Necroptosis in Cholangiocarcinoma

 ,
,
Abstract
1. Introduction
2. Overview on Types of Cell Death
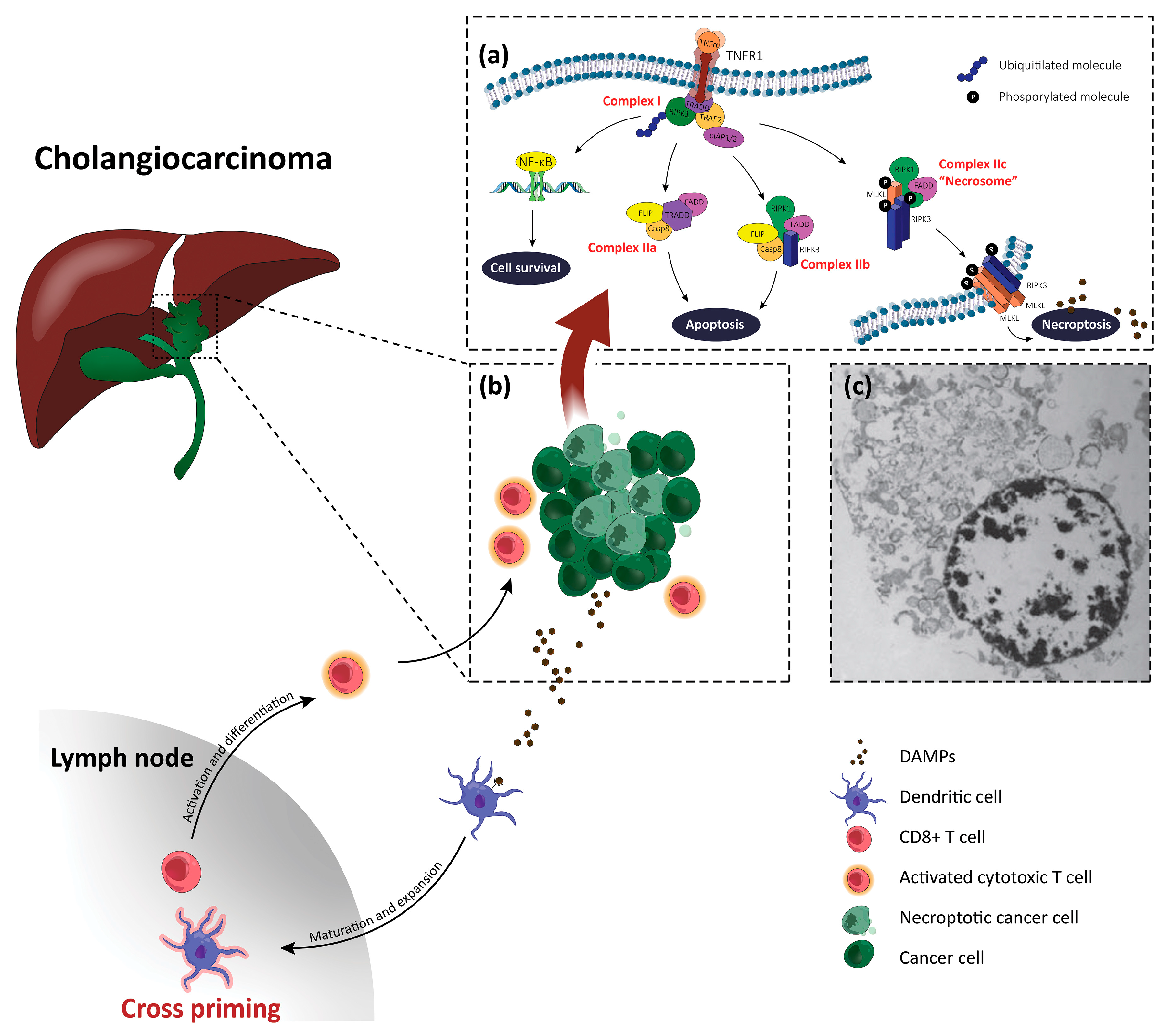
3. Necroptosis
4. Overview on Necroptosis in Liver Disease
5. Necroptosis and Cholangiocarcinoma
6. Necroptosis-Based Therapies for Cholangiocarcinoma
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIH | autoimmune hepatitis |
| AIHI | acetaminophen-induced hepatic injury |
| ALT | alanine transaminase |
| ATP | adenosine triphosphate |
| BDL | bile duct ligation |
| CCA | cholangiocarcinoma |
| CD | cell death |
| cIAP | cellular inhibitor of apoptosis protein |
| DAMPs | damage-associated molecular patterns |
| FADD | Fas-associated protein with death domain |
| FLIP | FLICE-inhibitory protein |
| HCC | hepatocellular carcinoma |
| iCCA | intrahepatic CCA |
| IRI | ischemia/reperfusion injury |
| miR-21 | microRNA-21 |
| MLKL | mixed lineage kinase domain-like |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NCCD | Nomenclature Committee on Cell Death |
| Nec-1 | necrostatin-1 |
| NF-κB | nuclear factor-kappa B |
| PolyI:C | polyinosinic:polycytidylic acid |
| RCD | regulated cell death |
| RIPK3 | receptor-interacting serine/threonine-protein kinase 3 |
| ROS | reactive oxygen species |
| Smac | second mitochondrial activator of caspases |
| TAK1 | transforming growth factor β-activated kinase 1 |
| TLRs | toll-like receptors |
| TNFα | tumor necrosis factor α |
| TNFR1 | TNFα receptor 1 |
| TRADD | TNFR1-associated death domain |
References
- Zhou, W.; Yuan, J. Necroptosis in health and diseases. Semin. Cell. Dev. Biol. 2014, 35, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Koh, D.H. Necroptosis signaling in liver diseases: An update. Pharmacol. Res. 2019, 148, 104439. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Kepp, O.; Chan, F.K.; Kroemer, G. Necroptosis: Mechanisms and relevance to disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Yuan, J.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Vanden Berghe, T.; Vandenabeele, P. Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 2012, 19, 75–86. [Google Scholar] [CrossRef]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Liu, C.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef]
- Liang, Y.Y.; Rainprecht, D.; Eichmair, E.; Messner, B.; Oehler, R. Serum-dependent processing of late apoptotic cells and their immunogenicity. Apoptosis 2015, 20, 1444–1456. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Kroemer, G.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef]
- Krysko, D.V.; Agostinis, P.; Krysko, O.; Garg, A.D.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164. [Google Scholar] [CrossRef]
- Kroemer, G.; El-Deiry, W.S.; Golstein, P.; Peter, M.E.; Vaux, D.; Vandenabeele, P.; Zhivotovsky, B.; Blagosklonny, M.V.; Malorni, W.; Melino, G.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005, 12, 1463–1467. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Melino, G.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Kroemer, G.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Kroemer, G.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Sasaki, M.; Nakanuma, Y. Stress-induced cellular responses and cell death mechanisms during inflammatory cholangiopathies. Clin. Res. Hepatol. Gastroenterol. 2017, 41, 129–138. [Google Scholar] [CrossRef]
- Yu, X.; Deng, Q.; Bode, A.M.; Dong, Z.; Cao, Y. The role of necroptosis, an alternative form of cell death, in cancer therapy. Expert Rev. Anticancer Ther. 2013, 13, 883–893. [Google Scholar] [CrossRef]
- Shi, S.; Verstegen, M.M.A.; Mezzanotte, L.; de Jonge, J.; Löwik, C.W.G.M.; van der Laan, L.J.W. Necroptotic cell death in liver transplantation and underlying diseases: Mechanisms and clinical perspective. Liver Transpl. 2019, 25, 1091–1104. [Google Scholar] [CrossRef]
- Moriwaki, K.; Chan, F.K. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev. 2014, 25, 167–174. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 2010, 11, 700–714. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat. Immunol. 2011, 12, 1143–1149. [Google Scholar] [CrossRef]
- Lalaoui, N.; Brumatti, G. Relevance of necroptosis in cancer. Immunol. Cell Biol. 2017, 95, 137–145. [Google Scholar] [CrossRef]
- Khan, N.; Lawlor, K.E.; Murphy, J.M.; Vince, J.E. More to life than death: Molecular determinants of necroptotic and non-necroptotic RIP3 kinase signaling. Curr. Opin. Immunol. 2014, 26, 76–89. [Google Scholar] [CrossRef]
- Moriwaki, K.; Chan, F.K. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef]
- Qin, X.; Ma, D.; Tan, Y.; Wang, H.; Cai, Z. The role of necroptosis in cancer: A double-edged sword? Biochim. Biophys. Acta. Rev. Cancer 2019, 1871, 259–266. [Google Scholar] [CrossRef]
- Jiao, D.; Cai, Z.; Choksi, S.; Ma, D.; Choe, M.; Kwon, H.; Baik, J.Y.; Rowan, B.G.; Liu, C.; Liu, Z.G. Necroptosis of tumor cells leads to tumor necrosis and promotes tumor metastasis. Cell Res. 2018, 28, 868–870. [Google Scholar] [CrossRef]
- Strilic, B.; Yang, L.; Albarrán-Juárez, J.; Wachsmuth, L.; Han, K.; Müller, U.C.; Pasparakis, M.; Offermanns, S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature 2016, 536, 215–218. [Google Scholar] [CrossRef]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Miller, G.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef]
- Groborz, K.; Gonzalez Ramirez, M.L.; Snipas, S.J.; Salvesen, G.S.; Drąg, M.; Poręba, M. Exploring the prime site in caspases as a novel chemical strategy for understanding the mechanisms of cell death: A proof of concept study on necroptosis in cancer cells. Cell Death Differ. 2020, 27, 451–465. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Wu, B.; Guo, Y.S.; Zhou, Y.H.; Fu, Z.G.; Xu, B.Q.; Li, J.H.; Jing, L.; Jiang, J.L.; Chen, Z.N.; et al. Necrostatin-1 reduces intestinal inflammation and colitis-associated tumorigenesis in mice. Am. J. Cancer Res. 2015, 5, 3174–3185. [Google Scholar]
- Takemura, R.; Takaki, H.; Okada, S.; Shime, H.; Akazawa, T.; Oshiumi, H.; Matsumoto, M.; Teshima, T.; Seya, T. PolyI:C-induced, TLR3/RIP3-dependent necroptosis backs up immune effector-mediated tumor elimination in vivo. Cancer Immunol. Res. 2015, 3, 902–914. [Google Scholar] [CrossRef]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; Barreira da Silva, R.; Reis e Sousa, C.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8+ T cells. Science 2015, 350, 328–334. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Milutinovic, S.; Dickson, K.M.; Ho, W.C.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; Barker, P.A. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 2008, 30, 689–700. [Google Scholar] [CrossRef]
- Dayal, D.; Martin, S.M.; Limoli, C.L.; Spitz, D.R. Hydrogen peroxide mediates the radiation-induced mutator phenotype in mammalian cells. Biochem. J. 2008, 413, 185–191. [Google Scholar] [CrossRef]
- Kang, Y.J.; Bang, B.R.; Han, K.H.; Hong, L.; Shim, E.J.; Ma, J.; Lerner, R.A.; Otsuka, M. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat. Commun. 2015, 6, 8371. [Google Scholar] [CrossRef]
- Vucur, M.; Reisinger, F.; Gautheron, J.; Janssen, J.; Roderburg, C.; Cardenas, D.V.; Kreggenwinkel, K.; Koppe, C.; Hammerich, L.; Luedde, T.; et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep. 2013, 4, 776–790. [Google Scholar] [CrossRef]
- Krishna-Subramanian, S.; Singer, S.; Armaka, M.; Banales, J.M.; Holzer, K.; Schirmacher, P.; Walczak, H.; Kollias, G.; Pasparakis, M.; Kondylis, V. RIPK1 and death receptor signaling drive biliary damage and early liver tumorigenesis in mice with chronic hepatobiliary injury. Cell Death Differ. 2019, 26, 2710–2726. [Google Scholar] [CrossRef]
- Seehawer, M.; Heinzmann, F.; D’Artista, L.; Harbig, J.; Roux, P.F.; Hoenicke, L.; Dang, H.; Klotz, S.; Robinson, L.; Zender, L.; et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature 2018, 562, 69–75. [Google Scholar] [CrossRef]
- Schmidt, S.V.; Seibert, S.; Walch-Rückheim, B.; Vicinus, B.; Kamionka, E.M.; Pahne-Zeppenfeld, J.; Solomayer, E.F.; Kim, Y.J.; Bohle, R.M.; Smola, S. RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1α release, and efficient paracrine dendritic cell activation. Oncotarget 2015, 6, 8635–8647. [Google Scholar] [CrossRef]
- Koo, G.; Morgan, M.J.; Lee, D.; Kim, W.; Yoon, J.; Koo, J.S.; Kim, S.I.; Kim, S.J.; Son, M.K.; Kim, Y.S.; et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015, 25, 707–725. [Google Scholar] [CrossRef]
- Feng, X.; Song, Q.; Yu, A.; Tang, H.; Peng, Z.; Wang, X. Receptor-interacting protein kinase 3 is a predictor of survival and plays a tumor suppressive role in colorectal cancer. Neoplasma 2015, 62, 592–601. [Google Scholar] [CrossRef]
- Ertao, Z.; Jianhui, C.; Kang, W.; Zhijun, Y.; Hui, W.; Chuangqi, C.; Changjiang, Q.; Sile, C.; Yulong, H.; Shirong, C. Prognostic value of mixed lineage kinase domain-like protein expression in the survival of patients with gastric cancer. Tumour Biol. 2016, 37, 13679–13685. [Google Scholar] [CrossRef]
- He, L.; Peng, K.; Liu, Y.; Xiong, J.; Zhu, F. Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. Onco. Targets Ther. 2013, 6, 1539–1543. [Google Scholar]
- Nugues, A.L.; El Bouazzati, H.; Hetuin, D.; Berthon, C.; Loyens, A.; Bertrand, E.; Jouy, N.; Idziorek, T.; Quesnel, B. RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis. 2014, 5, e1384. [Google Scholar] [CrossRef]
- Geserick, P.; Wang, J.; Schilling, R.; Horn, S.; Harris, P.A.; Bertin, J.; Gough, P.J.; Feoktistova, M.; Leverkus, M. Absence of RIPK3 predicts necroptosis resistance in malignant melanoma. Cell Death Dis. 2015, 6, e1884. [Google Scholar] [CrossRef]
- McCormick, K.D.; Ghosh, A.; Trivedi, S.; Wang, L.; Coyne, C.B.; Ferris, R.L.; Sarkar, S.N. Innate immune signaling through differential RIPK1 expression promote tumor progression in head and neck squamous cell carcinoma. Carcinogenesis 2016, 37, 522–529. [Google Scholar] [CrossRef]
- Bozec, D.; Iuga, A.C.; Roda, G.; Dahan, S.; Yeretssian, G. Critical function of the necroptosis adaptor RIPK3 in protecting from intestinal tumorigenesis. Oncotarget 2016, 7, 46384–46400. [Google Scholar] [CrossRef]
- Li, X.; Guo, J.; Ding, A.; Qi, W.; Zhang, P.; Lv, J.; Qiu, W.S.; Sun, Z.Q. Association of mixed lineage kinase domain-like protein expression with prognosis in patients with colon cancer. Technol. Cancer Res. Treat. 2017, 16, 428–434. [Google Scholar] [CrossRef]
- Park, S.; Hatanpaa, K.J.; Xie, Y.; Mickey, B.E.; Madden, C.J.; Raisanen, J.M.; Ramnarain, D.B.; Xiao, G.; Saha, D.; Habib, A.A.; et al. The receptor interacting protein 1 inhibits p53 induction through NF-κB activation and confers a worse prognosis in glioblastoma. Cancer Res. 2009, 69, 2809–2816. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, W.; Xu, X.; Li, B.; He, W.; Padilla, M.T.; Jang, J.H.; Nyunoya, T.; Amin, S.; Lin, Y.; et al. RIP1 potentiates BPDE-induced transformation in human bronchial epithelial cells through catalase-mediated suppression of excessive reactive oxygen species. Carcinogenesis 2013, 34, 2119–2128. [Google Scholar] [CrossRef]
- Buchheit, C.L.; Rayavarapu, R.R.; Schafer, Z.T. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin. Cell. Dev. Biol. 2012, 23, 402–411. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Vucur, M.; Luedde, T. Life is fragile: FMRP controls cell death in liver disease. Gut 2020, 69, 2–3. [Google Scholar] [CrossRef]
- Dara, L.; Liu, Z.X.; Kaplowitz, N. Questions and controversies: The role of necroptosis in liver disease. Cell Death Dis. 2016, 2, 16089. [Google Scholar] [CrossRef]
- Dara, L.; Johnson, H.; Suda, J.; Win, S.; Gaarde, W.; Han, D.; Kaplowitz, N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology 2015, 62, 1847–1857. [Google Scholar] [CrossRef]
- Aizawa, S.; Brar, G.; Tsukamoto, H. Cell death and liver disease. Gut Liver 2020, 14, 20–29. [Google Scholar] [CrossRef]
- Khoury, M.K.; Gupta, K.; Franco, S.R.; Liu, B. Necroptosis in the pathophysiology of disease. Am. J. Pathol. 2020, 190, 272–285. [Google Scholar] [CrossRef]
- Takemoto, K.; Hatano, E.; Iwaisako, K.; Takeiri, M.; Noma, N.; Ohmae, S.; Toriguchi, K.; Tanabe, K.; Tanaka, H.; Asagiri, M.; et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 2014, 4, 777–787. [Google Scholar] [CrossRef]
- Zhang, Y.F.; He, W.; Zhang, C.; Liu, X.J.; Lu, Y.; Wang, H.; Zhang, Z.H.; Chen, X.; Xu, D.X. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol. Lett. 2014, 225, 445–453. [Google Scholar] [CrossRef]
- Li, J.X.; Feng, J.M.; Wang, Y.; Li, X.H.; Chen, X.X.; Su, Y.; Shen, Y.Y.; Chen, Y.; Xiong, B.; Miao, Z.H.; et al. The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 2014, 5, e1278. [Google Scholar] [CrossRef]
- Jaeschke, H.; Ramachandran, A.; Chao, X.; Ding, W.X. Emerging and established modes of cell death during acetaminophen-induced liver injury. Arch. Toxicol. 2019, 93, 3491–3502. [Google Scholar] [CrossRef]
- Ramachandran, A.; McGill, M.R.; Xie, Y.; Ni, H.M.; Ding, W.X.; Jaeschke, H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013, 58, 2099–2108. [Google Scholar] [CrossRef]
- Deutsch, M.; Graffeo, C.S.; Rokosh, R.; Pansari, M.; Ochi, A.; Levie, E.M.; Van Heerden, E.; Tippens, D.M.; Greco, S.; Miller, G.; et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015, 6, e1759. [Google Scholar] [CrossRef]
- Gunther, C.; He, G.W.; Kremer, A.E.; Murphy, J.M.; Petrie, E.J.; Amann, K.; Vandenabeele, P.; Linkermann, A.; Poremba, C.; Wirtz, S.; et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J. Clin. Invest. 2016, 126, 4346–4360. [Google Scholar] [CrossRef]
- Tian, R.D.; Chen, Y.Q.; He, Y.H.; Tang, Y.J.; Chen, G.M.; Yang, F.W.; Li, Y.; Huang, W.G.; Chen, H.; Lin, S.D.; et al. Phosphorylation of eIF2α mitigates endoplasmic reticulum stress and hepatocyte necroptosis in acute liver injury. Ann. Hepatol. 2020, 19, 79–87. [Google Scholar] [CrossRef]
- Roychowdhury, S.; McCullough, R.L.; Sanz-Garcia, C.; Saikia, P.; Alkhouri, N.; Matloob, A.; Pollard, K.A.; McMullen, M.R.; Croniger, C.M.; Nagy, L.E. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology 2016, 64, 1518–1533. [Google Scholar] [CrossRef]
- Gautheron, J.; Vucur, M.; Schneider, A.T.; Severi, I.; Roderburg, C.; Roy, S.; Bartneck, M.; Schrammen, P.; Diaz, M.B.; Luedde, T.; et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat. Commun. 2016, 7, 11869. [Google Scholar] [CrossRef]
- Xu, H.; Du, X.; Liu, G.; Huang, S.; Du, W.; Zou, S.; Tang, D.; Fan, C.; Xie, Y.; Fu, X.; et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol. Metab. 2019, 23, 14–23. [Google Scholar] [CrossRef]
- Majdi, A.; Aoudjehane, L.; Ratziu, V.; Islam, T.; Afonso, M.B.; Conti, F.; Mestiri, T.; Lagouge, M.; Foufelle, F.; Gautheron, J.; et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J. Hepatol. 2020, 72, 627–635. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Carvalho, T.; Caridade, M.; Borralho, P.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin. Sci. 2015, 129, 721–739. [Google Scholar] [CrossRef]
- Gautheron, J.; Vucur, M.; Luedde, T. Necroptosis in nonalcoholic steatohepatitis. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 264–265. [Google Scholar] [CrossRef]
- Gautheron, J.; Vucur, M.; Reisinger, F.; Cardenas, D.V.; Roderburg, C.; Koppe, C.; Kreggenwinkel, K.; Schneider, A.T.; Bartneck, M.; Luedde, T.; et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol. Med. 2014, 6, 1062–1074. [Google Scholar] [CrossRef]
- Wang, S.; Ni, H.M.; Dorko, K.; Kumer, S.C.; Schmitt, T.M.; Nawabi, A.; Komatsu, M.; Huang, H.; Ding, W.X. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016, 7, 17681–17698. [Google Scholar] [CrossRef]
- Roychowdhury, S.; McMullen, M.R.; Pisano, S.G.; Liu, X.; Nagy, L.E. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013, 57, 1773–1783. [Google Scholar] [CrossRef]
- Zhang, B.; Li, M.D.; Yin, R.; Liu, Y.; Yang, Y.; Mitchell-Richards, K.A.; Nam, J.H.; Li, R.; Wang, L.; Yang, X.; et al. O-GlcNAc transferase suppresses necroptosis and liver fibrosis. JCI Insight 2019, 4, 127709. [Google Scholar] [CrossRef]
- Zhou, Y.; Dai, W.; Lin, C.; Wang, F.; He, L.; Shen, M.; Chen, P.; Wang, C.; Lu, J.; Guo, C.; et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013, 2013, 706156. [Google Scholar] [CrossRef]
- Arshad, M.I.; Piquet-Pellorce, C.; Filliol, A.; L’Helgoualc’h, A.; Lucas-Clerc, C.; Jouan-Lanhouet, S.; Dimanche-Boitrel, M.T.; Samson, M. The chemical inhibitors of cellular death, PJ34 and Necrostatin-1, down-regulate IL-33 expression in liver. J. Mol. Med. 2015, 93, 867–878. [Google Scholar] [CrossRef]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Dimanche-Boitrel, M.T.; et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Diff. 2012, 19, 2003–2014. [Google Scholar] [CrossRef]
- Suda, J.; Dara, L.; Yang, L.; Aghajan, M.; Song, Y.; Kaplowitz, N.; Liu, Z.X. Knockdown of RIPK1 markedly exacerbates murine immune-mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of NFkappaB. J. Immunol. 2016, 197, 3120–3129. [Google Scholar] [CrossRef]
- Filliol, A.; Piquet-Pellorce, C.; Le Seyec, J.; Farooq, M.; Genet, V.; Lucas-Clerc, C.; Bertin, J.; Gough, P.J.; Dimanche-Boitrel, M.T.; Samson, M.; et al. RIPK1 protects from TNF-α-mediated liver damage during hepatitis. Cell Death Dis. 2016, 7, e2462. [Google Scholar] [CrossRef]
- Lim, E.J.; El Khobar, K.; Chin, R.; Earnest-Silveira, L.; Angus, P.W.; Bock, C.T.; Nachbur, U.; Silke, J.; Torresi, J. Hepatitis C virus-induced hepatocyte cell death and protection by inhibition of apoptosis. J. Gen. Virol. 2014, 95, 2204–2215. [Google Scholar] [CrossRef]
- Bittel, M.; Kremer, A.E.; Stürzl, M.; Wirtz, S.; Stolzer, I.; Neurath, M.F.; Ballon, G.; Günther, C. Modulation of the extrinsic cell death signaling pathway by viral Flip induces acute-death mediated liver failure. Cell Death Dis. 2019, 10, 878. [Google Scholar] [CrossRef]
- Haga, S.; Kanno, A.; Ozawa, T.; Morita, N.; Asano, M.; Ozaki, M. Detection of necroptosis in ligand-mediated and hypoxiainduced injury of hepatocytes using a novel optic probe detecting receptor-interacting protein (RIP)1/RIP3 binding. Oncol. Res. 2018, 26, 503–513. [Google Scholar] [CrossRef]
- Hong, J.M.; Kim, S.J.; Lee, S.M. Role of necroptosis in autophagy signaling during hepatic ischemia and reperfusion. Toxicol. Appl. Pharmacol. 2016, 308, 1–10. [Google Scholar] [CrossRef]
- Liss, K.H.H.; McCommis, K.S.; Chambers, K.T.; Pietka, T.A.; Schweitzer, G.G.; Park, S.L.; Nalbantoglu, I.; Weinheimer, C.J.; Hall, A.M.; Finck, B.N. The impact of diet-induced hepatic steatosis in a murine model of hepatic ischemia/reperfusion injury. Liver Transpl. 2018, 24, 908–921. [Google Scholar] [CrossRef]
- Yang, F.; Shang, L.; Wang, S.; Liu, Y.; Ren, H.; Zhu, W.; Shi, X. TNFα-mediated necroptosis aggravates ischemia-reperfusion injury in the fatty liver by regulating the inflammatory response. Oxid Med. Cell Longev. 2019, 2019, 2301903. [Google Scholar] [CrossRef]
- Ni, H.M.; Chao, X.; Kaseff, J.; Deng, F.; Wang, S.; Shi, Y.H.; Li, T.; Ding, W.X.; Jaeschke, H. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)-Mixed Lineage Kinase Domain-Like protein (MLKL)-mediated necroptosis contributes to ischemia-reperfusion injury of steatotic livers. Am. J. Pathol. 2019, 189, 1363–1374. [Google Scholar] [CrossRef]
- Rosentreter, D.; Funken, D.; Reifart, J.; Mende, K.; Rentsch, M.; Khandoga, A. RIP1-dependent programmed necrosis is negatively regulated by caspases during hepatic ischemia-reperfusion. Shock 2015, 44, 72–76. [Google Scholar] [CrossRef]
- Saeed, W.K.; Jun, D.W.; Jang, K.; Chae, Y.J.; Lee, J.S.; Kang, H.T. Does necroptosis have a crucial role in hepatic ischemia-reperfusion injury? PLoS ONE 2017, 12, e0184752. [Google Scholar] [CrossRef]
- Salas-Silva, S.; Simoni-Nieves, A.; Lopez-Ramirez, J.; Bucio, L.; Gómez-Quiroz, L.E.; Gutiérrez-Ruiz, M.C.; Roma, M.G. Cholangiocyte death in ductopenic cholestatic cholangiopathies: Mechanistic basis and emerging therapeutic strategies. Life Sci. 2019, 218, 324–339. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Ofengeim, D.; Carvalho, T.; Amaral, J.D.; Gaspar, M.M.; Cortez-Pinto, H.; Castro, R.E.; Rodrigues, C.M.; et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis. 2016, 7, e2390. [Google Scholar] [CrossRef]
- Koppe, C.; Verheugd, P.; Gautheron, J.; Reisinger, F.; Kreggenwinkel, K.; Roderburg, C.; Quagliata, L.; Terracciano, L.; Gassler, N.; Luedde, T.; et al. IκB kinaseα/β control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell-death mediator receptor-interacting protein kinase 1. Hepatology 2016, 64, 1217–1231. [Google Scholar] [CrossRef]
- Gujral, J.S.; Liu, J.; Farhood, A.; Jaeschke, H. Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology 2004, 40, 998–1007. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Simao, A.L.; Gaspar, M.M.; Carvalho, T.; Borralho, P.; Bañales, J.M.; Castro, R.E.; Rodrigues, C.M.P. miRNA-21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ. 2018, 25, 857–872. [Google Scholar] [CrossRef]
- Squadroni, M.; Tondulli, L.; Gatta, G.; Mosconi, S.; Beretta, G.; Labianca, R. Cholangiocarcinoma. Crit. Rev. Oncol. Hematol. 2017, 116, 11–31. [Google Scholar] [CrossRef]
- Bartella, I.; Dufour, J.F. Clinical diagnosis and staging of intrahepatic cholangiocarcinoma. J. Gastrointestin. Liver Dis. 2015, 24, 481–489. [Google Scholar] [CrossRef]
- Kayhanian, H.; Smyth, E.C.; Braconi, C. Emerging molecular targets and therapy for cholangiocarcinoma. World J. Gastrointest. Oncol. 2017, 9, 268–280. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Alvaro, D.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Bridgewater, J.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Pellino, A.; Loupakis, F.; Cadamuro, M.; Dadduzio, V.; Fassan, M.; Guido, M.; Cillo, U.; Indraccolo, S.; Fabris, L. Precision medicine in cholangiocarcinoma. Trans. Gastroenterol. Hepatol. 2018, 3, 40. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Kudo, M.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Cadamuro, M.; Brivio, S.; Spirli, C.; Joplin, R.E.; Strazzabosco, M.; Fabris, L. Autocrine and paracrine mechanisms promoting chemoresistance in cholangiocarcinoma. Int. J. Mol. Sci. 2017, 18, 149. [Google Scholar] [CrossRef]
- Morton, S.D.; Cadamuro, M.; Brivio, S.; Vismara, M.; Stecca, T.; Massani, M.; Bassi, N.; Furlanetto, A.; Joplin, R.E.; Strazzabosco, M.; et al. Leukemia inhibitory factor protects cholangiocarcinoma cells from drug-induced apoptosis via a PI3K/AKT-dependent Mcl-1 activation. Oncotarget 2015, 6, 26052–26064. [Google Scholar] [CrossRef]
- Saeed, W.K.; Jun, D.W. Viewpoint: Necroptosis influences the type of liver cancer via changes of hepatic microenvironment. Hepatobiliary Surg. Nutr. 2019, 8, 549–551. [Google Scholar] [CrossRef]
- Sacchi, D.; Sarcognato, S.; Cillo, U.; Gringeri, E.; Fabris, L.; Di Giunta, M.; Nicolè, L.; Guzzardo, V.; Fassina, A.; Guido, M. Necroptosis is associated with a better survival in intrahepatic cholangiocarcinoma. Dig. Liver Dis. 2019, 51, e2–e3. [Google Scholar] [CrossRef]
- Yang, C.; Sun, P.; Deng, M.; Loughran, P.; Li, W.; Yi, Z.; Li, S.; Zhang, X.; Fan, J.; Scott, M.J.; et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis. 2019, 10, 481. [Google Scholar] [CrossRef]
- Degterev, A.; Ofengeim, D.; Yuan, J. Targeting RIPK1 for the treatment of human diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 9714–9722. [Google Scholar] [CrossRef]
- Najafov, A.; Chen, H.; Yuan, J. Necroptosis and cancer. Trends Cancer 2017, 3, 294–301. [Google Scholar] [CrossRef]
- Brumatti, G.; Ma, C.; Lalaoui, N.; Nguyen, N.; Navarro, M.; Tanzer, M.C.; Richmond, J.; Ghisi, M.; Salmon, J.M.; Silke, J.; et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci. Trans. Med. 2016, 8, 339ra69. [Google Scholar] [CrossRef]
- McCabe, K.E.; Bacos, K.; Lu, D.; Delaney, J.R.; Axelrod, J.; Potter, M.D.; Vamos, M.; Wong, V.; Cosford, N.D.; Stupack, D.G.; et al. Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis. 2014, 5, e1496. [Google Scholar] [CrossRef]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xie, L.; DeWitt, J.P.; Chen, Y. Cancer therapy in the necroptosis era. Cell Death Differ. 2016, 23, 748–756. [Google Scholar] [CrossRef]
- Han, W.; Li, L.; Qiu, S.; Lu, Q.; Pan, Q.; Gu, Y.; Luo, J.; Hu, X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol. Cancer Ther. 2007, 6, 1641–1649. [Google Scholar] [CrossRef]
- Yu, X.; Deng, Q.; Li, W.; Xiao, L.; Luo, X.; Liu, X.; Yang, L.; Peng, S.; Ding, Z.; Cao, Y.; et al. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFα and ROS production. Oncotarget 2015, 6, 1995–2008. [Google Scholar] [CrossRef]
- Li, Y.; Tian, X.; Liu, X.; Gong, P. Bufalin inhibits human breast cancer tumorigenesis by inducing cell death through the ROS-mediated RIP1/RIP3/PARP-1 pathways. Carcinogenesis 2018, 39, 700–707. [Google Scholar] [CrossRef]
- Akara-Amornthum, P.; Lomphithak, T.; Choksi, S.; Tohtong, R.; Jitkaew, S. Key necroptotic proteins are required for Smac mimetic-mediated sensitization of cholangiocarcinoma cells to TNF-α and chemotherapeutic gemcitabine-induced necroptosis. PLoS ONE 2020, 15, e0227454. [Google Scholar] [CrossRef]
- Xu, B.; Xu, M.; Tian, Y.; Yu, Q.; Zhao, Y.; Chen, X.; Mi, P.; Cao, H.; Zhang, B.; Hu, T.; et al. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Dis. 2017, 3, 16096. [Google Scholar] [CrossRef]


{kind=link}
| Pro-Tumoral Effects | Anti-Tumoral Effects |
|---|---|
| Necroptosis results in chronic inflammation and eventually tumorigenesis [32]. | Cross-priming of cytotoxic CD8+ T cells enhances tumor clearance [33,34]. |
| DAMP-induced inflammation activates the NF-κB pathway, inhibits apoptosis, and increases genomic instability [35,36]. | RIPK3-induced activation of natural killer T cells promotes anti-tumor immunity [37]. |
| Necroptosis of endothelial cells and extracellular matrix induces metastasis [29]. | Necroptosis in apoptosis-resistant cancer cells inhibits carcinogenesis [38,39]. |
| A necroptosis-dominant microenvironment induces lineage commitment towards intrahepatic CCA [40]. | RIPK3 expression levels may predict the efficacy of cancer immunotherapy [41]. |
| Necroptosis induces the production of molecules that promote an immune suppressive tumor microenvironment [30]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarcognato, S.; de Jong, I.E.M.; Fabris, L.; Cadamuro, M.; Guido, M. Necroptosis in Cholangiocarcinoma. Cells 2020, 9, 982. https://doi.org/10.3390/cells9040982
Sarcognato S, de Jong IEM, Fabris L, Cadamuro M, Guido M. Necroptosis in Cholangiocarcinoma. Cells. 2020; 9(4):982. https://doi.org/10.3390/cells9040982
Chicago/Turabian StyleSarcognato, Samantha, Iris E. M. de Jong, Luca Fabris, Massimiliano Cadamuro, and Maria Guido. 2020. "Necroptosis in Cholangiocarcinoma" Cells 9, no. 4: 982. https://doi.org/10.3390/cells9040982
APA StyleSarcognato, S., de Jong, I. E. M., Fabris, L., Cadamuro, M., & Guido, M. (2020). Necroptosis in Cholangiocarcinoma. Cells, 9(4), 982. https://doi.org/10.3390/cells9040982