CUL4-DDB1-CRBN E3 Ubiquitin Ligase Regulates Proteostasis of ClC-2 Chloride Channels: Implication for Aldosteronism and Leukodystrophy

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. cDNA Constructs
2.2. Preparation of Animal Samples
2.3. Cell Culture and DNA Transfection
2.4. RNA Interference
2.5. Immunoblotting
2.6. Co-Immunoprecipitation
2.7. Cycloheximide Chase
2.8. Protein Ubiquitination Analyses
2.9. Immunofluorescence
2.10. Cell Surface Biotinylation
2.11. Statistical Analyses
3. Results
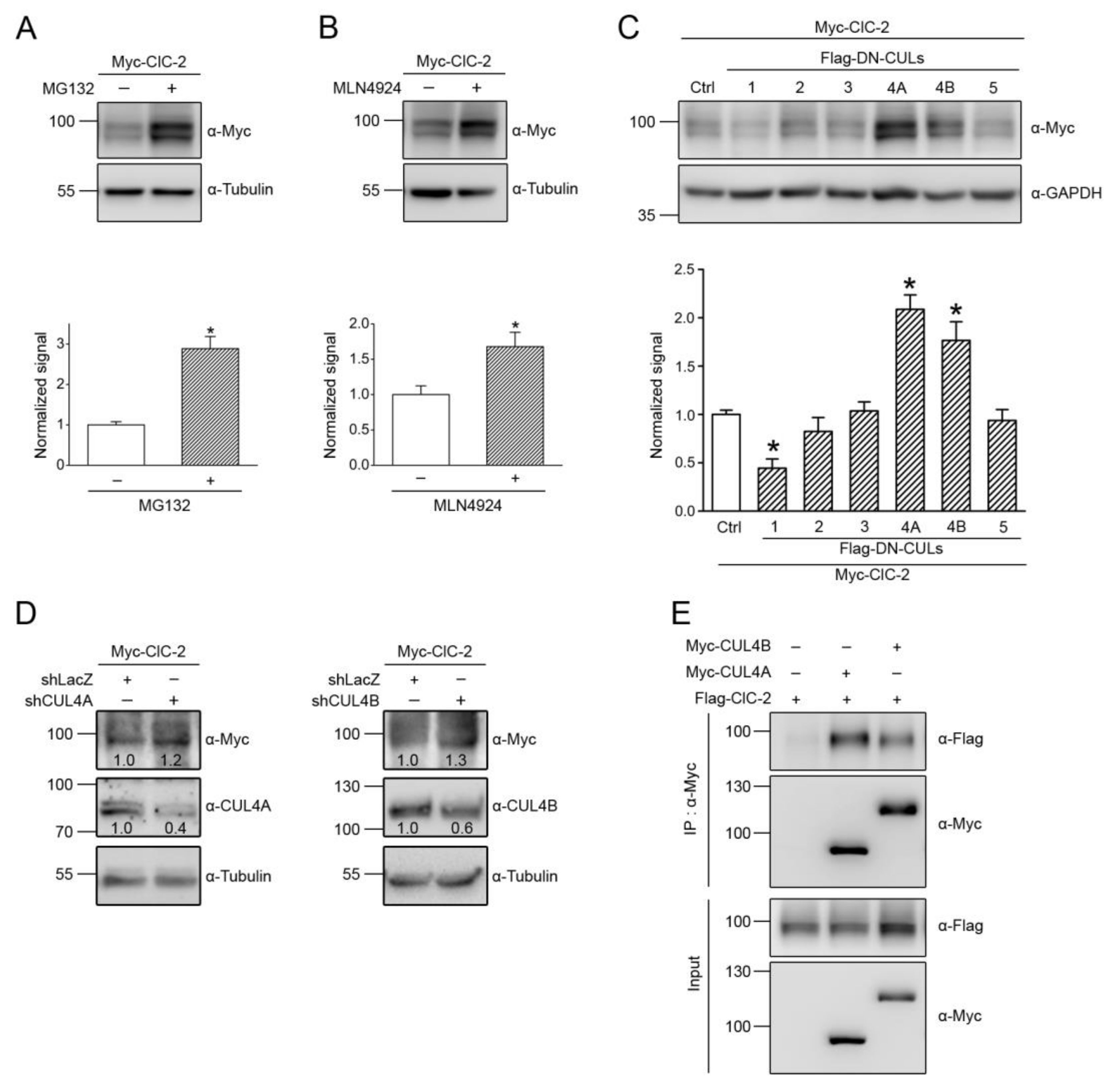
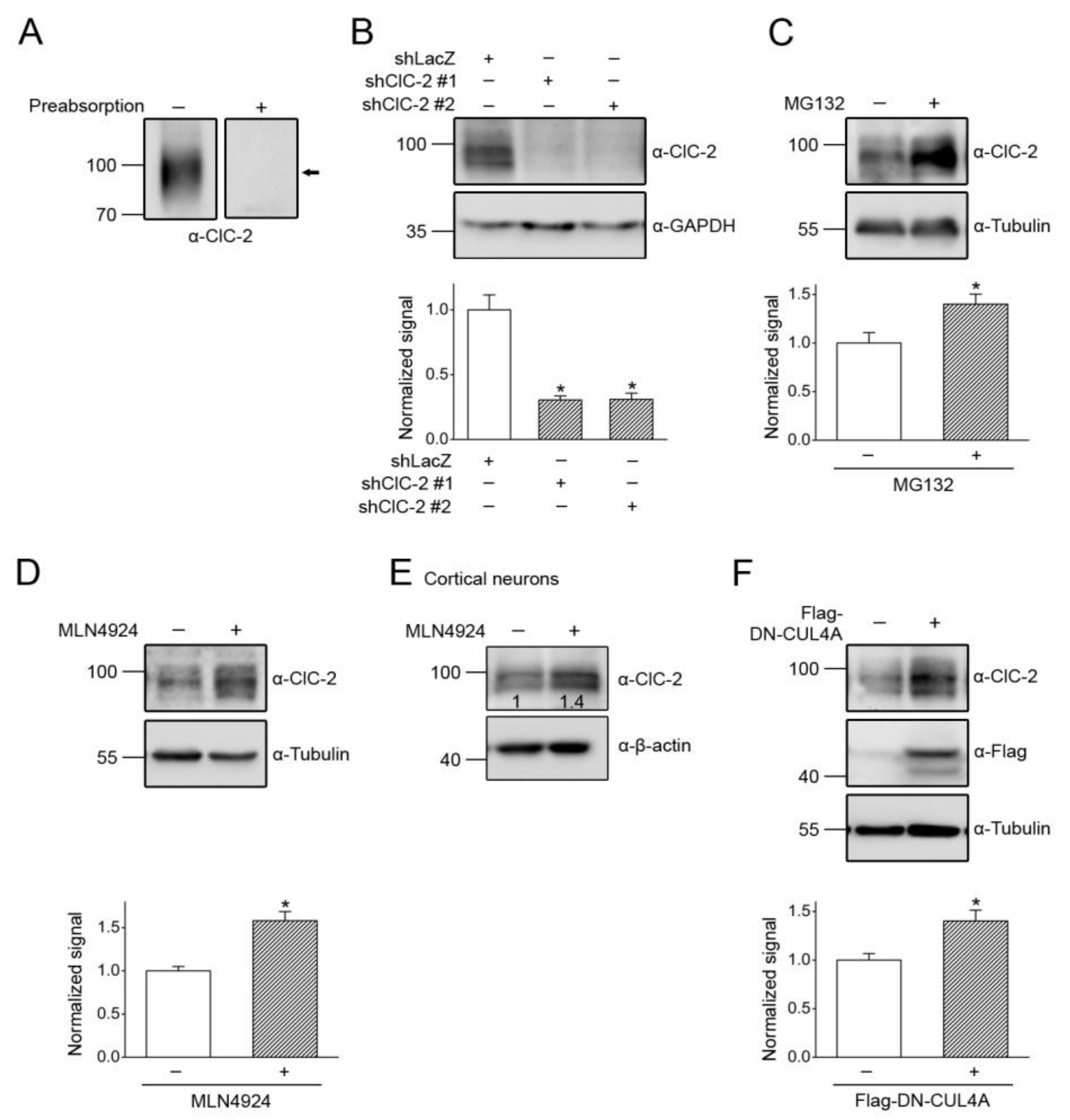
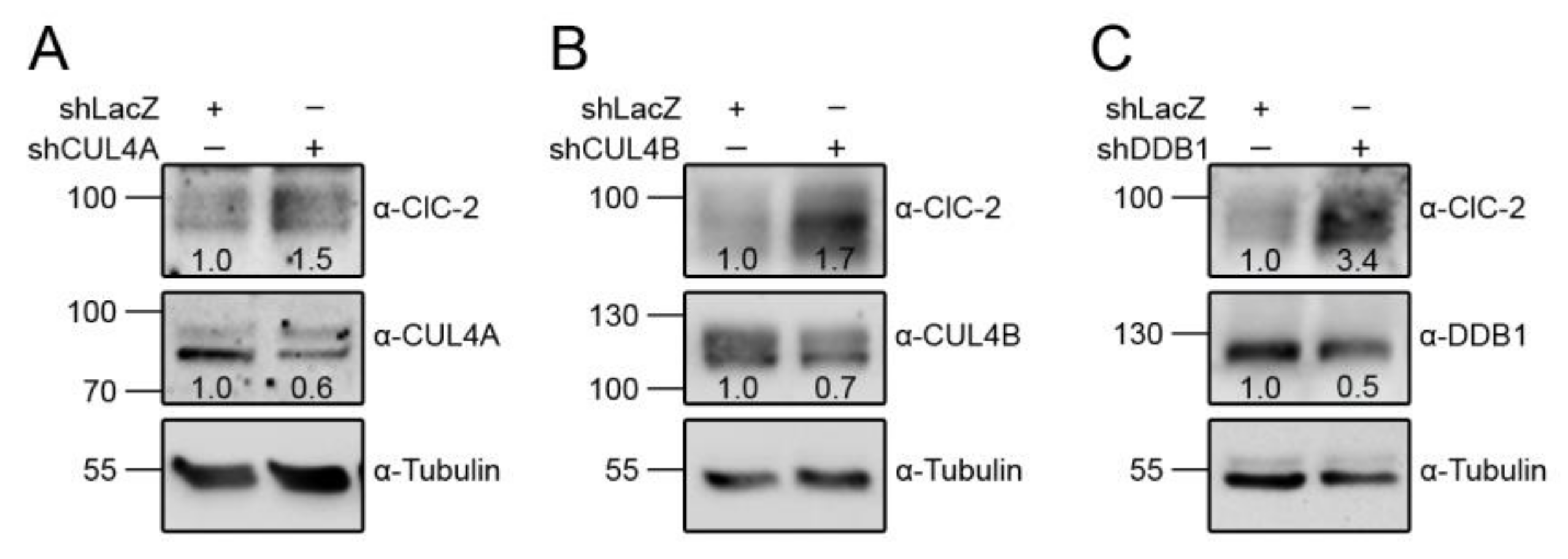
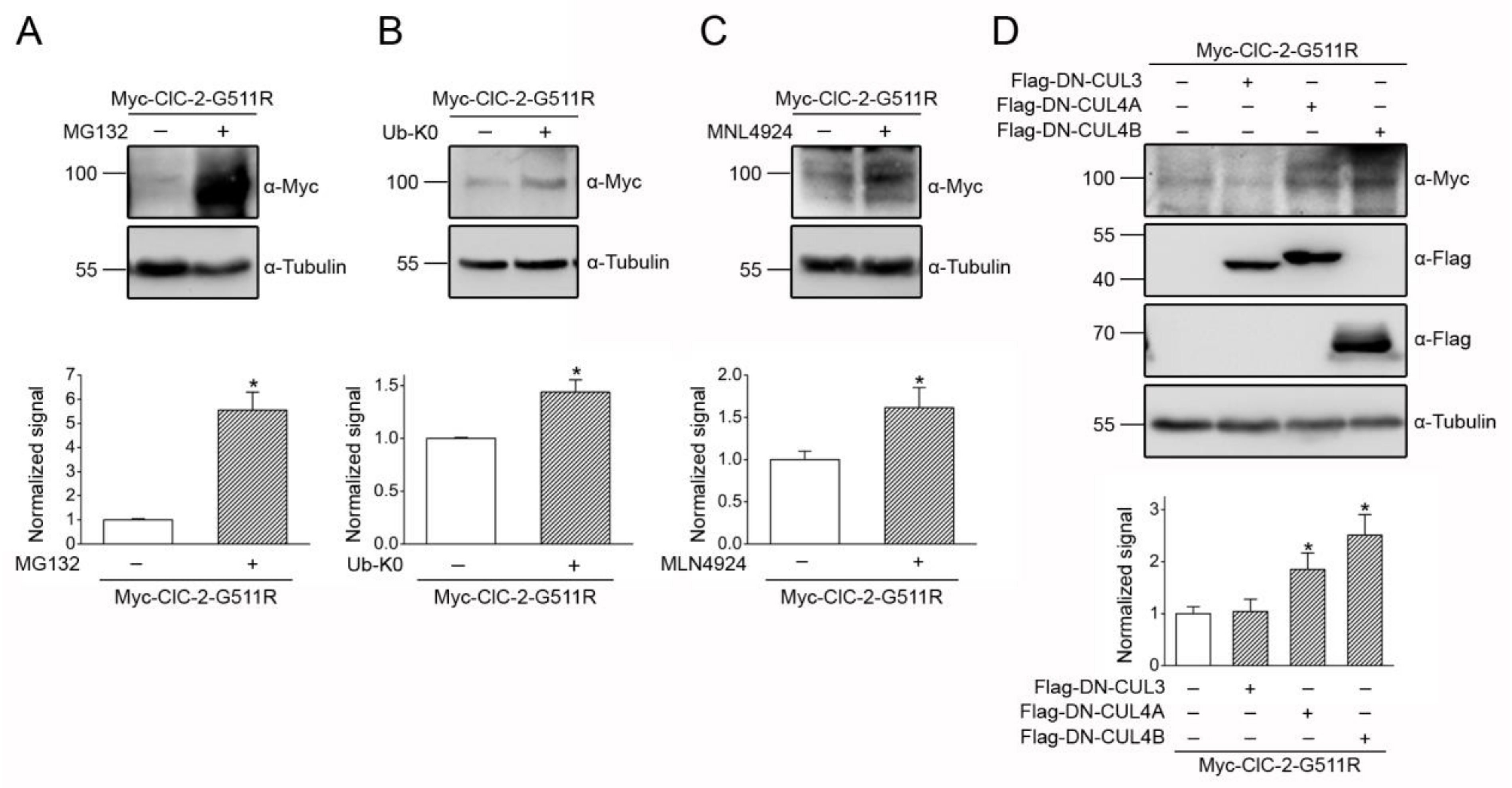
3.1. Proteasomal Degradation of ClC-2 is Mediated by Cullin 4 E3 Ubiquitin Ligase
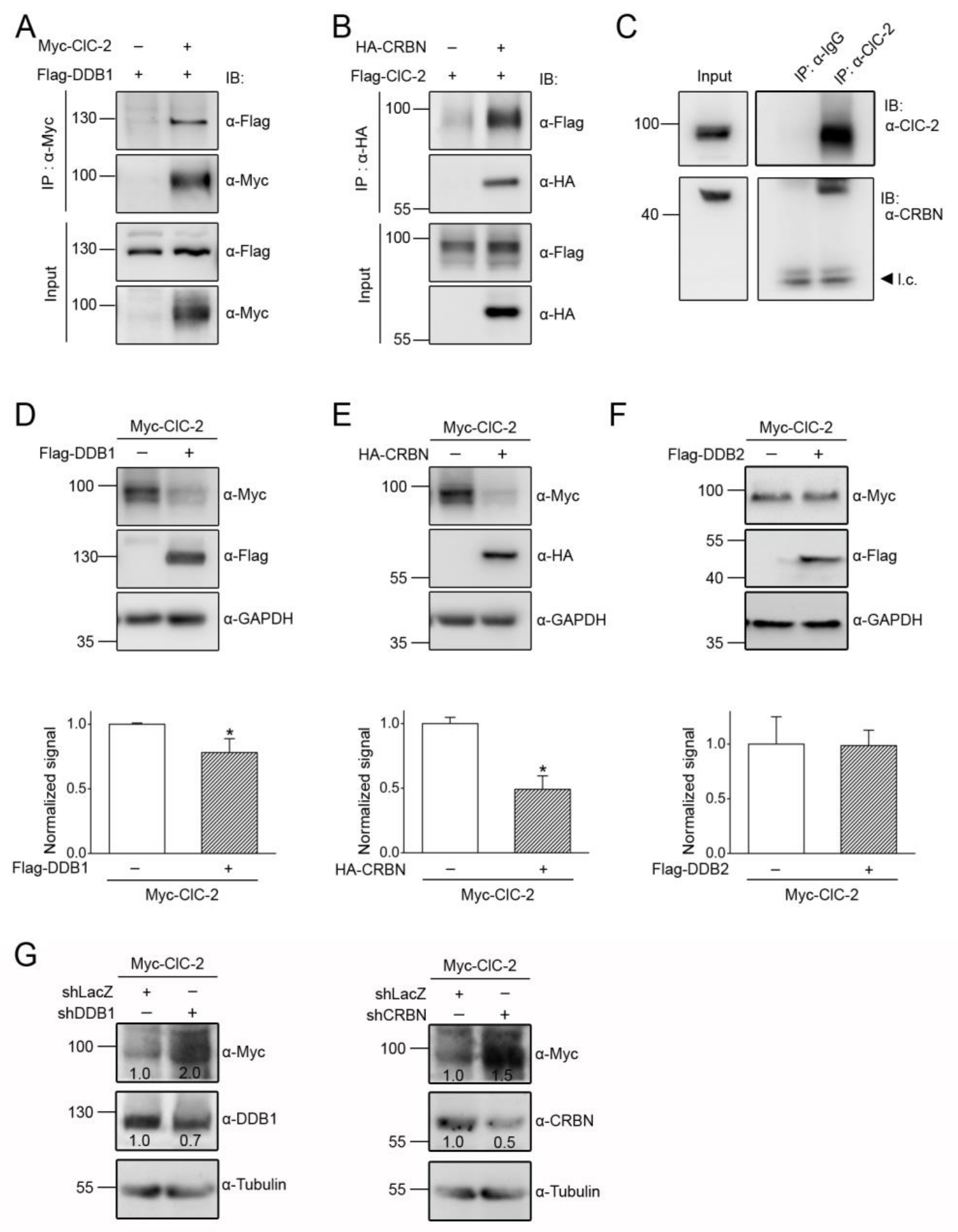
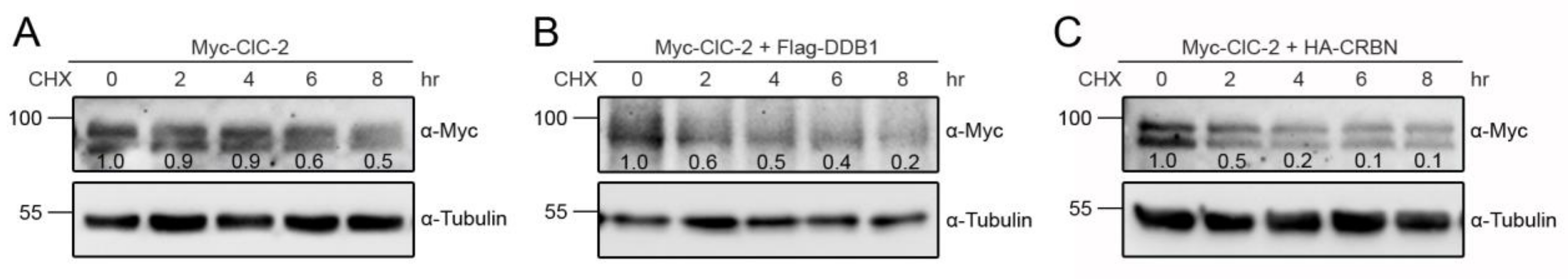
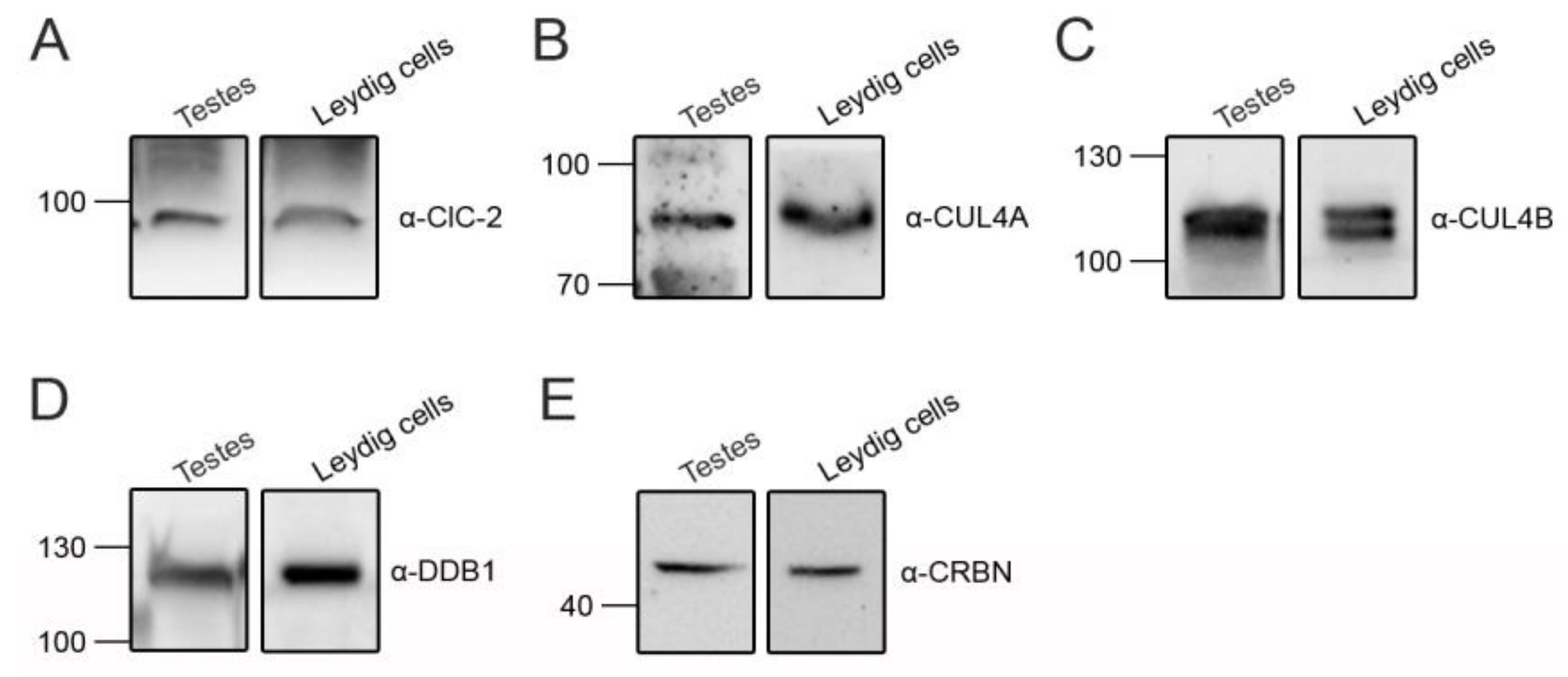
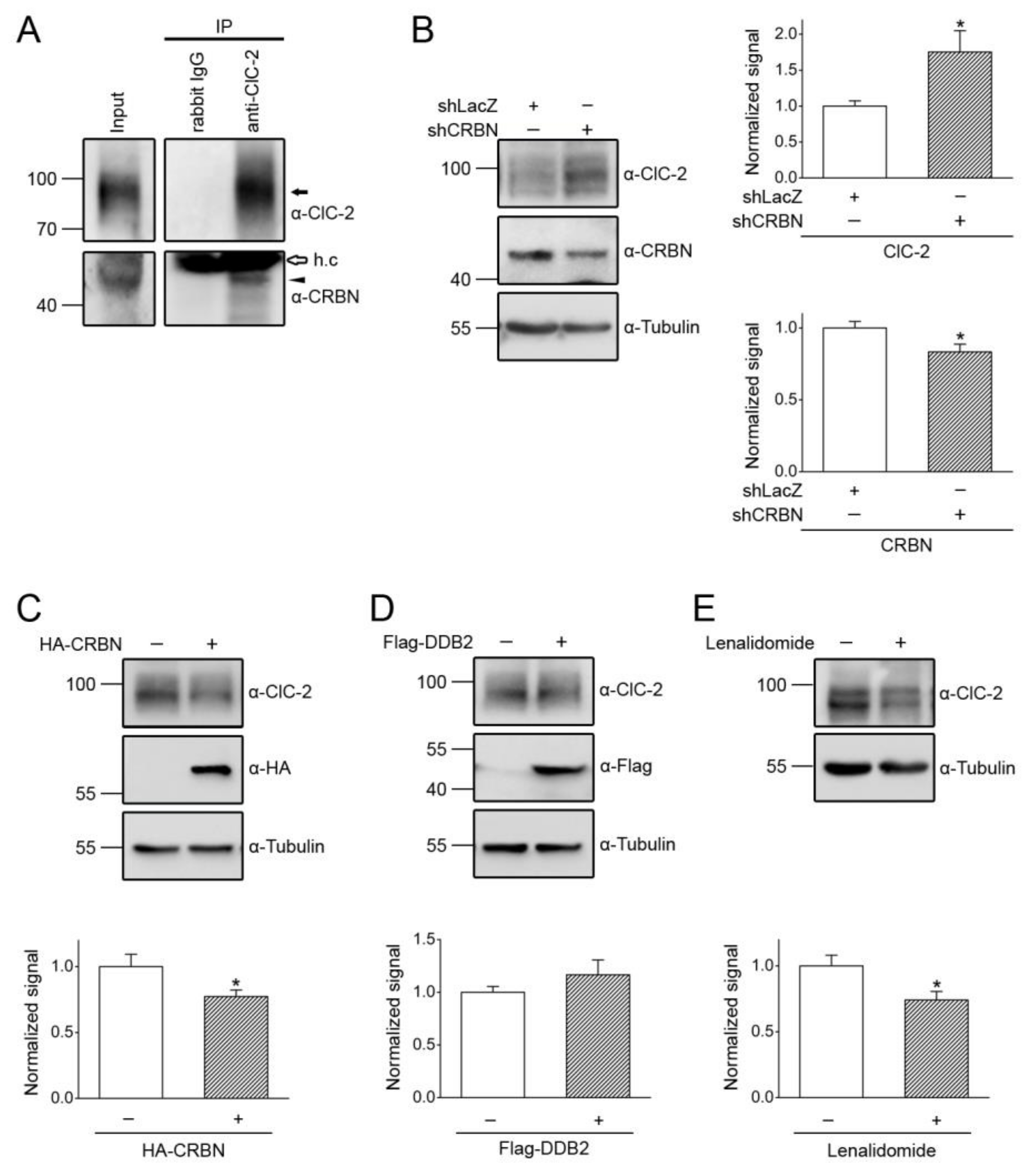
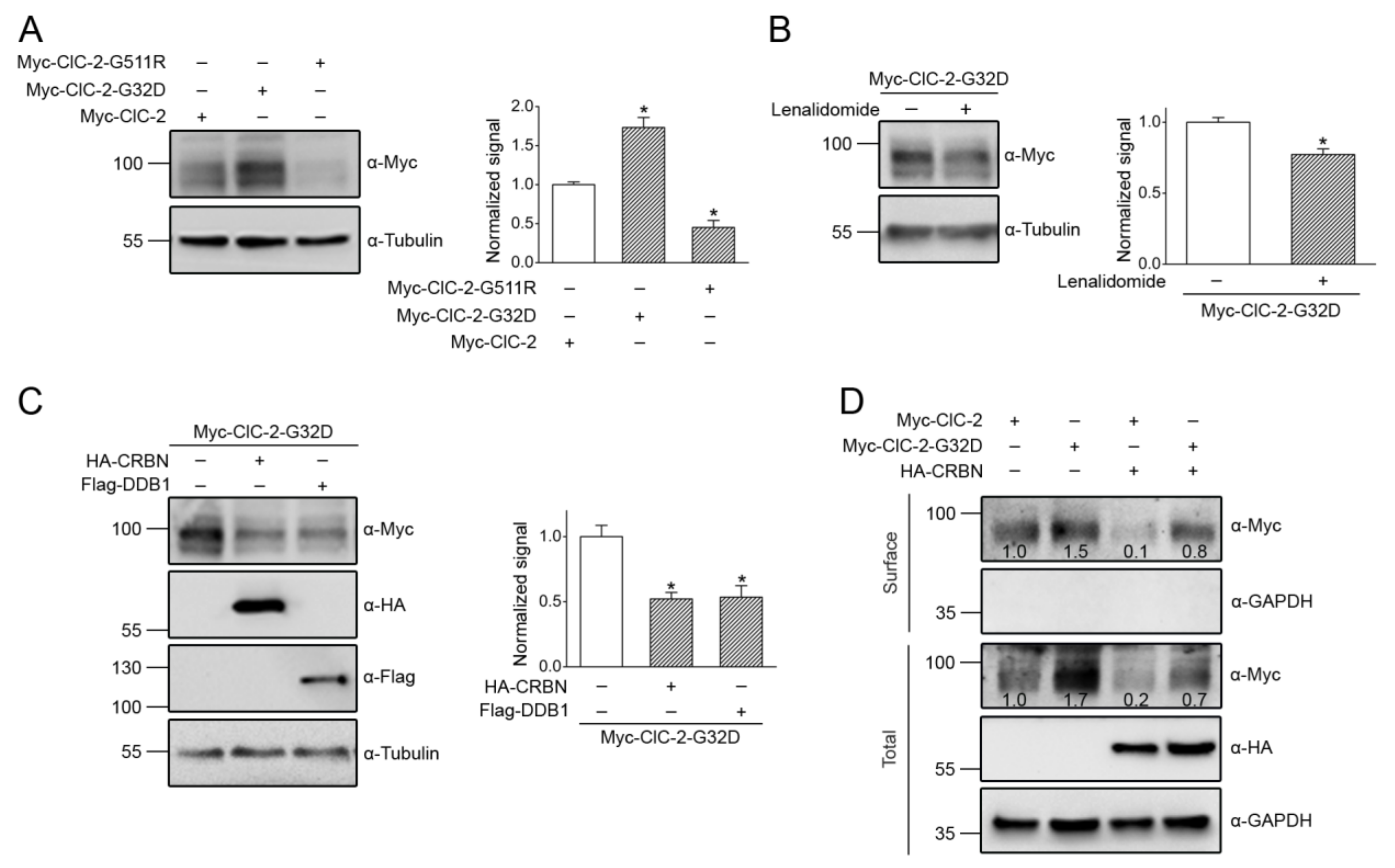
3.2. Cereblon Serves as the Substrate Receptor Protein of CUL4 E3 Ligase Complex for ClC-2 Degradation
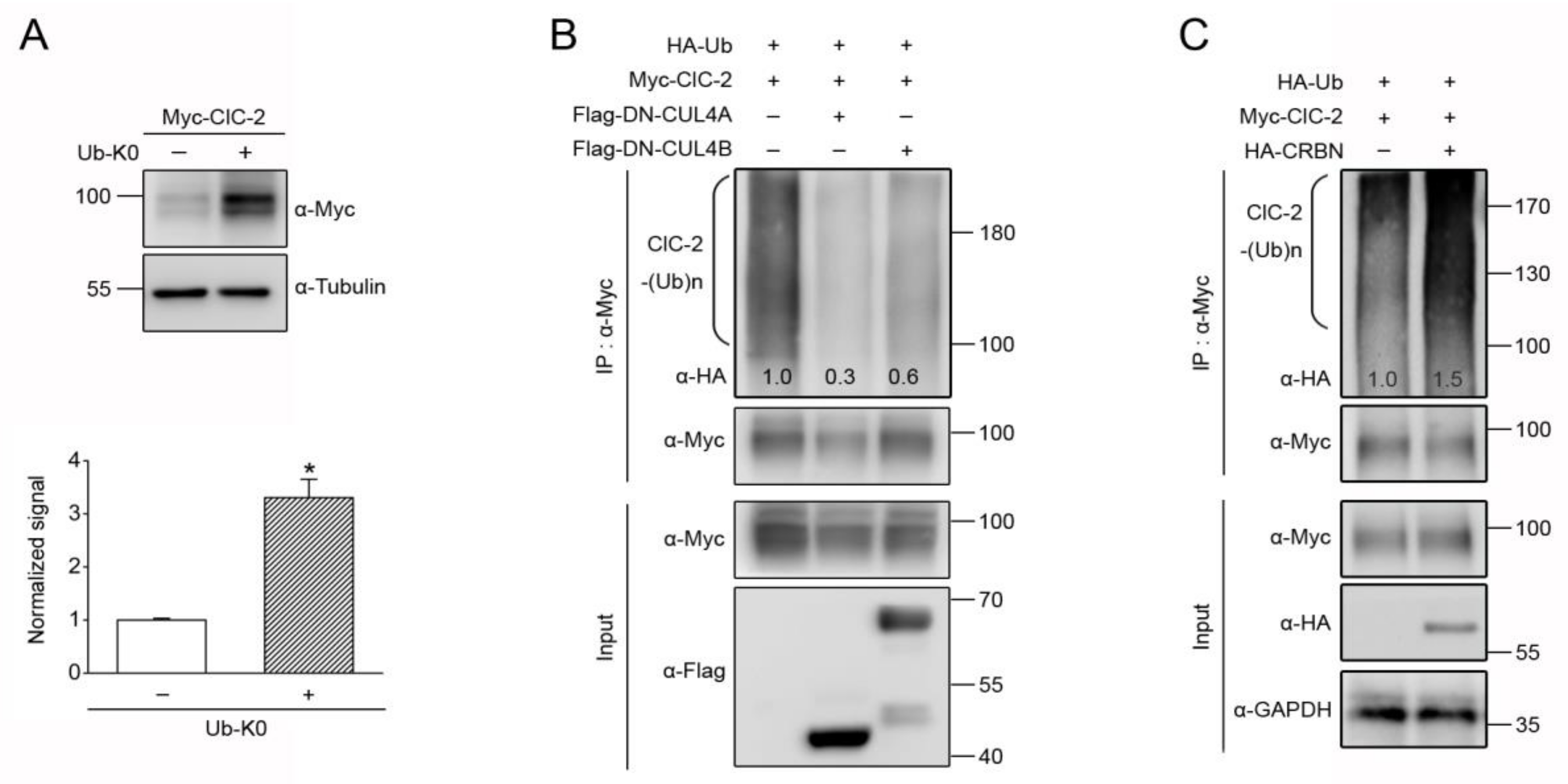
3.3. CUL4 E3 Ligase Mediates Polyubiquitinaion of ClC-2
3.4. CUL4 E3 Ligase Regulates Endogenous ClC-2 Degradation
3.5. Correction of Disease-Associated ClC-2 Proteostasis Anomaly by Modifying CUL4 E3 Ligase Activity
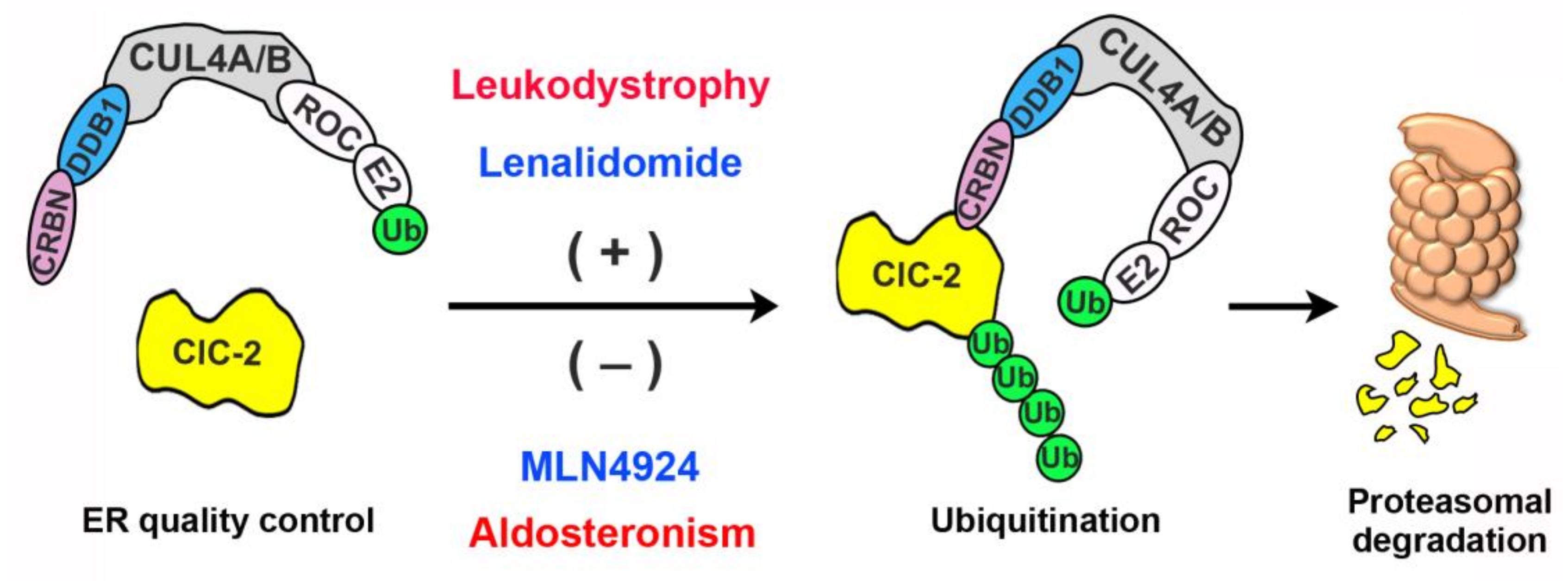
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Cl− | chloride |
| CRBN | cereblon |
| CUL | cullin |
| DDB | damage-specific DNA binding protein |
| D-PBS | Dulbecco’s phosphate buffered saline |
| DIV | days in vitro |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DTT | dithiothreitol |
| ER | endoplasmic reticulum |
| FBS | Fetal bovine serum |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase |
| HEK | human embryonic kidney |
| IP | immunoprecipitation |
| MLC1 | megalencephalic leukoencephalopathy with subcortical cysts 1 |
| PBS | Phosphate buffered saline |
| PMSF | phenylmethylsulfonyl fluoride |
| RING | really interesting new gene |
| Ub | ubiquitin |
| Ub-K0 | lysine-less ubiquitin |
| WT | wild-type. |
References
- Cid, L.P.; Montrose-Rafizadeh, C.; Smith, D.I.; Guggino, W.B.; Cutting, G.R. Cloning of a putative human voltage-gated chloride channel (CIC-2) cDNA widely expressed in human tissues. Hum. Mol. Genet. 1995, 4, 407–413. [Google Scholar] [CrossRef]
- Thiemann, A.; Grunder, S.; Pusch, M.; Jentsch, T.J. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature 1992, 356, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Sik, A.; Smith, R.L.; Freund, T.F. Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience 2000, 101, 51–65. [Google Scholar] [CrossRef]
- Gyomorey, K.; Yeger, H.; Ackerley, C.; Garami, E.; Bear, C.E. Expression of the chloride channel ClC-2 in the murine small intestine epithelium. Am. J. Physiol. Cell Physiol. 2000, 279, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, T.J.; Pusch, M. CLC Chloride Channels and Transporters: Structure, Function, Physiology, and Disease. Physiol. Rev. 2018, 98, 1493–1590. [Google Scholar] [CrossRef] [PubMed]
- Grunder, S.; Thiemann, A.; Pusch, M.; Jentsch, T.J. Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature 1992, 360, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Jordt, S.E.; Jentsch, T.J. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 1997, 16, 1582–1592. [Google Scholar] [CrossRef]
- Nehrke, K.; Arreola, J.; Nguyen, H.V.; Pilato, J.; Richardson, L.; Okunade, G.; Baggs, R.; Shull, G.E.; Melvin, J.E. Loss of hyperpolarization-activated Cl(-) current in salivary acinar cells from Clcn2 knockout mice. J. Biol. Chem. 2002, 277, 23604–23611. [Google Scholar] [CrossRef] [Green Version]
- Bösl, M.R.; Stein, V.; Hübner, C.; Zdebik, A.A.; Jordt, S.E.; Mukhopadhyay, A.K.; Davidoff, M.S.; Holstein, A.-F.; Jentsch, T.J. Male germ cells and photoreceptors, both dependent on close cell-cell interactions, degenerate upon ClC-2 Cl(-) channel disruption. EMBO J. 2001, 20, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Blanz, J.; Schweizer, M.; Auberson, M.; Maier, H.; Muenscher, A.; Hubner, C.A.; Jentsch, T.J. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci. 2007, 27, 6581–6589. [Google Scholar] [CrossRef] [Green Version]
- Schewe, J.; Seidel, E.; Forslund, S.; Marko, L.; Peters, J.; Muller, D.N.; Fahlke, C.; Stölting, G.; Scholl, U. Elevated aldosterone and blood pressure in a mouse model of familial hyperaldosteronism with ClC-2 mutation. Nat. Commun. 2019, 10, 5155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göppner, C.; Orozco, I.J.; Hoegg-Beiler, M.B.; Soria, A.H.; Hübner, C.A.; Fernandes-Rosa, F.L.; Boulkroun, S.; Zennaro, M.-C.; Jentsch, T.J. Pathogenesis of hypertension in a mouse model for human CLCN2 related hyperaldosteronism. Nat. Commun. 2019, 10, 4678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholl, U.I.; Stölting, G.; Schewe, J.; Thiel, A.; Tan, H.; Nelson-Williams, C.; Vichot, A.A.; Jin, S.C.; Loring, E.; Untiet, V.; et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet. 2018, 50, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Rosa, F.L.; Daniil, G.; Orozco, I.J.; Göppner, C.; El Zein, R.; Jain, V.; Boulkroun, S.; Jeunemaitre, X.; Amar, L.; Lefebvre, H.; et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet. 2018, 50, 355–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowasser, M.; Wolley, M.; Wu, A.; Gordon, R.D.; Schewe, J.; Stolting, G.; Scholl, U.I. Pathogenesis of Familial Hyperaldosteronism Type II: New Concepts Involving Anion Channels. Curr. Hypertens. Rep. 2019, 21, 31. [Google Scholar] [CrossRef]
- Depienne, C.; Bugiani, M.; Dupuits, C.; Galanaud, D.; Touitou, V.; Postma, N.; van Berker, C.; Polder, E.; Tollard, E.; Darios, F.; et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: An observational analytical study. Lancet Neurol. 2013, 12, 659–668. [Google Scholar] [CrossRef]
- Guo, Z.; Lu, T.; Peng, L.; Cheng, H.; Peng, F.; Li, J.; Lu, Z.; Chen, S.; Qiu, W. CLCN2-related leukoencephalopathy: A case report and review of the literature. BMC Neurol. 2019, 19, 156. [Google Scholar] [CrossRef]
- Gaitán-Peñas, H.; Apaja, P.M.; Arnedo, T.; Castellanos, A.; Elorza-Vidal, X.; Soto, D.; Gasull, X.; Lukacs, G.L.; Estévez, R. Leukoencephalopathy-causing CLCN2 mutations are associated with impaired Cl (-) channel function and trafficking. J. Physiol. 2017, 595, 6993–7008. [Google Scholar] [CrossRef] [Green Version]
- Jeworutzki, E.; López-Hernández, T.; Capdevila-Nortes, X.; Sirisi, S.; Bengtsson, L.; Montolio, M.; Zifarelli, G.; Arnedo, T.; Müller, C.S.; Schulte, U.; et al. GlialCAM, a protein defective in a leukodystrophy, serves as a ClC-2 Cl (-) channel auxiliary subunit. Neuron 2012, 73, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Hoegg-Beiler, M.B.; Sirisi, S.; Orozco, I.J.; Ferrer, I.; Hohensee, S.; Auberson, M.; Gödde, K.; Vilches, C.; de Heredia, M.L.; Nunes, V.; et al. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat. Commun. 2014, 5, 3475. [Google Scholar] [CrossRef] [Green Version]
- Sirisi, S.; Elorza-Vidal, X.; Arnedo, T.; Armand-Ugón, M.; Callejo, G.; Capdevila-Nortes, X.; López-Hernández, T.; Schulte, U.; Barrallo-Gimeno, A.; Nunes, V.; et al. Depolarization causes the formation of a ternary complex between GlialCAM, MLC1 and ClC-2 in astrocytes: Implications in megalencephalic leukoencephalopathy. Hum. Mol. Genet. 2017, 26, 2436–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bella, D.; Pareyson, D.; Savoiardo, M.; Farina, L.; Ciano, C.; Caldarazzo, S.; Sagnelli, A.; Bonato, S.; Nava, S.; Bresolin, N.; et al. Subclinical leukodystrophy and infertility in a man with a novel homozygous CLCN2 mutation. Neurology 2014, 83, 1217–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [Green Version]
- Bagola, K.; Mehnert, M.; Jarosch, E.; Sommer, T. Protein dislocation from the ER. Biochim. Biophys. Acta 2011, 1808, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Guerriero, C.J.; Brodsky, J.L. The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 2012, 92, 537–576. [Google Scholar] [CrossRef]
- Claessen, J.H.; Kundrat, L.; Ploegh, H.L. Protein quality control in the ER: Balancing the ubiquitin checkbook. Trends Cell Biol. 2012, 22, 22–32. [Google Scholar] [CrossRef]
- Banker, G.; Goslin, K. Culturing Nerve Cells; MIT Press: Cambridge, MA, USA, 1998. [Google Scholar]
- Jeng, C.J.; Chang, C.C.; Tang, C.Y. Differential localization of rat Eag1 and Eag2 K+ channels in hippocampal neurons. Neuroreport 2005, 16, 229–233. [Google Scholar] [CrossRef]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Deshaies, R.J.; Joazeiro, C.A. RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 2009, 78, 399–434. [Google Scholar] [CrossRef] [PubMed]
- MacGurn, J.A.; Hsu, P.C.; Emr, S.D. Ubiquitin and membrane protein turnover: From cradle to grave. Annu. Rev. Biochem. 2012, 81, 231–259. [Google Scholar] [CrossRef] [PubMed]
- Duda, D.M.; Scott, D.C.; Calabrese, M.F.; Zimmerman, E.S.; Zheng, N.; Schulman, B.A. Structural regulation of cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 2011, 21, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarikas, A.; Hartmann, T.; Pan, Z.Q. The cullin protein family. Genome Biol. 2011, 12, 220. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, E.S.; Schulman, B.A.; Zheng, N. Structural assembly of cullin-RING ubiquitin ligase complexes. Curr. Opin. Struct. Biol. 2010, 20, 714–721. [Google Scholar] [CrossRef] [Green Version]
- Brownell, J.E.; Sintchak, M.D.; Gavin, J.M.; Liao, H.; Bruzzese, F.J.; Bump, N.J.; Soucy, T.A.; Milhollen, M.A.; Yang, X.; Burkhardt, A.L.; et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: The NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell 2010, 37, 102–111. [Google Scholar] [CrossRef]
- Soucy, T.A.; Smith, P.G.; Milhollen, M.A.; Berger, A.J.; Gavin, J.M.; Adhikari, S.; Brownell, J.E.; Burke, K.E.; Cardin, D.P.; Critchley, S.; et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 2009, 458, 732–736. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Kobayashi, R.; Grewal, S.I. Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat. Cell Biol. 2005, 7, 1007–1013. [Google Scholar] [CrossRef]
- Jin, J.; Ang, X.L.; Shirogane, T.; Wade Harper, J. Identification of substrates for F-box proteins. Methods Enzymol. 2005, 399, 287–309. [Google Scholar] [PubMed]
- Jackson, S.; Xiong, Y. CRL4s: The CUL4-RING E3 ubiquitin ligases. Trends. Biochem. Sci. 2009, 34, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iovine, B.; Iannella, M.L.; Bevilacqua, M.A. Damage-specific DNA binding protein 1 (DDB1): A protein with a wide range of functions. Int. J. Biochem. Cell Biol. 2011, 43, 1664–1667. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Zhou, P. DCAFs, the missing link of the CUL4-DDB1 ubiquitin ligase. Mol. Cell 2007, 26, 775–780. [Google Scholar] [CrossRef]
- He, Y.J.; McCall, C.M.; Hu, J.; Zeng, Y.; Xiong, Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes. Dev. 2006, 20, 2949–2954. [Google Scholar] [CrossRef] [Green Version]
- Higa, L.A.; Wu, M.; Ye, T.; Kobayashi, R.; Sun, H.; Zhang, H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat. Cell Biol. 2006, 8, 1277–1283. [Google Scholar] [CrossRef]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [Green Version]
- Hohberger, B.; Enz, R. Cereblon is expressed in the retina and binds to voltage-gated chloride channels. FEBS Lett. 2009, 583, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Abriel, H.; Staub, O. Ubiquitylation of ion channels. Physiology 2005, 20, 398–407. [Google Scholar] [CrossRef]
- Kravtsova-Ivantsiv, Y.; Ciechanover, A. Non-canonical ubiquitin-based signals for proteasomal degradation. J. Cell Sci. 2012, 125, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volk, S.; Wang, M.; Pickart, C.M. Chemical and genetic strategies for manipulating polyubiquitin chain structure. Methods Enzymol. 2005, 399, 3–20. [Google Scholar] [PubMed]
- Bloom, J.; Amador, V.; Bartolini, F.; DeMartino, G.; Pagano, M. Proteasome-mediated degradation of p21 via N-terminal ubiquitinylation. Cell 2003, 115, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N.A.; Huhtaniemi, I.T. Testicular cell lines. Mol. Cell Endocrinol. 2004, 228, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Zirkin, B.R.; Papadopoulos, V. Leydig cells: Formation, function, and regulation. Biol. Reprod. 2018, 99, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Krönke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.; Middleton, R.E.; Sun, H.; Naniong, M.; Ott, C.J.; Mitsiades, C.S.; Wong, K.-K.; Bradner, J.E.; Kaelin, W.G. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Science 2014, 343, 305–309. [Google Scholar] [CrossRef] [Green Version]
- Palmada, M.; Dieter, M.; Boehmer, C.; Waldegger, S.; Lang, F. Serum and glucocorticoid inducible kinases functionally regulate ClC-2 channels. Biochem. Biophys. Res. Commun. 2004, 321, 1001–1006. [Google Scholar] [CrossRef]
- Chen, T.Y. Structure and function of clc channels. Annu. Rev. Physiol. 2005, 67, 809–839. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Poet, M.; Fuhrmann, J.C.; Zdebik, A.A. Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Annu. Rev. Physiol. 2005, 67, 779–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, C.J.; Fu, S.J.; You, C.Y.; Peng, Y.J.; Hsiao, C.T.; Chen, T.Y.; Tang, C.Y. Defective Gating and Proteostasis of Human ClC-1 Chloride Channel: Molecular Pathophysiology of Myotonia Congenita. Front. Neurol. 2020, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.A.; Peng, Y.J.; Hu, M.C.; Huang, J.J.; Chien, Y.C.; Wu, J.T.; Chen, T.Y.; Tang, C.Y. The Cullin 4A/B-DDB1-Cereblon E3 Ubiquitin Ligase Complex Mediates the Degradation of CLC-1 Chloride Channels. Sci. Rep. 2015, 5, 10667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hipp, M.S.; Park, S.H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends. Cell. Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef]
- Lindquist, S.L.; Kelly, J.W. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: Progress and prognosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004507. [Google Scholar] [CrossRef]
- Tao, Y.X.; Conn, P.M. Pharmacoperones as Novel Therapeutics for Diverse Protein Conformational Diseases. Physiol. Rev. 2018, 98, 697–725. [Google Scholar] [CrossRef]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Kortum, K.M.; Zhu, Y.X.; Shi, C.X.; Jedlowski, P.; Stewart, A.K. Cereblon binding molecules in multiple myeloma. Blood Rev. 2015, 29, 329–334. [Google Scholar] [CrossRef]
- Soucy, T.A.; Dick, L.R.; Smith, P.G.; Milhollen, M.A.; Brownell, J.E. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes. Cancer 2010, 1, 708–716. [Google Scholar] [CrossRef] [Green Version]
- McMillin, D.W.; Jacobs, H.M.; Delmore, J.E.; Buon, L.; Hunter, Z.R.; Monrose, V.; Yu, J.; Smith, P.G.; Richardson, P.G.; Anderson, K.C.; et al. Molecular and cellular effects of NEDD8-activating enzyme inhibition in myeloma. Mol. Cancer Ther. 2012, 11, 942–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Nakatani, T.; Kamitani, T. Inhibition of NEDD8-conjugation pathway by novel molecules: Potential approaches to anticancer therapy. Mol. Oncol. 2012, 6, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.J.; Li, J. Proteasome Inhibitors: Harnessing Proteostasis to Combat Disease. Molecules 2020, 25, 671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcaterra, N.E.; Hoeppner, D.J.; Wei, H.; Jaffe, A.E.; Maher, B.J.; Barrow, J.C. Schizophrenia-Associated hERG channel Kv11.1-3.1 Exhibits a Unique Trafficking Deficit that is Rescued Through Proteasome Inhibition for High Throughput Screening. Sci. Rep. 2016, 6, 19976. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, S.-J.; Hu, M.-C.; Peng, Y.-J.; Fang, H.-Y.; Hsiao, C.-T.; Chen, T.-Y.; Jeng, C.-J.; Tang, C.-Y. CUL4-DDB1-CRBN E3 Ubiquitin Ligase Regulates Proteostasis of ClC-2 Chloride Channels: Implication for Aldosteronism and Leukodystrophy. Cells 2020, 9, 1332. https://doi.org/10.3390/cells9061332
Fu S-J, Hu M-C, Peng Y-J, Fang H-Y, Hsiao C-T, Chen T-Y, Jeng C-J, Tang C-Y. CUL4-DDB1-CRBN E3 Ubiquitin Ligase Regulates Proteostasis of ClC-2 Chloride Channels: Implication for Aldosteronism and Leukodystrophy. Cells. 2020; 9(6):1332. https://doi.org/10.3390/cells9061332
Chicago/Turabian StyleFu, Ssu-Ju, Meng-Chun Hu, Yi-Jheng Peng, Hsin-Yu Fang, Cheng-Tsung Hsiao, Tsung-Yu Chen, Chung-Jiuan Jeng, and Chih-Yung Tang. 2020. "CUL4-DDB1-CRBN E3 Ubiquitin Ligase Regulates Proteostasis of ClC-2 Chloride Channels: Implication for Aldosteronism and Leukodystrophy" Cells 9, no. 6: 1332. https://doi.org/10.3390/cells9061332
APA StyleFu, S.-J., Hu, M.-C., Peng, Y.-J., Fang, H.-Y., Hsiao, C.-T., Chen, T.-Y., Jeng, C.-J., & Tang, C.-Y. (2020). CUL4-DDB1-CRBN E3 Ubiquitin Ligase Regulates Proteostasis of ClC-2 Chloride Channels: Implication for Aldosteronism and Leukodystrophy. Cells, 9(6), 1332. https://doi.org/10.3390/cells9061332