
2.6. Date Analysis, Identification of DMRs, and Functional Enrichment Analysis
2.7. Bisulfite Sequencing Polymerase Chain Reaction (BSP)
3. Results
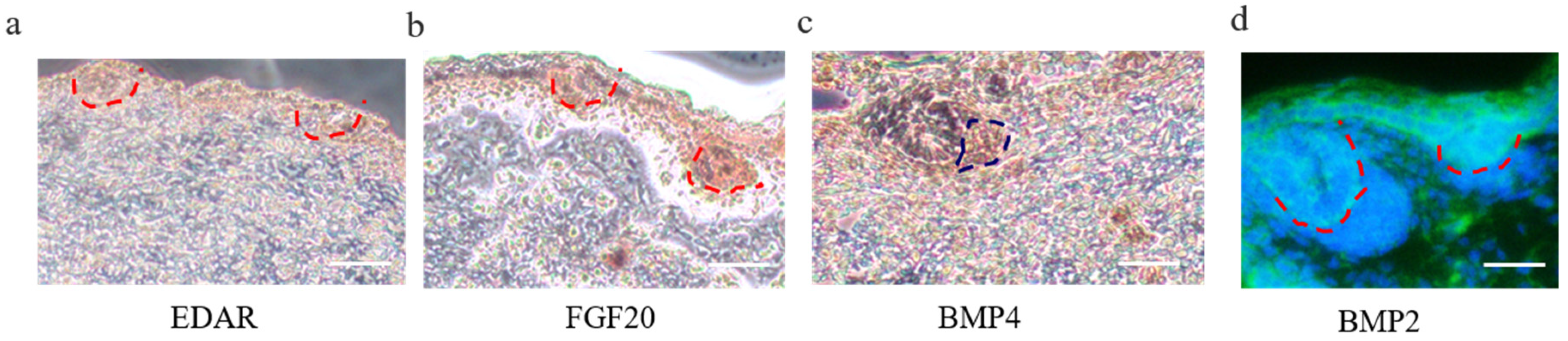
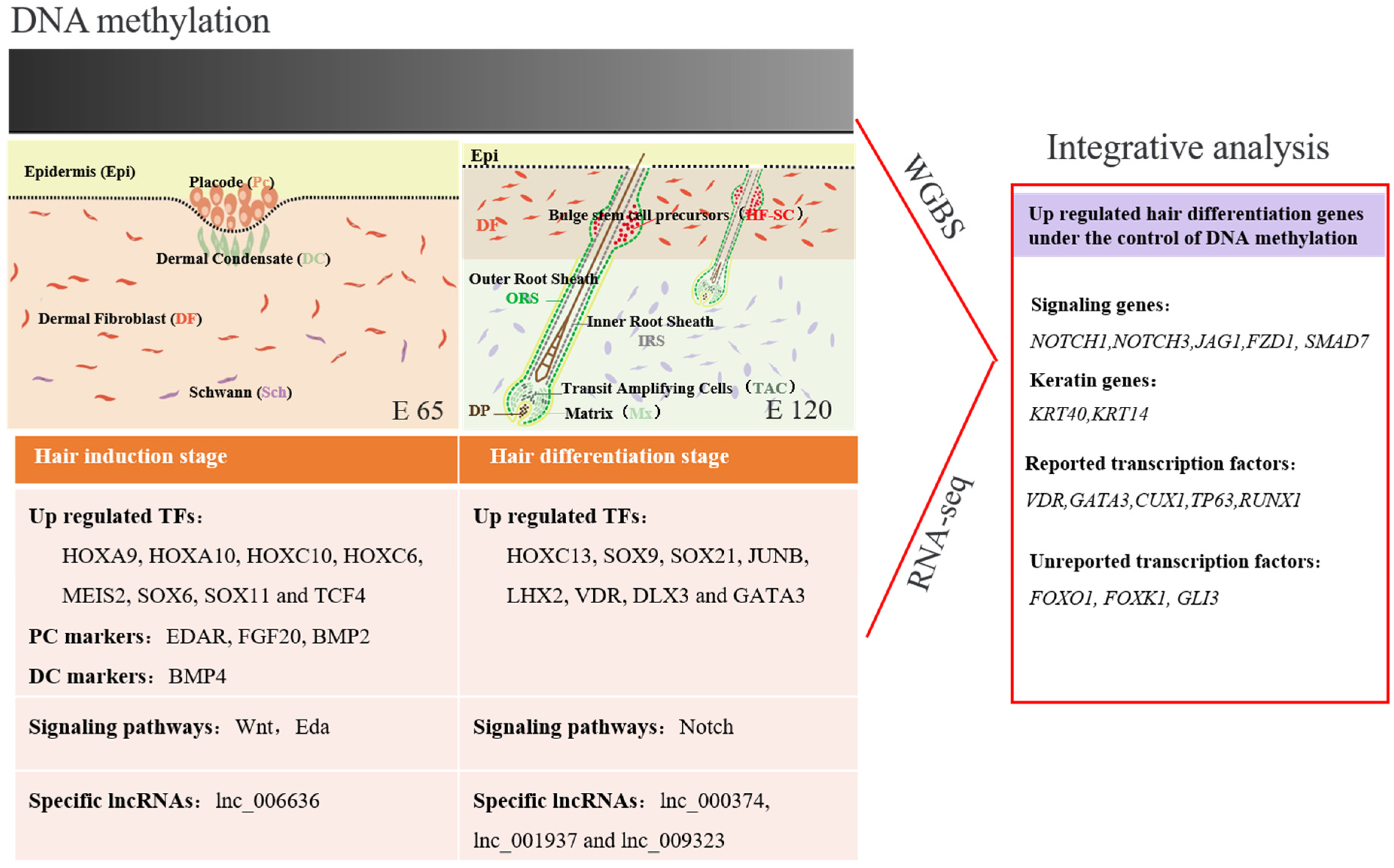
3.1. The Morphology of Hair Follicle Induction and Differentiation Stages in Cashmere Goats
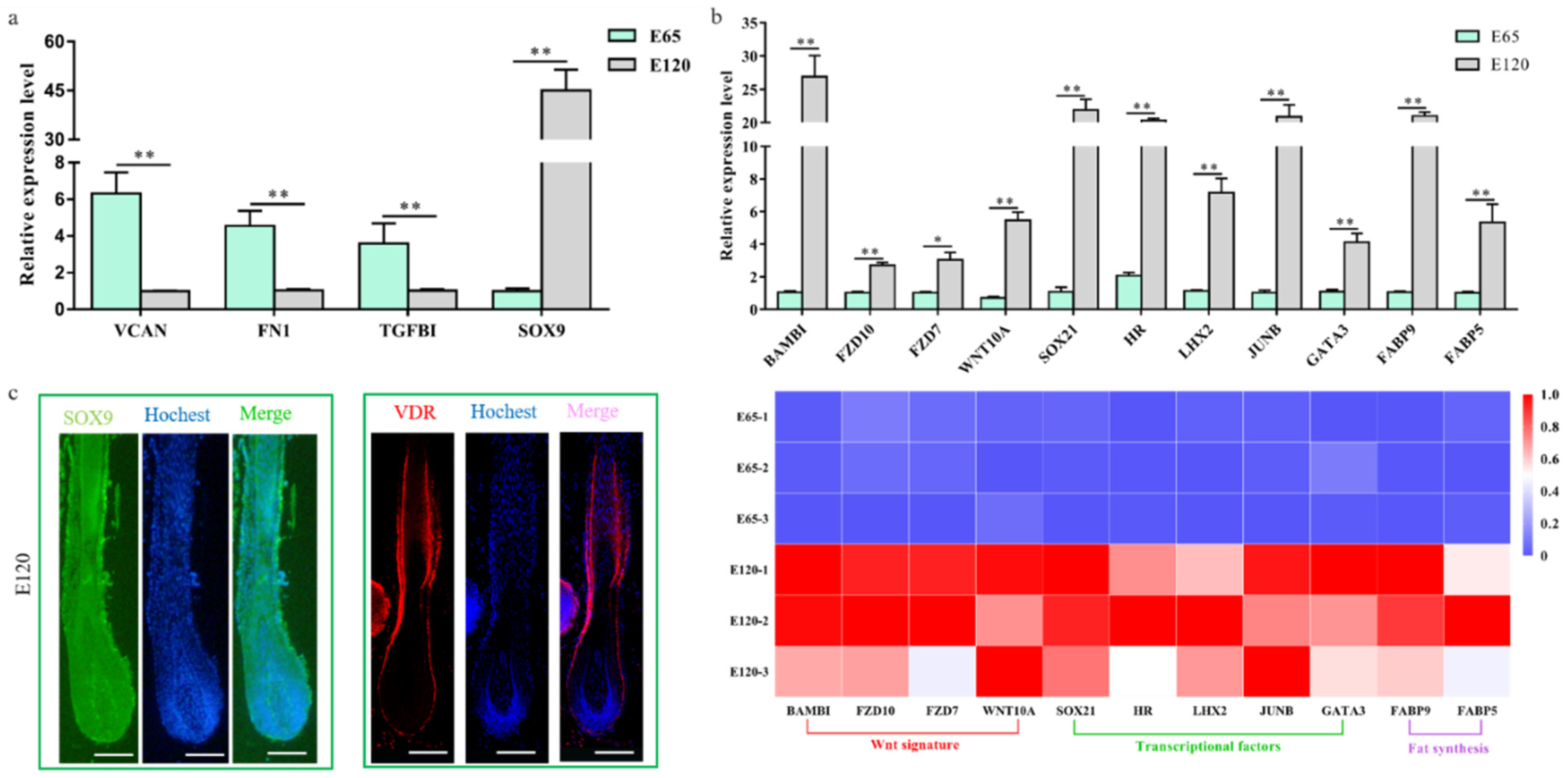
3.2. Defining Distinct Molecular Signatures of Hair Follicle Induction and Differentiation
3.3. Wnt Signal in Hair Follicle Induction and Differentiation
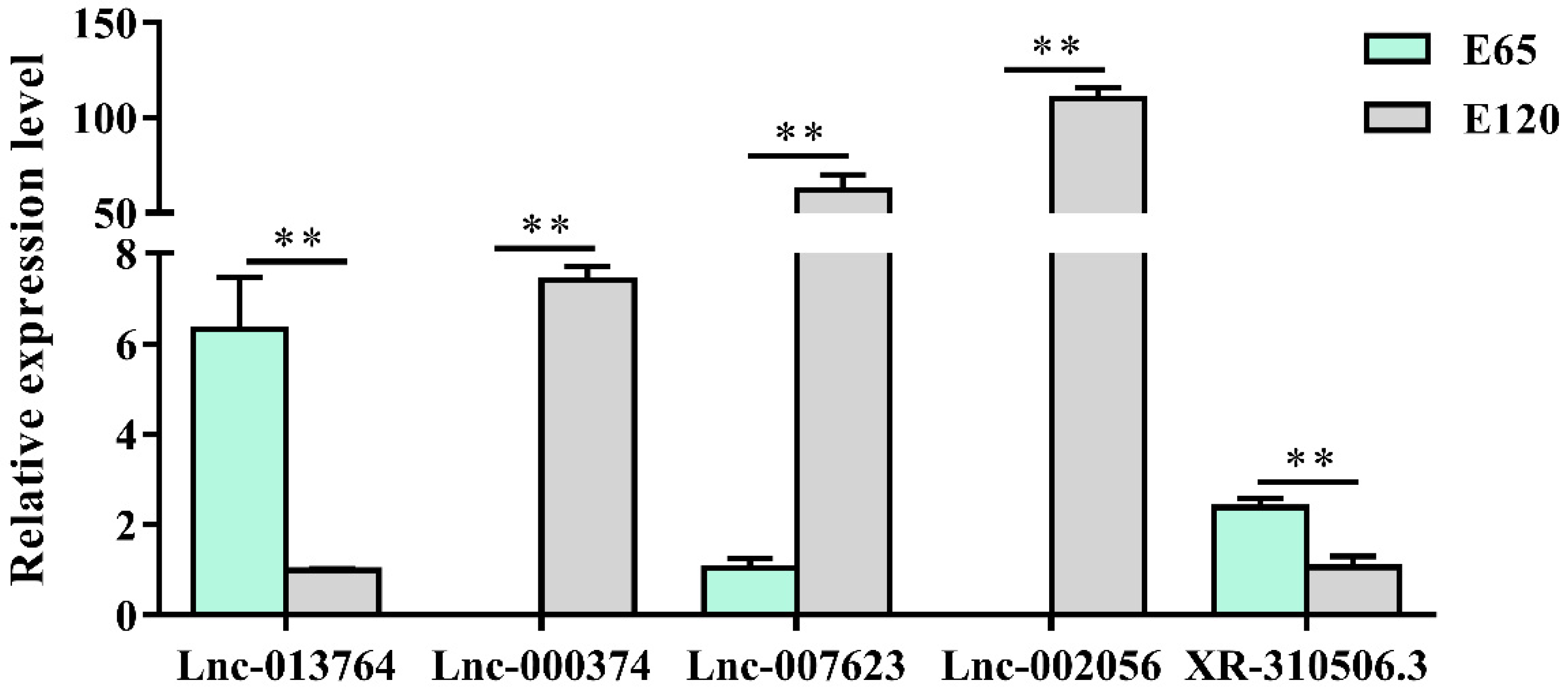
3.4. LncRNA Analysis of Skin Hair Follicle Development
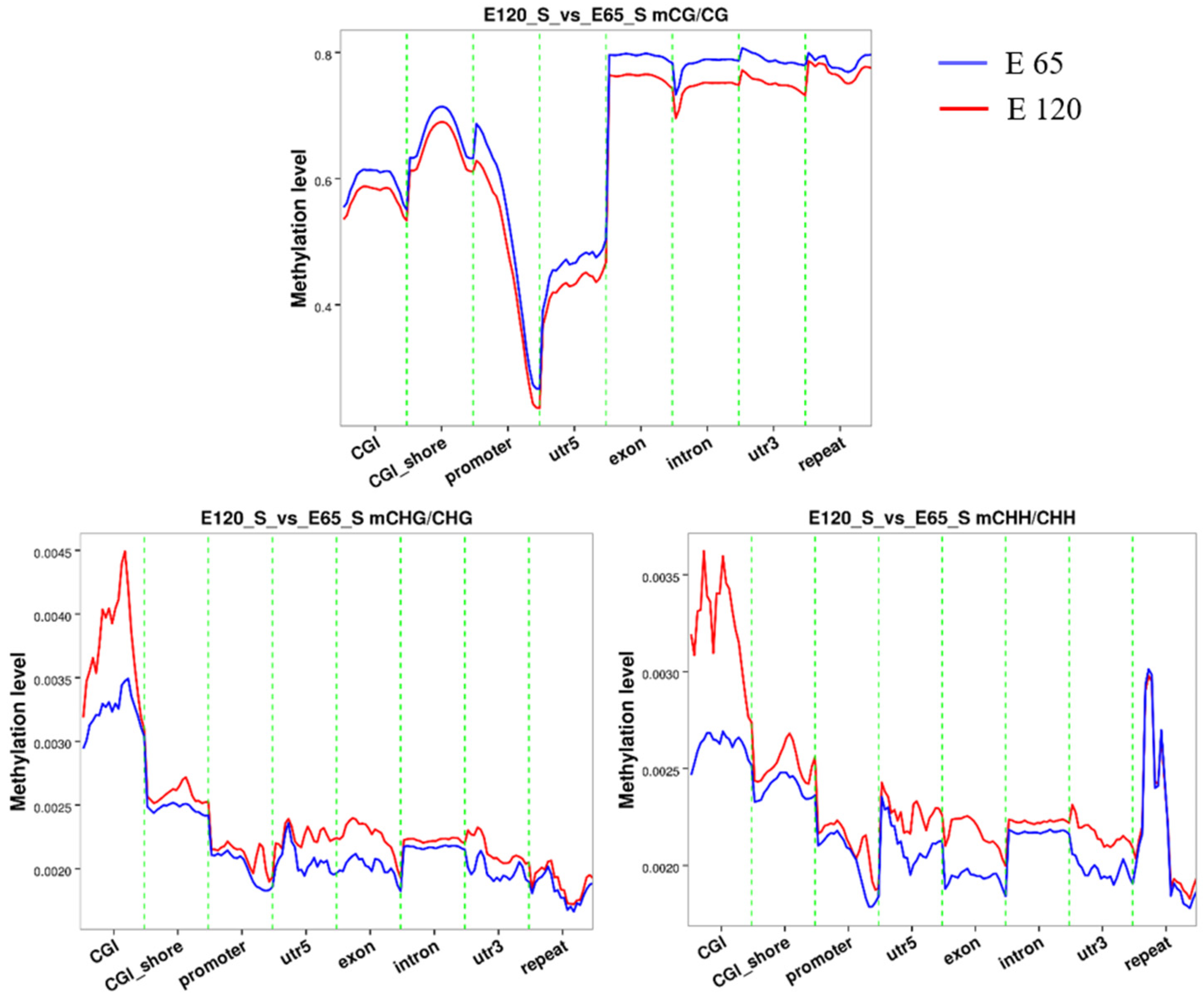
3.5. Genome DNA Methylation of Hair Induction and Differentiation during Morphogenesis
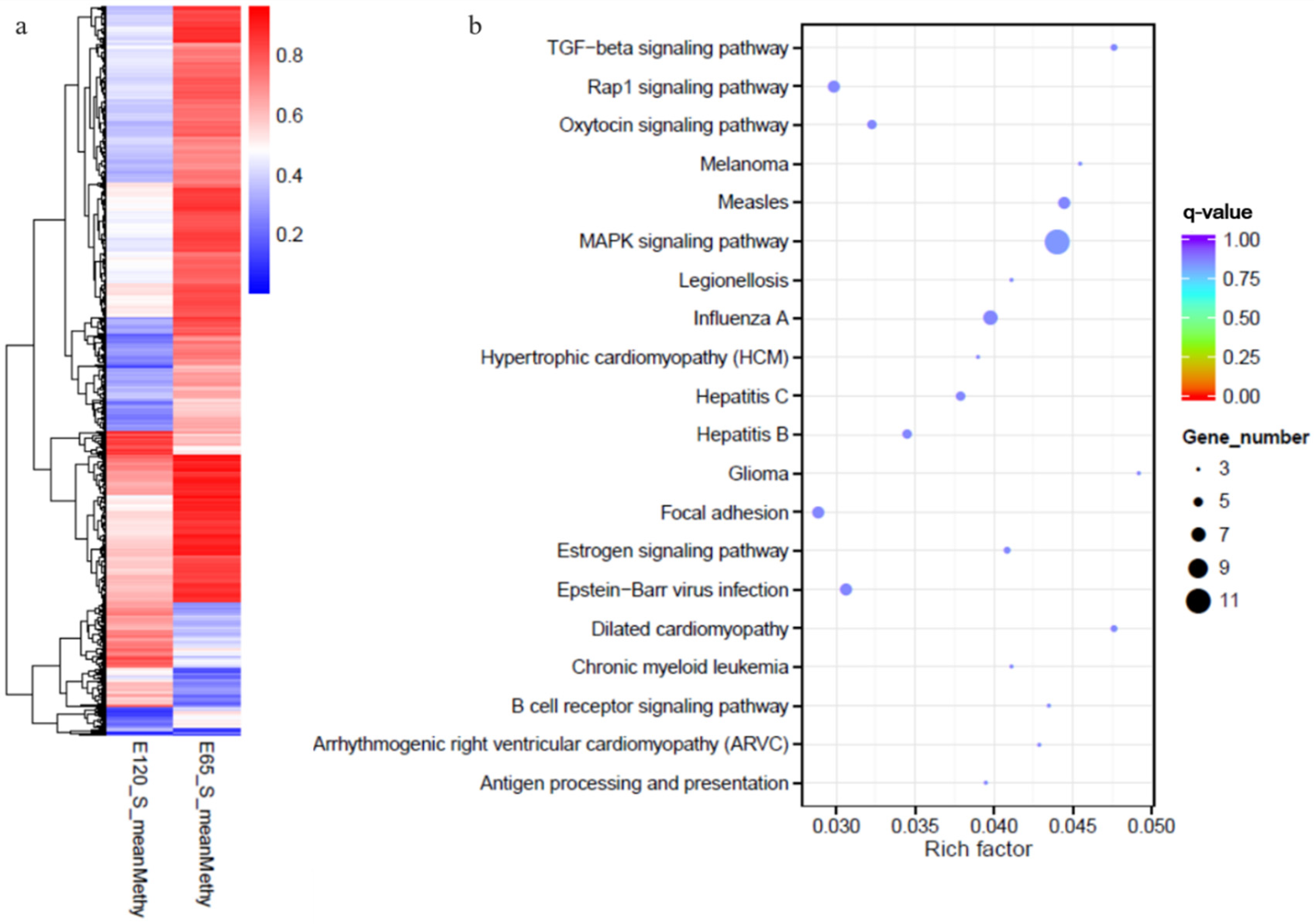
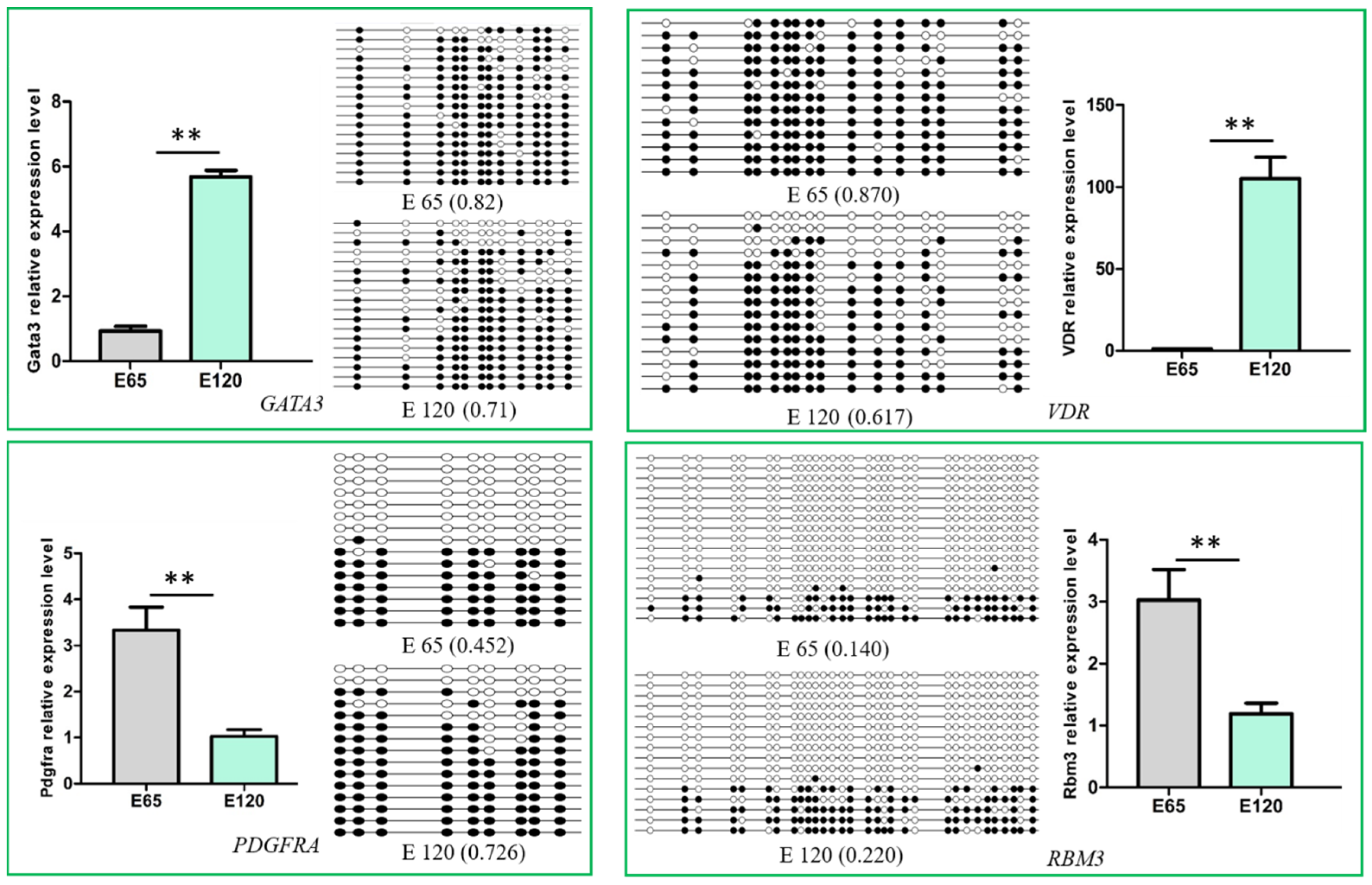
3.6. Integrated Analysis of WGBS and mRNA-seq Data
3.7. Potential lncRNA that Could Take Part in DNA Methylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Accession Numbers
Appendix A
References
- Paus, R.; Cotsarelis, G. The biology of hair follicles. N. Engl. J. Med. 1999, 341, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.R.; Schmidt-Ullrich, R.; Paus, R. The hair follicle as a dynamic miniorgan. Curr. Biol. 2009, 19, R132–R142. [Google Scholar] [CrossRef] [Green Version]
- Ge, W.; Wang, S.-H.; Sun, B.; Zhang, Y.-L.; Shen, W.; Khatib, H.; Wang, X. Melatonin promotes Cashmere goat (Capra hircus) secondary hair follicle growth: A view from integrated analysis of long non-coding and coding RNAs. Cell Cycle 2018, 17, 1255–1267. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Ge, W.; Luo, Z.; Guo, Y.; Jiao, B.; Qu, L.; Zhang, Z.; Wang, X. Integrated analysis of coding genes and non-coding RNAs during hair follicle cycle of cashmere goat (Capra hircus). BMC Genom. 2017, 18, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, S.E. Molecular mechanisms regulating hair follicle development. J. Investig. Dermatol. 2002, 118, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Kishimoto, J. Molecular Control of Epithelial–Mesenchymal Interactions During Hair Follicle Cycling. J. Investig. Dermatol. Symp. Proc. 2003, 8, 46–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt-Ullrich, R.; Paus, R. Molecular principles of hair follicle induction and morphogenesis. BioEssays 2005, 27, 247–261. [Google Scholar] [CrossRef]
- Biggs, L.C.; Mikkola, M.L. Early inductive events in ectodermal appendage morphogenesis. Semin. Cell Dev. Biol. 2014, 25–26, 11–21. [Google Scholar] [CrossRef]
- Chen, D.; Jarrell, A.; Guo, C.; Lang, R.; Atit, R. Dermal beta-catenin activity in response to epidermal Wnt ligands is required for fibroblast proliferation and hair follicle initiation. Development 2012, 139, 1522–1533. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Tumbar, T. Hairy tale of signaling in hair follicle development and cycling. Semin. Cell Dev. Biol. 2012, 23, 906–916. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tredget, E.E.; Wu, Y. Dynamic signals for hair follicle development and regeneration. Stem Cells Dev. 2012, 21, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.H.; Narhi, K.; Lindfors, P.H.; Haara, O.; Yang, L.; Ornitz, D.M.; Mikkola, M.L. Fgf20 governs formation of primary and secondary dermal condensations in developing hair follicles. Genes Dev. 2013, 27, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, K.W.; Saxena, N.; Heitman, N.; Grisanti, L.; Srivastava, D.; Muraro, M.J.; Jacob, T.; Sennett, R.; Wang, Z.; Su, Y.; et al. Dermal Condensate Niche Fate Specification Occurs Prior to Formation and Is Placode Progenitor Dependent. Dev Cell 2019, 48, 32–48.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesler, A.L.; Veniaminova, N.A.; Lull, M.V.; Wong, S.Y. Hair Follicle Terminal Differentiation Is Orchestrated by Distinct Early and Late Matrix Progenitors. Cell Rep. 2017, 19, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Avigad Laron, E.; Aamar, E.; Enshell-Seijffers, D. The Mesenchymal Niche of the Hair Follicle Induces Regeneration by Releasing Primed Progenitors from Inhibitory Effects of Quiescent Stem Cells. Cell Rep. 2018, 24, 909–921.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asakawa, K.; Toyoshima, K.E.; Tsuji, T. Functional Hair Follicle Regeneration by the Rearrangement of Stem Cells. Methods Mol. Biol. 2017, 1597, 117–134. [Google Scholar] [PubMed]
- Zhang, Y.; Wang, L.; Li, Z.; Chen, D.; Han, W.; Wu, Z.; Shang, F.; Hai, E.; Wei, Y.; Su, R.; et al. Transcriptome profiling reveals transcriptional and alternative splicing regulation in the early embryonic development of hair follicles in the cashmere goat. Sci. Rep. 2019, 9, 17735. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.D.; Wells, K.L.; Matthaus, F.; Painter, K.J.; Ho, W.; Riddell, J.; Johansson, J.A.; Ford, M.J.; Jahoda, C.A.B.; Klika, V.; et al. Hierarchical patterning modes orchestrate hair follicle morphogenesis. PLoS Biol. 2017, 15, e2002117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bak, S.S.; Kwack, M.H.; Shin, H.S.; Kim, J.C.; Kim, M.K.; Sung, Y.K. Restoration of hair-inductive activity of cultured human follicular keratinocytes by co-culturing with dermal papilla cells. Biochem. Biophys. Res. Commun. 2018, 505, 360–364. [Google Scholar] [CrossRef]
- Nakamura, M.; Schneider, M.R.; Schmidt-Ullrich, R.; Paus, R. Mutant laboratory mice with abnormalities in hair follicle morphogenesis, cycling, and/or structure: An update. Exp. Dermatol. 2013, 69, 6–29. [Google Scholar] [CrossRef]
- Sennett, R.; Wang, Z.; Rezza, A.; Grisanti, L.; Roitershtein, N.; Sicchio, C.; Mok, K.W.; Heitman, N.; Clavel, C.; Ma’Ayan, A. An Integrated Transcriptome Atlas of Embryonic Hair Follicle Progenitors, Their Niche, and the Developing Skin. Dev. Cell 2015, 34, 577–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, W.; Ines, H.; Stadler, M.B.; Liliana, R.; Svante, P.B.; Michael, R.; Dirk, S. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar]
- Reik, W.; Dean, W. DNA methylation and mammalian epigenetics. Electrophoresis 2001, 22, 2838–2843. [Google Scholar] [CrossRef]
- Baubec, T.; Schubeler, D. Genomic patterns and context specific interpretation of DNA methylation. Curr. Opin. Genet. Dev. 2014, 25, 85–92. [Google Scholar] [CrossRef]
- Senner, C.E. The role of DNA methylation in mammalian development. Reprod. Biomed. Online 2011, 22, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465. [Google Scholar] [CrossRef]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Zhong, T.; Liu, Z.; Ding, Y.; Guan, W.; He, X.; Pu, Y.; Jiang, L.; Ma, Y.; Zhao, Q. Integrated Analysis of Methylome and Transcriptome Changes Reveals the Underlying Regulatory Signatures Driving Curly Wool Transformation in Chinese Zhongwei Goats. Front. Genet. 2019, 10, 1263. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Li, Y.; Zhou, G.; Gao, Y.; Ma, S.; Chen, Y.; Song, J.; Wang, X. Whole-genome bisulfite sequencing of goat skins identifies signatures associated with hair cycling. BMC Genom. 2018, 19, 638. [Google Scholar] [CrossRef]
- Li, J.; Jiang, T.X.; Hughes, M.W.; Wu, P.; Yu, J.; Widelitz, R.B.; Fan, G.; Chuong, C.M. Progressive alopecia reveals decreasing stem cell activation probability during aging of mice with epidermal deletion of DNA methyltransferase 1. J. Investig. Dermatol. 2012, 132, 2681–2690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, F.; Pandey, G.K.; Mondal, T.; Enroth, S.; Redrup, L.; Gyllensten, U.; Kanduri, C. Long noncoding RNA-mediated maintenance of DNA methylation and transcriptional gene silencing. Development 2012, 139, 2792–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, H.L.; Quinn, J.J.; Yang, Y.W.; Thornburg, C.K.; Chang, H.Y.; Stadler, H.S. LncRNA-HIT Functions as an Epigenetic Regulator of Chondrogenesis through Its Recruitment of p100/CBP Complexes. PLoS Genet. 2015, 11, e1005680. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Xie, R.; Gu, P.; Huang, M.; Han, J.; Dong, W.; Xie, W.; Wang, B.; He, W.; Zhong, G.; et al. Long Noncoding RNA LBCS Inhibits Self-Renewal and Chemoresistance of Bladder Cancer Stem Cells through Epigenetic Silencing of SOX2. Clin. Cancer Res. 2019, 25, 1389–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, B.; Zheng, Y.; Ma, S.; Xing, Q.; Wang, X.; Yang, B.; Yin, G.; Guan, F. Long noncoding RNA regulates hair follicle stem cell proliferation and differentiation through PI3K/AKT signal pathway. Mol. Med. Rep. 2018, 17, 5477–5483. [Google Scholar]
- Wang, S.; Luo, Z.; Zhang, Y.; Yuan, D.; Ge, W.; Wang, X. The inconsistent regulation of HOXC13 on different keratins and the regulation mechanism on HOXC13 in cashmere goat (Capra hircus). BMC Genom. 2018, 19, 630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tomann, P.; Andl, T.; Gallant, N.M.; Huelsken, J.; Jerchow, B.; Birchmeier, W.; Paus, R.; Piccolo, S.; Mikkola, M.L. Reciprocal Requirements for EDA/EDAR/NF-κB and Wnt/β-Catenin Signaling Pathways in Hair Follicle Induction: Developmental Cell. Dev. Cell 2009, 17, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.H.; Leimeister, C.; Gessler, M.; Kopan, R. Activation of the Notch pathway in the hair cortex leads to aberrant differentiation of the adjacent hair-shaft layers. Development 2000, 127, 2421–2432. [Google Scholar]
- Ouji, Y.; Yoshikawa, M.; Moriya, K.; Nishiofuku, M.; Matsuda, R.; Ishizaka, S. Wnt-10b, uniquely among Wnts, promotes epithelial differentiation and shaft growth. Biochem. Biophys. Res. Commun. 2008, 367, 299–304. [Google Scholar] [CrossRef]
- Rogers, G.E. Hair follicle differentiation and regulation. Int. J. Dev. Biol. 2004, 48, 163–170. [Google Scholar] [CrossRef]
- Jave-Suarez, L.F.; Winter, H.; Langbein, L.; Rogers, M.A.; Schweizer, J. HOXC13 is involved in the regulation of human hair keratin gene expression. J. Biol. Chem. 2002, 277, 3718–3726. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, C.K. GATA-3: An unexpected regulator of cell lineage determination in skin. Genes Dev. 2003, 17, 2108–2122. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Mehrani, T.; Millar, S.E.; Morasso, M.I. Dlx3 is a crucial regulator of hair follicle differentiation and cycling. Development 2008, 135, 3149–3159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, S.M.; Keough, R.A.; Rogers, G.E.; Powell, B.C. Regulation of a hair follicle keratin intermediate filament gene promoter. J. Cell Sci. 1998, 111 Pt 23, 3487. [Google Scholar]
- Powell, B.C.; Nesci, A.; Rogers, G.E. Regulation of keratin gene expression in hair follicle differentiation. Ann. N. Y. Acad. Sci. 1992, 642, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Vidal, V.P.I.; Marie-Christine, C.; Susanne, L.; George, C.; Pleasantine, M.; Chi-Chung, H.; Nicolas, O.; Jean-Paul, O.; Andreas, S. Sox9 is essential for outer root sheath differentiation and the formation of the hair stem cell compartment. Curr. Biol. 2005, 15, 1340–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Törnqvist, G.; Sandberg, A.; Hägglund, A.C.; Carlsson, L. Cyclic Expression of Lhx2 Regulates Hair Formation. PLoS Genet. 2010, 6, e1000904. [Google Scholar] [CrossRef] [Green Version]
- Ahn, Y. Signaling in tooth, hair, and mammary placodes. Curr. Top. Dev. Biol. 2014, 111, 421–459. [Google Scholar]
- Tsai, S.Y.; Sennett, R.; Rezza, A.; Clavel, C.; Grisanti, L.; Zemla, R.; Najam, S.; Rendl, M. Wnt/beta-catenin signaling in dermal condensates is required for hair follicle formation. Dev. Biol. 2014, 385, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Bänziger, C.; Soldini, D.; Schütt, C.; Zipperlen, P.; Hausmann, G.; Basler, K. Wntless, a Conserved Membrane Protein Dedicated to the Secretion of Wnt Proteins from Signaling Cells. Cell 2006, 125, 509–522. [Google Scholar] [CrossRef] [Green Version]
- Millar, S.E.; Willert, K.; Salinas, P.C.; Roelink, H.; Nusse, R.; Sussman, D.J.; Barsh, G.S. WNT signaling in the control of hair growth and structure. Dev. Biol. 1999, 207, 133–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Paus, R.; Muller-Rover, S.; Van Der Veen, C.; Maurer, M.; Eichmuller, S.; Ling, G.; Hofmann, U.; Foitzik, K.; Mecklenburg, L.; Handjiski, B. A comprehensive guide for the recognition and classification of distinct stages of hair follicle morphogenesis. J. Investig. Dermatol. 1999, 113, 523–532. [Google Scholar]
- Sundberg, J.P.; Peters, E.M.; Paus, R. Analysis of hair follicles in mutant laboratory mice. J. Investig. Dermatol. Symp. Proc. 2005, 10, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Saxena, N.; Mok, K.W.; Rendl, M. An Updated Classification of Hair Follicle Morphogenesis. Exp. Dermatol. 2019, 28, 332–344. [Google Scholar] [CrossRef] [Green Version]
- Mcdonald, B.J.; Hoey, W.A.; Hopkins, P.S. Cyclical fleece growth in cashmere goats. Aust. J. Agric. Res. 1987, 38, 597–609. [Google Scholar] [CrossRef]
- Srivastava, A.K.; Pispa, J.; Hartung, A.J.; Du, Y.; Ezer, S.; Jenks, T.; Shimada, T.; Pekkanen, M.; Mikkola, M.L.; Ko, M.S.; et al. The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-A) with collagenous domains. Proc. Natl. Acad. Sci. USA 1997, 94, 13069–13074. [Google Scholar] [CrossRef] [Green Version]
- Hao, F.; Yan, W.; Li, X.; Wang, H.; Wang, Y.; Hu, X.; Liu, X.; Liang, H.; Liu, D. Generation of Cashmere Goats Carrying anEDARGene Mutant Using CRISPR-Cas9-Mediated Genome Editing. Int. J. Biol. Sci. 2018, 14, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Andl, T.; Yang, S.H.; Teta, M.; Liu, F.; Seykora, J.T.; Tobias, J.W.; Piccolo, S.; Schmidt-Ullrich, R.; Nagy, A.; et al. Activation of beta-catenin signaling programs embryonic epidermis to hair follicle fate. Development 2008, 135, 2161–2172. [Google Scholar] [CrossRef] [Green Version]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT Signals Are Required for the Initiation of Hair Follicle Development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Wolf, R. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar]
- Ozkul, Y.; Galderisi, U. The Impact of Epigenetics on Mesenchymal Stem Cell Biology. J. Cell. Physiol. 2016, 231, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Dirk, S. Function and information content of DNA methylation. Nature 2015, 517, 321–326. [Google Scholar]
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Lilly, Z.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Sun, M.; Nie, F.; Wang, Y.; Zhang, Z.; Hou, J.; He, D.; Xie, M.; Wei, D.; Wang, Z.; Wang, J. LncRNA HOXA11-AS promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1 and DNMT1. Cancer Res. 2016, 76, 6299–6310. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; Chang, C.P. Long non-coding RNA and chromatin remodeling. RNA Biol. 2015, 12, 1094–1098. [Google Scholar] [CrossRef] [Green Version]
- St, L.G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar]
- Engreitz, J.M.; Pandyajones, A.; Mcdonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X-chromosome. Science 2013, 341, 767. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | E 120 FPKM | E 65 FPKM | Log 2 (Fold Change) | p-Value | E 120 Mean Methy | E 65 Mean Methy | Start | End |
|---|---|---|---|---|---|---|---|---|
| TP63 | 48.8 | 5.3 | 3.20 | 0.005 | 0.35 | 0.70 | 77226924 | 77227046 |
| VDR | 19.3 | 0.0 | Inf | 0.000 | 0.73 | 0.91 | 31960999 | 31961087 |
| GATA3 | 11.1 | 2.1 | 2.38 | 0.005 | 0.58 | 0.83 | 12388542 | 12388674 |
| CUX1 | 5.0 | 0.0 | 11.11 | 0.011 | 0.52 | 0.84 | 35639445 | 35639901 |
| RUNX1 | 3.1 | 1.0 | 1.59 | 0.002 | 0.65 | 0.19 | 146939482 | 146939629 |
| GLI3 | 5.1 | 3.1 | 0.72 | 0.003 | 0.44 | 0.88 | 41410654 | 41411108 |
| FOXO1 | 11.6 | 1.4 | 3.05 | 0.000 | 0.40 | 0.72 | 64694258 | 64694508 |
| FZD1 | 18.1 | 5.8 | 1.65 | 0.004 | 0.33 | 0.68 | 111902257 | 111902552 |
| NOTCH1 | 18.1 | 6.3 | 1.53 | 0.003 | 0.71 | 0.86 | 103351558 | 103351699 |
| NOTCH3 | 10.6 | 5.0 | 1.09 | 0.017 | 0.51 | 0.87 | 100777692 | 100777863 |
| SMAD7 | 7.6 | 1.4 | 2.42 | 0.005 | 0.36 | 0.80 | 48827145 | 48827355 |
| JAG1 | 33.5 | 8.3 | 2.02 | 0.001 | 0.33 | 0.79 | 3752377 | 3752803 |
| RORA | 23.5 | 4.2 | 2.49 | 0.003 | 0.55 | 0.87 | 53503152 | 53503499 |
| EGFR | 30.8 | 13.0 | 1.24 | 0.004 | 0.32 | 0.68 | 842607 | 843297 |
| FGFR2 | 3.2 | 0.9 | 1.78 | 0.007 | 0.44 | 0.70 | 10433298 | 10433462 |
| KRT40 | 28.6 | 0.1 | 8.43 | 0.000 | 0.39 | 0.79 | 40830782 | 40831215 |
| KRT14 | 1321.9 | 6.6 | 7.64 | 0.000 | 0.66 | 0.90 | 41440140 | 41440345 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Li, F.; Liu, J.; Zhang, Y.; Zheng, Y.; Ge, W.; Qu, L.; Wang, X. Integrative Analysis of Methylome and Transcriptome Reveals the Regulatory Mechanisms of Hair Follicle Morphogenesis in Cashmere Goat. Cells 2020, 9, 969. https://doi.org/10.3390/cells9040969
Wang S, Li F, Liu J, Zhang Y, Zheng Y, Ge W, Qu L, Wang X. Integrative Analysis of Methylome and Transcriptome Reveals the Regulatory Mechanisms of Hair Follicle Morphogenesis in Cashmere Goat. Cells. 2020; 9(4):969. https://doi.org/10.3390/cells9040969
Chicago/Turabian StyleWang, Shanhe, Fang Li, Jinwang Liu, Yuelang Zhang, Yujie Zheng, Wei Ge, Lei Qu, and Xin Wang. 2020. "Integrative Analysis of Methylome and Transcriptome Reveals the Regulatory Mechanisms of Hair Follicle Morphogenesis in Cashmere Goat" Cells 9, no. 4: 969. https://doi.org/10.3390/cells9040969
APA StyleWang, S., Li, F., Liu, J., Zhang, Y., Zheng, Y., Ge, W., Qu, L., & Wang, X. (2020). Integrative Analysis of Methylome and Transcriptome Reveals the Regulatory Mechanisms of Hair Follicle Morphogenesis in Cashmere Goat. Cells, 9(4), 969. https://doi.org/10.3390/cells9040969