The Histone Variant MacroH2A1 Regulates Key Genes for Myogenic Cell Fusion in a Splice-Isoform Dependent Manner

 ,
,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Isolation of Primary Myoblasts
2.2. Plasmids, Transfection and Retroviral Infection
2.3. Antibodies and Immunofluorescence
2.4. Image Data Analysis
2.5. Gene Expression Analysis
2.6. Chromatin immunoprecipitation, Sequencing and Data Analysis
3. Results
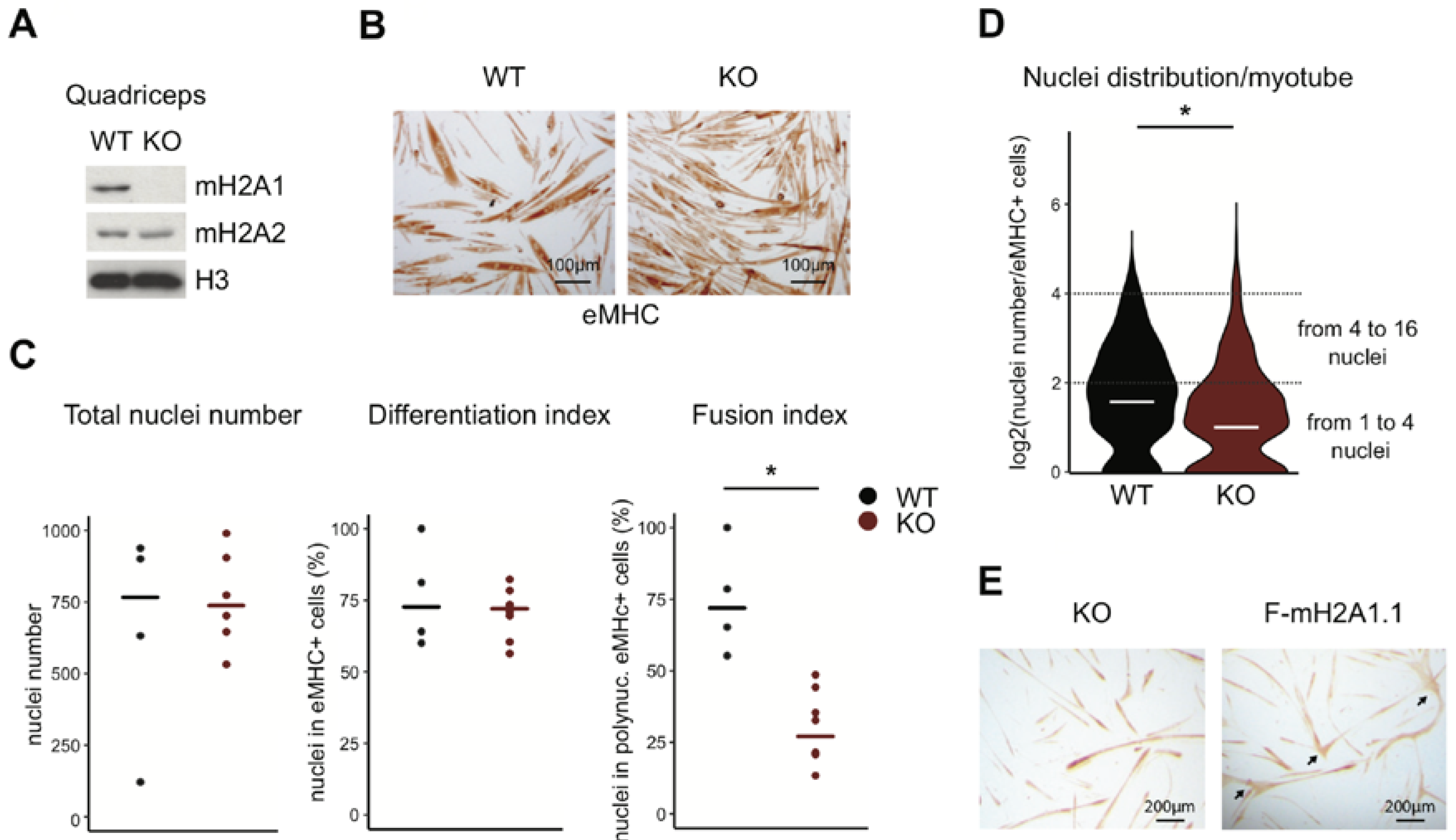
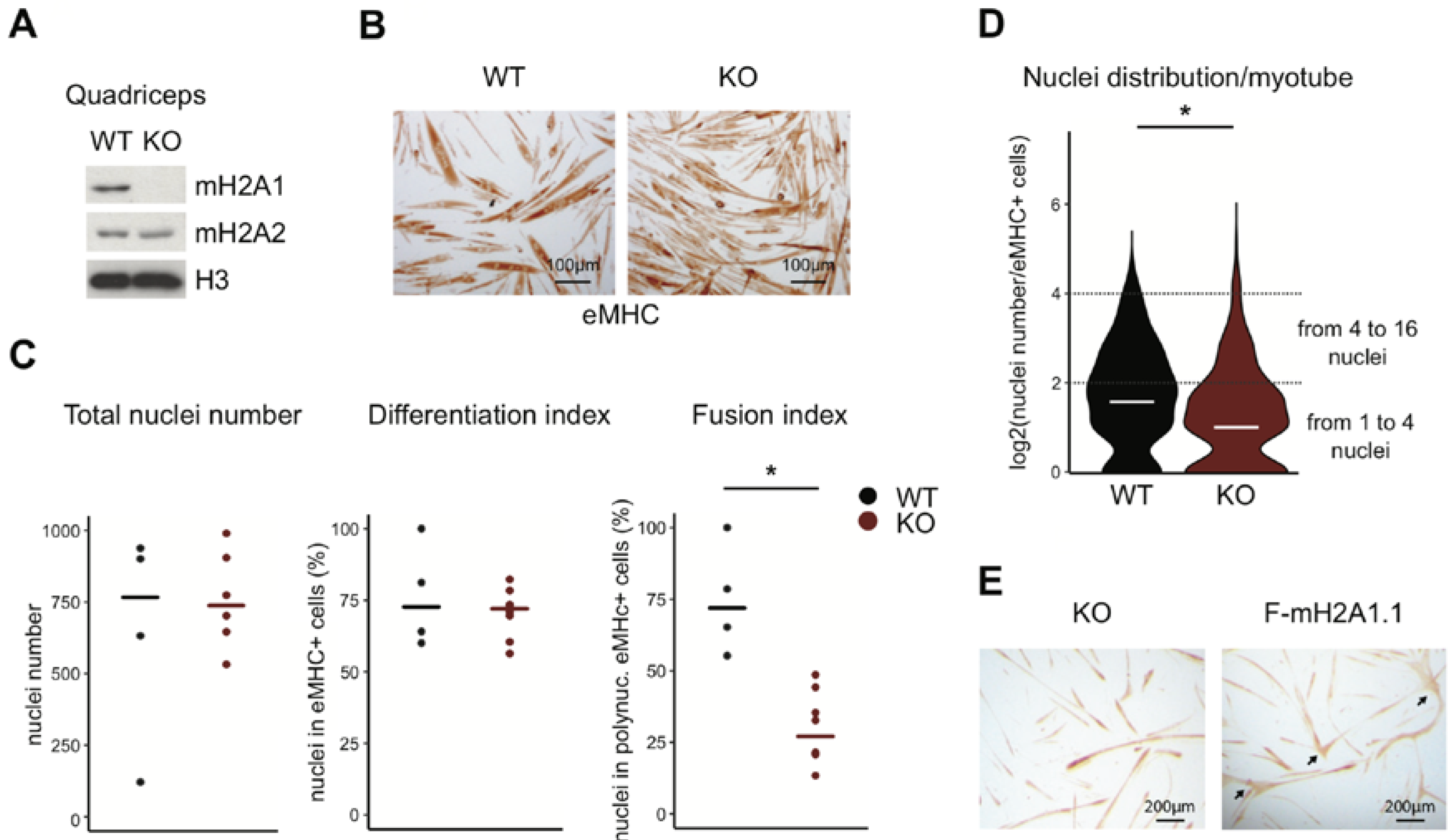
3.1. Loss of Total MacroH2A1 Reduces the Capacity of Primary Myoblasts to Fuse
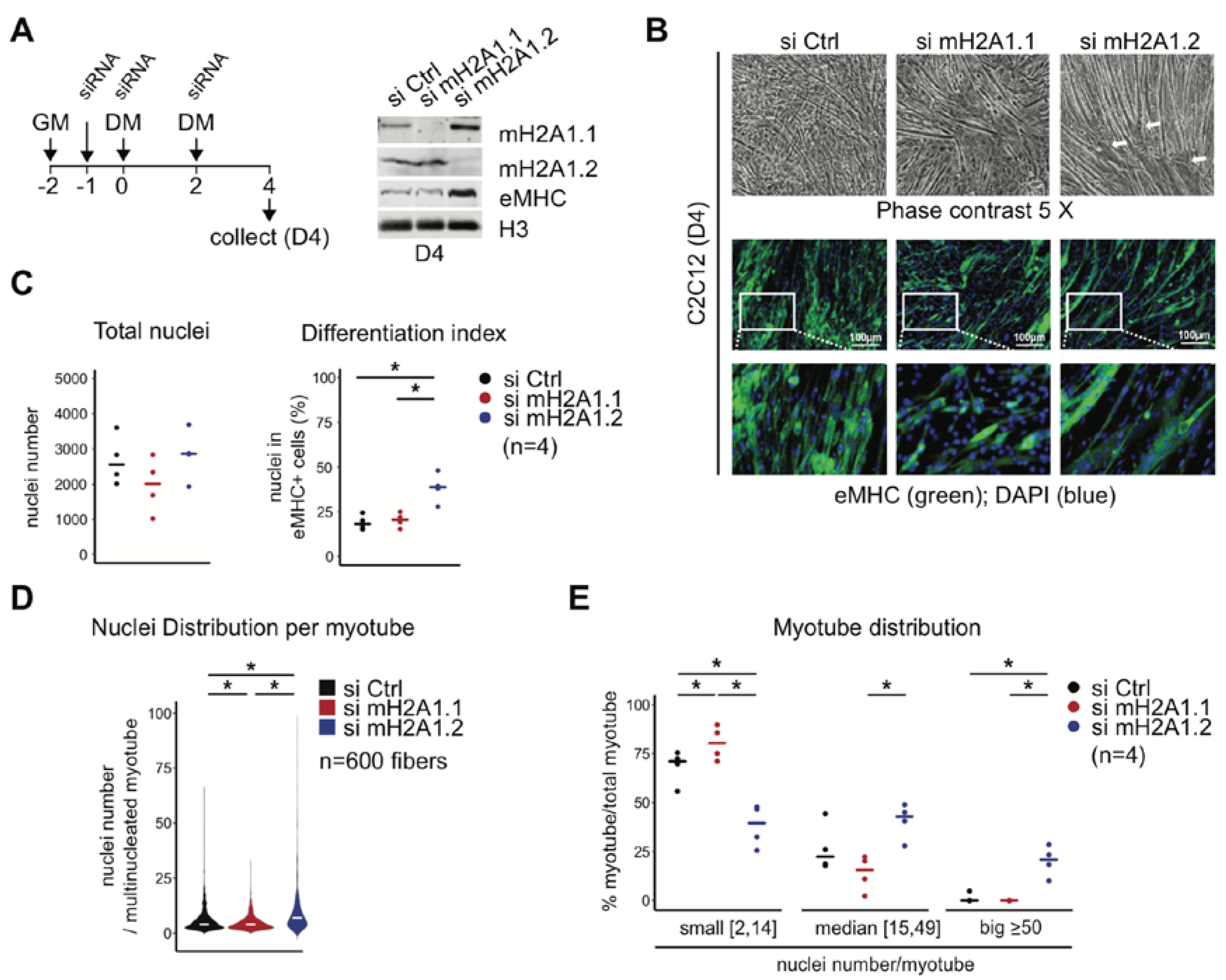
3.2. MacroH2A1 Isoforms Have an Opposing Effect on Cell Fusion—MacroH2A1.1 Promotes and MacroH2A1.2 Reduces It
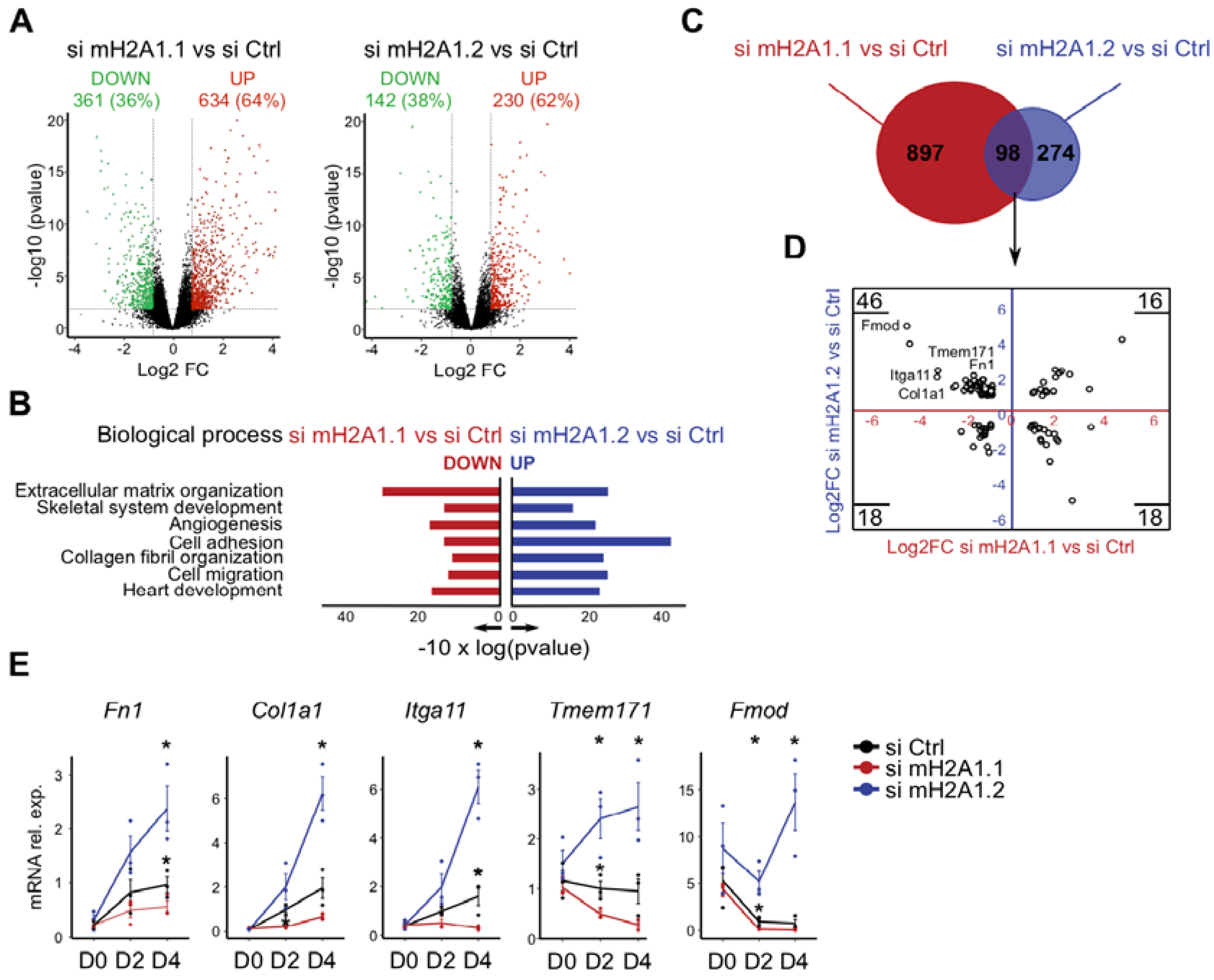
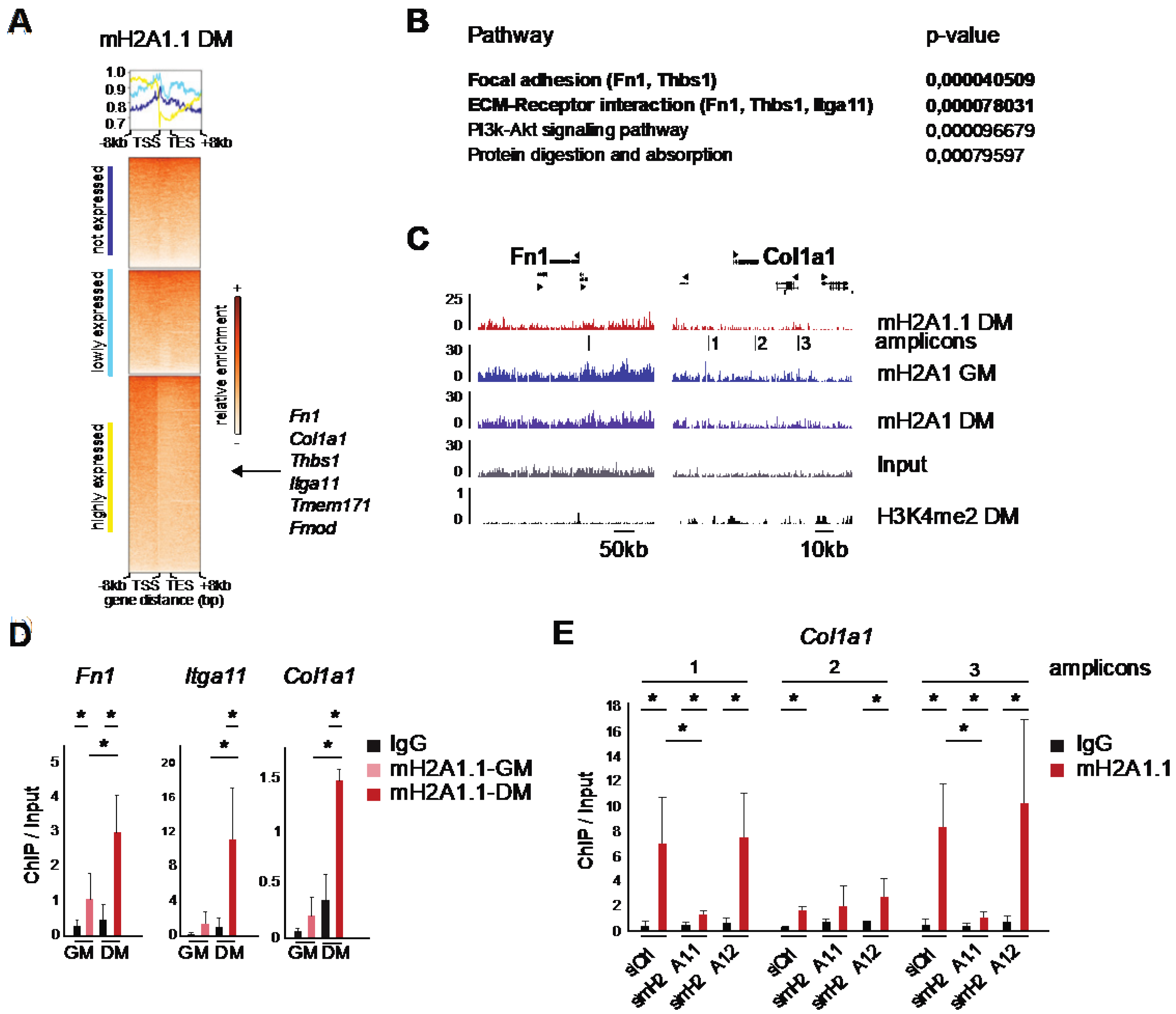
3.3. The Opposing Function of MacroH2A1 Isoforms Correlates with the Differential Regulation of a Subset of Genes Related to Extracellular Matrix and Adhesion
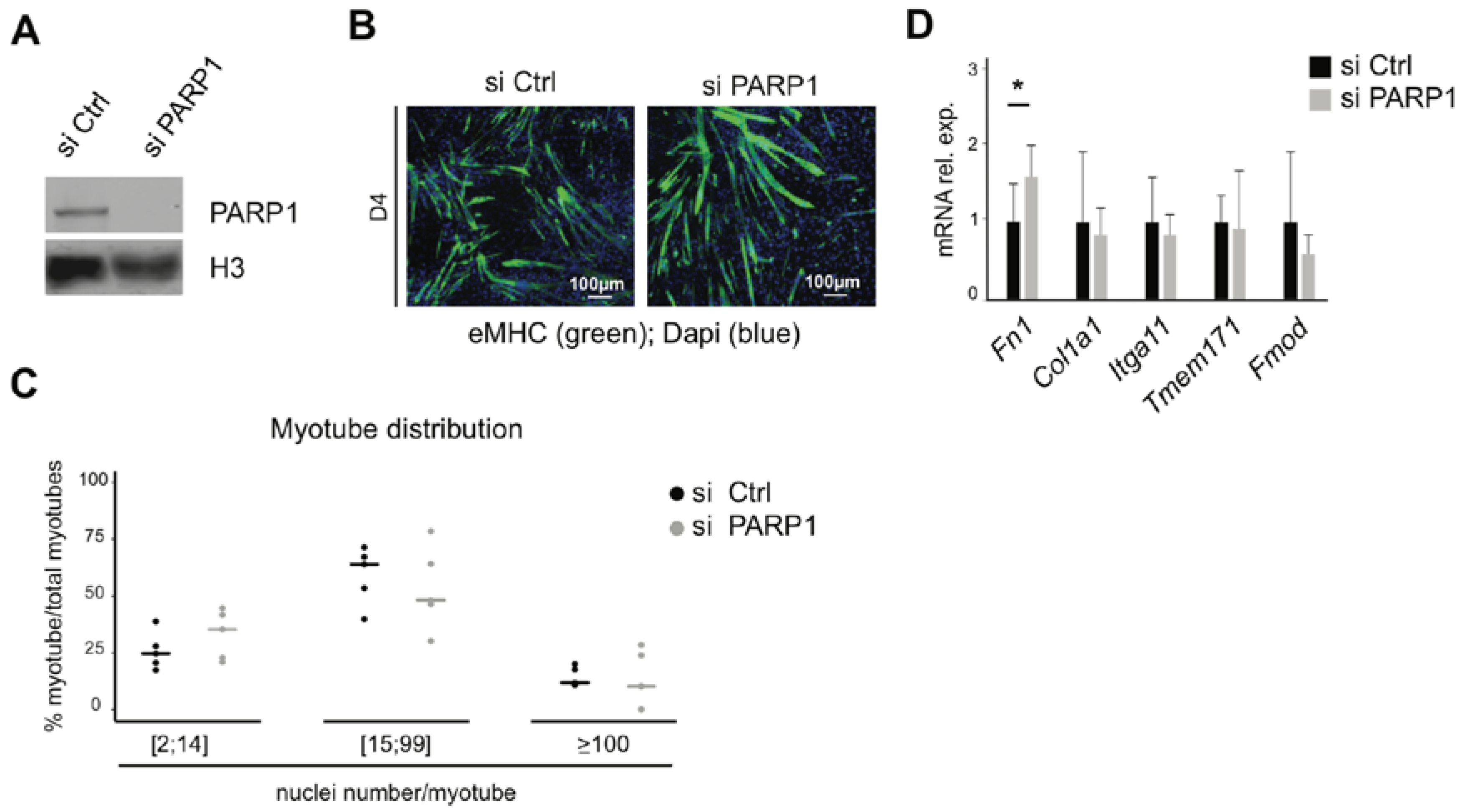
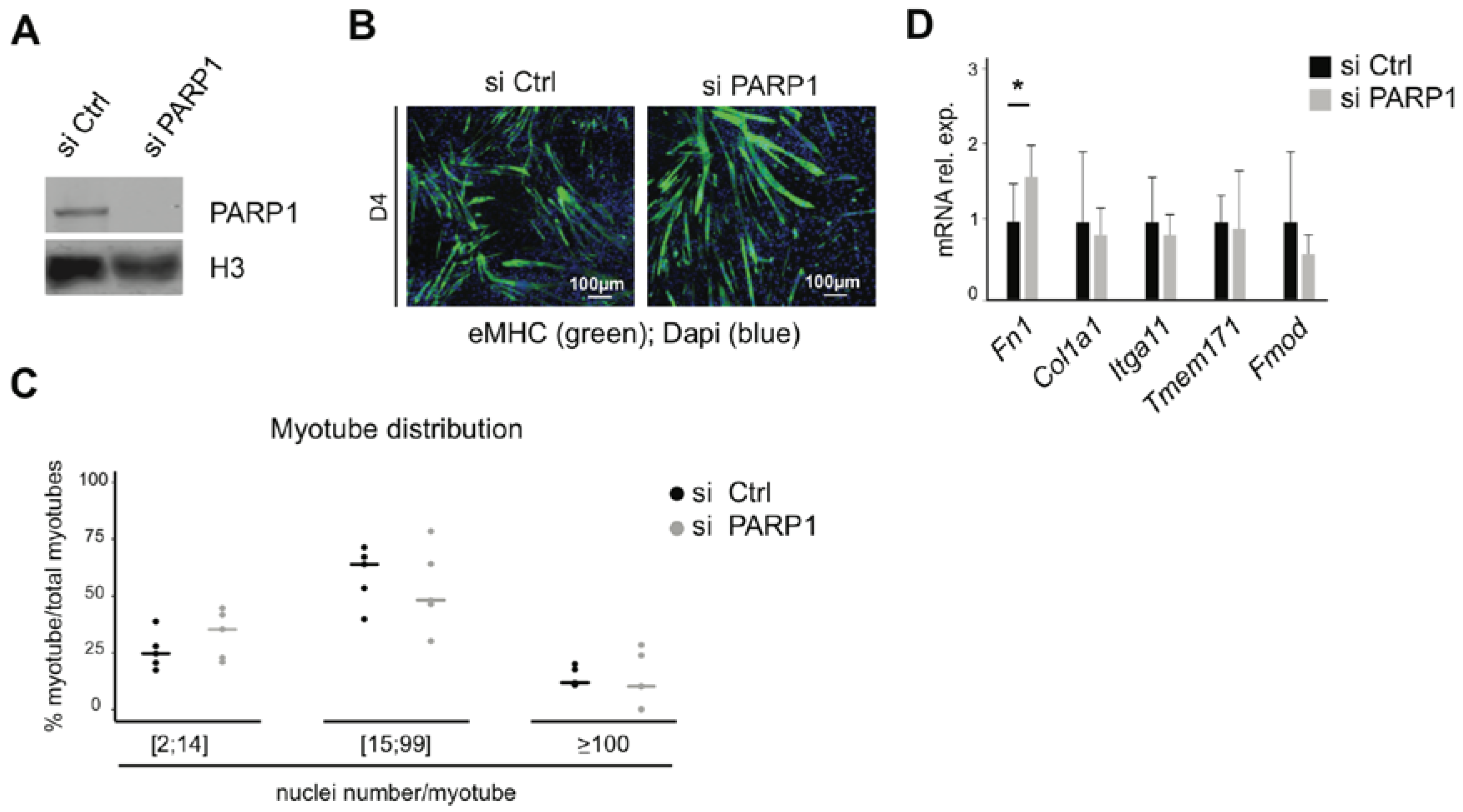
3.4. Knockdown of PARP1 Has Little Impact on Fusion and Expression of Related Genes
3.5. MacroH2A1.1 Becomes Enriched on Fusion-Relevant Genes During Differentiation
4. Discussion
4.1. How Can Splice Isoforms of Histone Variant MacroH2A1 Affect Gene Transcription?
4.2. The Relevance of Identified Genes for Cell Fusion?
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Application | Primer Name | Sequence (Genomic Position of Amplicon) |
|---|---|---|
| siRNA | scramble (ctrl) | CGUACGCGGAAUACUUCGATT |
| siRNA | macroH2A1.1#1 | CGACAAACACUGACUUCUATT |
| siRNA | macroH2A1.1#2 | CCGACAAACACUGACUUCUTT |
| siRNA | macroH2A1.2#1 | GCUUUGAGGUGGAGGCCAUTT |
| siRNA | macroH2A1.2#2 | UGACAUUGACCUUAAAGAUTT |
| siRNA | PARP1#1 | AGCUGAAGAAAGCGTGUUCTT |
| siRNA | PARP1#2 | AGGCGUGGCAGGCAAAGGCTT |
| qPCR | macroH2A1 F | GACGGTGAAAAACTGCTTGG |
| qPCR | macroH2A1 R | GGAGGAGGACATCGTGGAG |
| qPCR | macroH2A2 F | GCTGGAAGAGACCATCAAAAA |
| qPCR | macroH2A2 R | CGAAGTGAGCCGAGATGG |
| qPCR | macroH2A1 Exon5 | CCTACAGACGGCTTCACTGTC |
| qPCR | macroH2A1.2 Exon6 | GGTCAATGTCAGCATTGGTAGG |
| qPCR | macroH2A1.1 Exon7 | GTGTAGAAGTCAGTGTTTGTCG |
| qPCR | Rpl7 F | GAAGCTCATCTATGAGAAGGC |
| qPCR | Rpl7 R | AAGACGAAGGAGCTGCAGAAC |
| qPCR | Gapdh F | TGCACCACCAACTGCTTAG |
| qPCR | Gapdh R | GATGCAGGGATGATGTTC |
| qPCR | Rpl0 F | TTCATTGTGGGAGCAGAC |
| qPCR | Rpl0 R | CAGCAGTTTCTCCAGAGC |
| qPCR | Tmem171 F | AAACCCACCTTCCTATTCCAG |
| qPCR | Tmem171 R | ATGAACCCTGCCCAGAAATG |
| qPCR | Itga11 F | GGGAAACCTGTGGCTGAC |
| qPCR | Itga11 R | ATGAAGGGGCTGTGGAAC |
| qPCR | Parp1 F | CCTGAACAACGCAGACAGC |
| qPCR | Parp1 R | CGTTGTGCGTGGTAGCATGA |
| qPCR | Fmod F | TCCTCTGGGTCGCTCTACAT |
| qPCR | Fmod R | TGCCTCAGCTTGGAGAAGAC |
| qPCR | Col1a1 F | GCGAGTGCTGTGCTTTCTG |
| qPCR | Col1a1 R | GGTCCCTCGACTCCTACATCT |
| qPCR | Fn1 F | GACAACCGAGGAAACCTGCT |
| qPCR | Fn1 R | GATCCGGCTGAAGCACTTTG |
| Cloning | mH2A1.1_aa1_EcoRI F | GCGAATTCCATGTCGAGCCGCGGCGGGAAGAAG |
| Cloning | mH2A1.1_STOP_XhoI R | GGCTCGAGTCAGCCTAGTTGGCGTCCAGCTTGG |
| ChIP-qPCR | Fn1 F | TCCCCTCCATGACAACTAGGA (chr1:71,722,171-71,722,240) |
| ChIP-qPCR | Fn1 R | TTTTGACCCCAGGGATATGGC (chr1:71,722,171-71,722,240) |
| ChIP-qPCR | Itga11 F | GTTGGTTCTTTTGATATTCAGGCTC (chr9:62,524,168-62,524,259) |
| ChIP-qPCR | Itga11 R | ACCTTCAGGGCTAAGTTTCAGA (chr9:62,524,168-62,524,259) |
| ChIP-qPCR | Col1a1#1 F | GACGTCATGGCATGGAAAGC (chr11:94,784,360-94,784,821) |
| ChIP-qPCR | Col1a1#1 R | CTGGGATGAGGATCGCAGTG (chr11:94,784,360-94,784,821) |
| ChIP-qPCR | Col1a1#2 F | CCTCAGGCCCTTTACTGACC (chr11:94,804,479-94,805,581) |
| ChIP-qPCR | Col1a1#2 R | TGGAGCCAACCAATGGTCTC (chr11:94,804,479-94,805,581) |
| ChIP-qPCR | Col1a1#3 F | CAATTTGAACGGCACCTCCC (chr11:94,836,255-94,836,814) |
| ChIP-qPCR | Col1a1#3 R | AGCTAGGCCAGAGTCTAGGG (chr11:94,836,255-94,836,814) |
| Antibodies | Source |
|---|---|
| Control IgG | Abcam, ab-1791 |
| Anti-macroH2A1.1 | [26] |
| Anti-macroH2A1.2 | Cell Signaling, 4827S |
| Anti-macroH2A1.2 | Cell Signaling, 4827S |
| Anti-macroH2A1 | [14] |
| Anti-macroH2A2 | [14] |
| Anti-embryonic myosin heavy chain (eMHC) F1.652 | Santa Cruz, sc-53091 |
| Anti-Histone H3 | Abcam, ab-1791 |
| Anti-PARP-1 | Abcam Ab6079 or Trevigen 4338-MC-50 |
| Secondary anti-mouse and anti-rabbit conjugated to horseradish peroxidase | DakoCytomation |
References
- Buschbeck, M.; Hake, S.B. Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, S.; Gundimella, S.K.Y.; Caron, C.; Perche, P.-Y.; Pehrson, J.R.; Khochbin, S.; Luger, K. Structural characterization of the histone variant macroH2A. Mol. Cell. Biol. 2005, 25, 7616–7624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschbeck, M.; Di Croce, L. Approaching the molecular and physiological function of macroH2A variants. Epigenetics 2010, 5, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buschbeck, M.; Uribesalgo, I.; Wibowo, I.; Rué, P.; Martin, D.; Gutiérrez, A.; Morey, L.; Guigó, R.; López-Schier, H.; Di Croce, L. The histone variant macroH2A is an epigenetic regulator of key developmental genes. Nat. Struct. Mol. Biol. 2009, 16, 1074–1079. [Google Scholar] [CrossRef]
- Creppe, C.; Janich, P.; Cantarino, N.; Noguera, M.; Valero, V.; Musulen, E.; Douet, J.; Posavec, M.; Martin-Caballero, J.; Sumoy, L.; et al. MacroH2A1 Regulates the Balance between Self-Renewal and Differentiation Commitment in Embryonic and Adult Stem Cells. Mol. Cell. Biol. 2012, 32, 1442–1452. [Google Scholar] [CrossRef] [Green Version]
- Gaspar-Maia, A.; Qadeer, Z.A.; Hasson, D.; Ratnakumar, K.; Leu, N.A.; LeRoy, G.; Liu, S.; Costanzi, C.; Valle-Garcia, D.; Schaniel, C.; et al. MacroH2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 2013, 4, 1512–1565. [Google Scholar] [CrossRef] [Green Version]
- Pasque, V.; Gillich, A.; Garrett, N.; Gurdon, J.B. Histone variant macroH2A confers resistance to nuclear reprogramming. EMBO J. 2011, 30, 2373–2387. [Google Scholar] [CrossRef]
- Barrero, M.J.; Sesé, B.; Kuebler, B.; Bilic, J.; Boue, S.; Martí, M.; Belmonte, J.C.I. Macrohistone Variants Preserve Cell Identity by Preventing the Gain of H3K4me2 during Reprogramming to Pluripotency. CellReports 2013, 3, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I.; et al. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef]
- Corujo, D.; Buschbeck, M. Post-Translational Modifications of H2A Histone Variants and Their Role in Cancer. Cancers 2018, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, M.D.; Vatsellas, G.; Polyzos, A.; Mantouvalou, E.; Sianidis, G.; Maraziotis, I.; Agelopoulos, M.; Thanos, D. Composite macroH2A/NRF-1 Nucleosomes Suppress Noise and Generate Robustness in Gene Expression. CellReports 2015, 11, 1090–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelov, D.; Molla, A.; Perche, P.-Y.; Hans, F.; Côté, J.; Khochbin, S.; Bouvet, P.; Dimitrov, S. The histone variant macroH2A interferes with transcription factor binding and SWI/SNF nucleosome remodeling. Mol. Cell 2003, 11, 1033–1041. [Google Scholar] [CrossRef]
- Dell’Orso, S.; Wang, A.H.; Shih, H.-Y.; Saso, K.; Berghella, L.; Gutierrez-Cruz, G.; Ladurner, A.G.; O’Shea, J.J.; Sartorelli, V.; Zare, H. The Histone Variant MacroH2A1.2 Is Necessary for the Activation of Muscle Enhancers and Recruitment of the Transcription Factor Pbx1. CellReports 2016, 14, 1156–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douet, J.; Corujo, D.; Malinverni, R.; Renauld, J.; Sansoni, V.; Posavec Marjanović, M.; Cantariño, N.; Valero, V.; Mongelard, F.; Bouvet, P.; et al. MacroH2A histone variants maintain nuclear organization and heterochromatin architecture. J. Cell Sci. 2017, 130, 1570–1582. [Google Scholar] [CrossRef] [Green Version]
- Kustatscher, G.; Hothorn, M.; Pugieux, C.; Scheffzek, K.; Ladurner, A.G. Splicing regulates NAD metabolite binding to histone macroH2A. Nat. Struct. Mol. Biol. 2005, 12, 624–625. [Google Scholar] [CrossRef]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.K.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; Novikov, L.; Casill, A.D.; Park, J.W.; Gamble, M.J. MacroH2A1.1 and PARP-1 cooperate to regulate transcription by promoting CBP-mediated H2B acetylation. Nat. Struct. Mol. Biol. 2014, 21, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Ouararhni, K.; Hadj-Slimane, R.; Ait-Si-Ali, S.; Robin, P.; Mietton, F.; Harel-Bellan, A.; Dimitrov, S.; Hamiche, A. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 2006, 20, 3324–3336. [Google Scholar] [CrossRef] [Green Version]
- Posavec Marjanović, M.; Hurtado-Bagès, S.; Lassi, M.; Valero, V.; Malinverni, R.; Delage, H.; Navarro, M.; Corujo, D.; Guberovic, I.; Douet, J.; et al. MacroH2A1.1 regulates mitochondrial respiration by limiting nuclear NAD+ consumption. Nat. Struct. Mol. Biol. 2017, 24, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Hurtado-Bagès, S.; Knobloch, G.; Ladurner, A.G.; Buschbeck, M. The taming of PARP1 and its impact on NAD. Mol. Metab. 2020, 100950, in press. [Google Scholar] [CrossRef]
- Perdiguero, E.; Sousa-Victor, P.; Ballestar, E.; Muñoz-Cánoves, P. Epigenetic regulation of myogenesis. Epigenetics 2009, 4, 541–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulard, M.; Storck, S.; Cong, R.; Pinto, R.; Delage, H.; Bouvet, P. Histone variant macroH2A1 deletion in mice causes female-specific steatosis. Epigenetics Chromatin 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdiguero, E.; Ruiz-Bonilla, V.; Gresh, L.; Hui, L.; Ballestar, E.; Sousa-Victor, P.; Baeza-Raja, B.; Jardí, M.; Bosch-Comas, A.; Esteller, M.; et al. Genetic analysis of p38 MAP kinases in myogenesis: Fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J. 2007, 26, 1245–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardenne, E.; Pierredon, S.; Driouch, K.; Gratadou, L.; Lacroix-Triki, M.; Espinoza, M.P.; Zonta, E.; Germann, S.; Mortada, H.; Villemin, J.-P.; et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat. Struct. Mol. Biol. 2012, 19, 1139–1146. [Google Scholar] [CrossRef] [PubMed]
- Cantariño, N.; Fernández-Figueras, M.T.; Valero, V.; Musulen, E.; Malinverni, R.; Granada, I.; Goldie, S.J.; Martín-Caballero, J.; Douet, J.; Forcales, S.V.; et al. A cellular model reflecting the phenotypic heterogeneity of mutant HRASdriven squamous cell carcinoma. Int. J. Cancer 2016, 139, 1106–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Recoules, L.; Heurteau, A.; Raynal, F.; Moutahir, F.; Bejjani, F.; Jariel-Encontre, I.; Cuvier, O.; Lavigne, A.-C.; Bystricky, K. The histone variant macroH2A1.1 regulates gene expression by direct association with their transcription start site. bioRxiv 2020, in press. [Google Scholar]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Hernández-de-Diego, R.; Tarazona, S.; Martínez-Mira, C.; Balzano-Nogueira, L.; Furió-Tarí, P.; Pappas, G.J., Jr.; Conesa, A. PaintOmics 3: A web resource for the pathway analysis and visualization of multi-omics data. Nucleic Acids Res. 2018, 46, W503–W509. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.R.; Schroeder, M.; Fernandez, P.; Taubert, S.; Amati, B. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev. 2001, 15, 2069–2082. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, C.; Schones, D.E.; Zeng, C.; Cui, K.; Zhao, K.; Peng, W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009, 25, 1952–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Changolkar, L.N.; Costanzi, C.; Leu, N.A.; Chen, D.; McLaughlin, K.J.; Pehrson, J.R. Developmental Changes in Histone macroH2A1-Mediated Gene Regulation. Mol. Cell. Biol. 2007, 27, 2758–2764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Changolkar, L.N.; Singh, G.; Cui, K.; Berletch, J.B.; Zhao, K.; Disteche, C.M.; Pehrson, J.R. Genome-Wide Distribution of MacroH2A1 Histone Variants in Mouse Liver Chromatin. Mol. Cell. Biol. 2010, 30, 5473–5483. [Google Scholar] [CrossRef] [Green Version]
- Gamble, M.J.; Frizzell, K.M.; Yang, C.; Krishnakumar, R.; Kraus, W.L. The histone variant macroH2A1 marks repressed autosomal chromatin, but protects a subset of its target genes from silencing. Genes Dev. 2010, 24, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Filipescu, D.; Andrade, J.; Gaspar-Maia, A.; Ueberheide, B.; Bernstein, E. Transcription-associated histone pruning demarcates macroH2A chromatin domains. Nat. Struct. Mol. Biol. 2018, 25, 958–970. [Google Scholar] [CrossRef]
- Costanzi, C.; Pehrson, J.R. Histone macroH2A1 is concentrated in the inactive X chromosome of female mammals. Nature 1998, 393, 599–601. [Google Scholar] [CrossRef]
- Creppe, C.; Posavec, M.; Douet, J.; Buschbeck, M. MacroH2A in stem cells: A story beyond gene repression. Epigenomics 2012, 4, 221–227. [Google Scholar] [CrossRef]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, P.D.; Gamble, M.J. MacroH2A1 chromatin specification requires its docking domain and acetylation of H2B lysine 20. Nat. Commun. 2018, 9, 5143. [Google Scholar] [CrossRef]
- Gonzalez-Sandoval, A.; Towbin, B.D.; Kalck, V.; Cabianca, D.S.; Gaidatzis, D.; Hauer, M.H.; Geng, L.; Wang, L.; Yang, T.; Wang, X.; et al. Perinuclear Anchoring of H3K9-Methylated Chromatin Stabilizes Induced Cell Fate in C. elegans Embryos. Cell 2015, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlowski, M.; Corujo, D.; Hothorn, M.; Guberovic, I.; Mandemaker, I.K.; Blessing, C.; Sporn, J.; Gutierrez Triana, A.; Smith, R.; Portmann, T.; et al. MacroH2A histone variants limit chromatin plasticity through two distinct mechanisms. EMBO Rep. 2018, 19, e44445. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Shin, Y.; Lee, S.; Kim, M.; Punj, V.; Lu, J.F.; Shin, H.; Kim, K.; Ulmer, T.S.; Koh, J.; et al. Regulation of Breast Cancer-Induced Osteoclastogenesis by MacroH2A1.2 Involving EZH2-Mediated H3K27me3. CellReports 2018, 24, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.C.; Kustatscher, G.; Hothorn, T.; Collado, M.; Serrano, M.; Muley, T.; Schnabel, P.; Ladurner, A.G. Histone macroH2A isoforms predict the risk of lung cancer recurrence. Oncogene 2009, 28, 3423–3428. [Google Scholar] [CrossRef] [Green Version]
- Sporn, J.C.; Jung, B. Differential Regulation and Predictive Potential of MacroH2A1 Isoforms in Colon Cancer. AJPA 2012, 180, 2516–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ashraf, J.M.; Nahm, S.S.; Kim, Y.W.; Park, S.Y.; Choi, I. Fibromodulin: A master regulator of myostatin controlling progression of satellite cells through a myogenic program. FASEB J. 2016, 30, 2708–2719. [Google Scholar] [CrossRef]
- Grassot, V.; Da Silva, A.; Saliba, J.; Maftah, A.; Dupuy, F.; Petit, J.-M. Highlights of glycosylation and adhesion related genes involved in myogenesis. BMC Genom. 2014, 15, 621. [Google Scholar] [CrossRef] [Green Version]
- Grzelkowska-Kowalczyk, K. The Importance of Extracellular Matrix in Skeletal Muscle Development and Function. In Composition and Function of the Extracellular Matrix in the Human Body; InTechOpen: London, UK, 2016. [Google Scholar]
- Kim, T.; Ahmad, K.; Shaikh, S.; Jan, A.T.; Seo, M.-G.; Lee, E.J.; Choi, I. Dermatopontin in Skeletal Muscle Extracellular Matrix Regulates Myogenesis. Cells 2019, 8, 332. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hurtado-Bagès, S.; Posavec Marjanovic, M.; Valero, V.; Malinverni, R.; Corujo, D.; Bouvet, P.; Lavigne, A.-C.; Bystricky, K.; Buschbeck, M. The Histone Variant MacroH2A1 Regulates Key Genes for Myogenic Cell Fusion in a Splice-Isoform Dependent Manner. Cells 2020, 9, 1109. https://doi.org/10.3390/cells9051109
Hurtado-Bagès S, Posavec Marjanovic M, Valero V, Malinverni R, Corujo D, Bouvet P, Lavigne A-C, Bystricky K, Buschbeck M. The Histone Variant MacroH2A1 Regulates Key Genes for Myogenic Cell Fusion in a Splice-Isoform Dependent Manner. Cells. 2020; 9(5):1109. https://doi.org/10.3390/cells9051109
Chicago/Turabian StyleHurtado-Bagès, Sarah, Melanija Posavec Marjanovic, Vanesa Valero, Roberto Malinverni, David Corujo, Philippe Bouvet, Anne-Claire Lavigne, Kerstin Bystricky, and Marcus Buschbeck. 2020. "The Histone Variant MacroH2A1 Regulates Key Genes for Myogenic Cell Fusion in a Splice-Isoform Dependent Manner" Cells 9, no. 5: 1109. https://doi.org/10.3390/cells9051109
APA StyleHurtado-Bagès, S., Posavec Marjanovic, M., Valero, V., Malinverni, R., Corujo, D., Bouvet, P., Lavigne, A.-C., Bystricky, K., & Buschbeck, M. (2020). The Histone Variant MacroH2A1 Regulates Key Genes for Myogenic Cell Fusion in a Splice-Isoform Dependent Manner. Cells, 9(5), 1109. https://doi.org/10.3390/cells9051109