Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases


{kind=link}
Abstract
1. Introduction
2. Purinergic Signaling
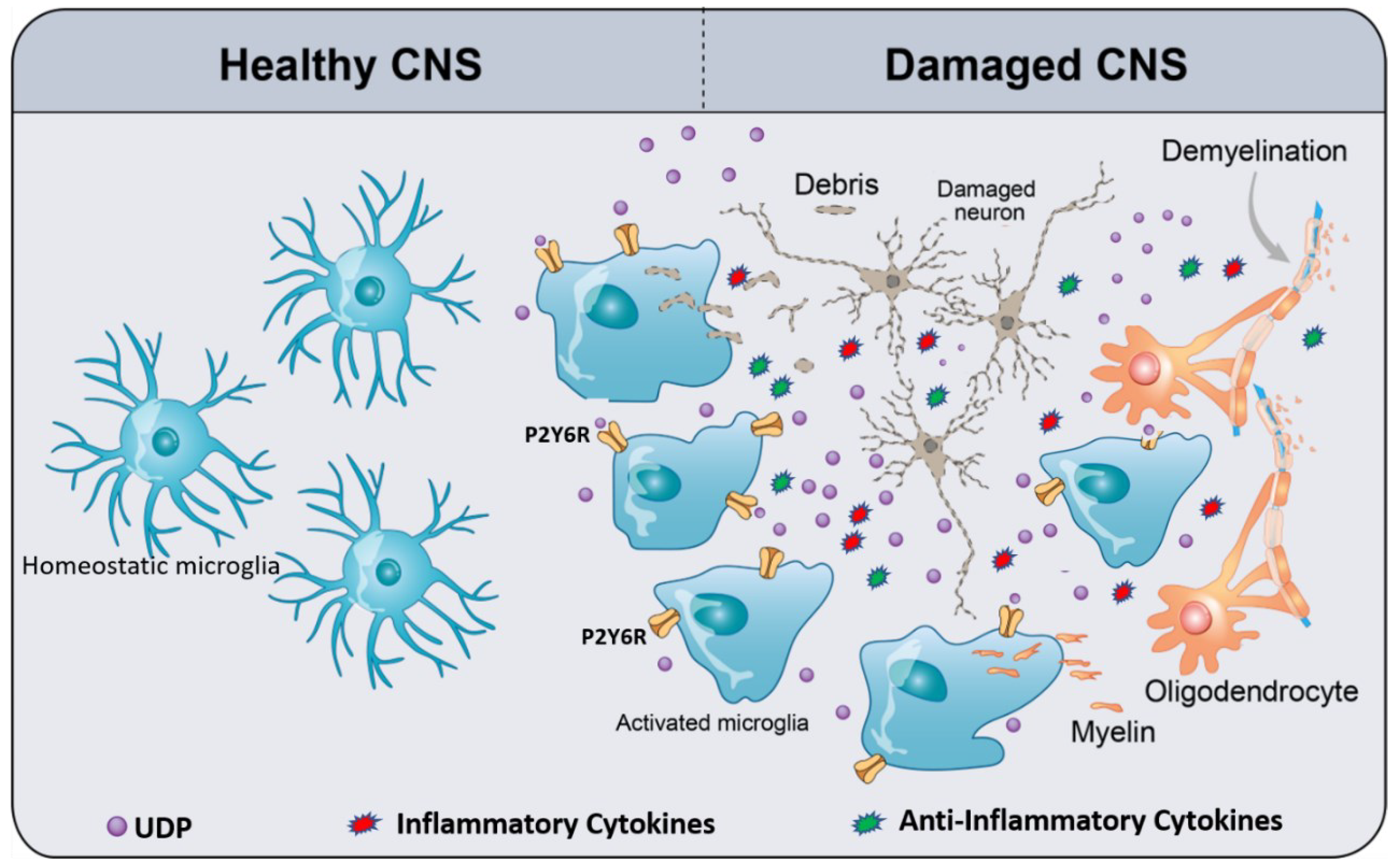
3. Microglia P2Y6R
4. Pathophysiology and Therapeutic Potential of P2Y6R
5. P2Y6R as a Potential Target in Neurological Diseases
5.1. Ischemic Stroke
5.2. Alzheimer’s Disease
5.3. Parkinson’s Disease
5.4. Radiation-Induced Brain Injury
5.5. Neuropathic Pain
6. Current Limitations and Prospects
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| Abbreviations | Definition |
| CNS | Central nervous system |
| DAMPs | Disease-associated molecular patterns |
| ATP | Adenosine triphosphate |
| UTP | Uridine triphosphate |
| GPCRs | G protein-coupled receptors |
| IP3 | Inositol trisphosphate |
| Il-6 | Interleukin-6 |
| LPS | Lipopolysaccharides |
| Rac-1 | Ras-related C3 botulinum toxin substrate 1 |
| AKT | Protein kinase B |
| PC-PLC | Phosphatidylcholine-specific phospholipase C |
| PKC | Protein kinase C |
| tMCAO | Transient middle cerebral artery occlusion |
| Iba1 | Ionized calcium-binding adaptor-1 |
| Aβ | Amyloid-Beta |
| SNpc | Substantia nigra pars compacta |
| NP | Neuropathic pain |
| CCI | Chronic constriction injury |
| IHC | Immunohistochemistry |
| PAMPs | Pathogen associated molecular patterns |
| TLRs | Toll-like receptors |
| ADP | Adenosine diphosphate |
| UDP | Uridine diphosphate |
| PLC | Phospholipase C |
| IL-1 | Interleukin-1 |
| TNF-α | Tumor necrosis factor |
| TGF-β | Transforming growth factor-beta |
| M-CSF | Macrophage colony-stimulating factor |
| CMML | Chronic myelomonocytic leukemia |
| PI-PLC | Phosphoinositide-specific phospholipase C |
| ERK | Extracellular signal-regulated kinases |
| AD | Alzheimer’s disease |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| PD | Parkinson’s disease |
| MLCK | Myosin light chain kinase |
| SD | Sprague–Dawley |
| WB | Western blot |
| NMDG1 | N-methyl-D-glucamine-1 |
References
- Herz, J.; Filiano, A.J.; Smith, A.; Yogev, N.; Kipnis, J. Myeloid Cells in the Central Nervous System. Immunity 2017, 46, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef]
- Ginhoux, F.; Garel, S. The mysterious origins of microglia. Nat. Neurosci. 2018, 21, 897–899. [Google Scholar] [CrossRef]
- Tay, T.L.; Savage, J.C.; Hui, C.W.; Bisht, K.; Tremblay, M.-È. Microglia across the lifespan: From origin to function in brain development, plasticity and cognition. J. Physiol. 2017, 595, 1929–1945. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharm. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Prinz, M. Microglia in steady state. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Anwar, S.; Rivest, S. Alzheimer’s disease: Microglia targets and their modulation to promote amyloid phagocytosis and mitigate neuroinflammation. Expert Opin. Ther. Targets 2020, 24, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Garaschuk, O.; Verkhratsky, A. Physiology of Microglia. Methods Mol. Biol. 2019, 2034, 27–40. [Google Scholar] [CrossRef]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- Kigerl, K.A.; de Rivero Vaccari, J.P.; Dietrich, W.D.; Popovich, P.G.; Keane, R.W. Pattern recognition receptors and central nervous system repair. Exp. Neurol. 2014, 258, 5–16. [Google Scholar] [CrossRef]
- Voet, S.; Prinz, M.; van Loo, G. Microglia in Central Nervous System Inflammation and Multiple Sclerosis Pathology. Trends Mol. Med. 2019, 25, 112–123. [Google Scholar] [CrossRef]
- Lecours, C.; Bordeleau, M.; Cantin, L.; Parent, M.; Paolo, T.D.; Tremblay, M.-È. Microglial Implication in Parkinson’s Disease: Loss of Beneficial Physiological Roles or Gain of Inflammatory Functions? Front. Cell. Neurosci. 2018, 12, 282. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.-Q.; Ma, X.-T.; Hu, Z.-W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.-J.; Tian, D.-S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933. [Google Scholar] [CrossRef]
- Haukedal, H.; Freude, K. Implications of Microglia in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. J. Mol. Biol. 2019, 431, 1818–1829. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Mariani, M.M.; Kielian, T. Microglia in infectious diseases of the central nervous system. J. Neuroimmune Pharm. 2009, 4, 448–461. [Google Scholar] [CrossRef]
- Nau, R.; Ribes, S.; Djukic, M.; Eiffert, H. Strategies to increase the activity of microglia as efficient protectors of the brain against infections. Front. Cell. Neurosci. 2014, 8, 138. [Google Scholar] [CrossRef] [PubMed]
- Färber, K.; Kettenmann, H. Purinergic signaling and microglia. Pflug. Arch. Eur. J. Physiol. 2006, 452, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic nerves. Pharmacol. Rev. 1972, 24, 509–581. [Google Scholar]
- Lüthje, J. Origin, metabolism and function of extracellular adenine nucleotides in the blood. Klin. Wochenschr. 1989, 67, 317–327. [Google Scholar] [CrossRef]
- Burnstock, G. Discovery of purinergic signalling, the initial resistance and current explosion of interest. Br. J. Pharmacol. 2012, 167, 238–255. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purinergic receptors. J. Theor. Biol. 1976, 62, 491–503. [Google Scholar] [CrossRef]
- Burnstock, G.; Approach, H.A.M. A basis for distinguishing two types of purinergic receptor. In Cell Membrane Receptors for Drugs and Hormone: A Multidisciplinary Approach; Raven Press: New York, NY, USA, 1978; pp. 107–118. [Google Scholar]
- Burnstock, G.; Kennedy, C. Is there a basis for distinguishing two types of P2-purinoceptor? Gen. Pharmacol. 1985, 16, 433–440. [Google Scholar] [CrossRef]
- Saul, A.; Hausmann, R.; Kless, A.; Nicke, A. Heteromeric assembly of P2X subunits. Front. Cell. Neurosci. 2013, 7, 250. [Google Scholar] [CrossRef]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.-M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, C.; Jacobson, K.A.; et al. International Union of Pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef]
- Trautmann, A. Extracellular ATP in the immune system: More than just a “danger signal”. Sci. Signal. 2009, 2, pe6. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2, 2398212818817494. [Google Scholar] [CrossRef]
- Oliveira-Giacomelli, Á.; Naaldijk, Y.; Sardá-Arroyo, L.; Gonçalves, M.C.B.; Corrêa-Velloso, J.; Pillat, M.M.; de Souza, H.D.N.; Ulrich, H. Purinergic Receptors in Neurological Diseases With Motor Symptoms: Targets for Therapy. Front. Pharmacol. 2018, 9, 325. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.-D.; Ding, J.-Q.; Xiao, Q.; Chen, S.-D. P2Y6 receptor and immunoinflammation. Neurosci. Bull. 2009, 25, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.E. Cellular distribution and functions of P2 receptor subtypes in different systems. Int. Rev. Cytol. 2004, 240, 31–304. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, J.; Wang, N.; Hao, G.; Sun, J. Characterization of UDP-Activated Purinergic Receptor P2Y6 Involved in Japanese Flounder Paralichthys olivaceus Innate Immunity. Int. J. Mol. Sci. 2018, 19, 2095. [Google Scholar] [CrossRef] [PubMed]
- Korcok, J.; Raimundo, L.N.; Du, X.; Sims, S.M.; Dixon, S.J. P2Y6 nucleotide receptors activate NF-kappaB and increase survival of osteoclasts. J. Biol. Chem. 2005, 280, 16909–16915. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Matsuoka, I. Inhibition of P2Y6 receptor-mediated phospholipase C activation and Ca(2+) signalling by prostaglandin E2 in J774 murine macrophages. Eur. J. Pharmacol. 2015, 749, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Turner, J.; Fountain, S.J. P2Y(2) and P2Y(6) receptor activation elicits intracellular calcium responses in human adipose-derived mesenchymal stromal cells. Purinergic Signal. 2018, 14, 371–384. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Inoue, K.; Koizumi, S.; Kataoka, A.; Tozaki-Saitoh, H.; Tsuda, M. P2Y(6)-Evoked Microglial Phagocytosis. Int. Rev. Neurobiol. 2009, 85, 159–163. [Google Scholar] [CrossRef]
- Inoue, K. UDP facilitates microglial phagocytosis through P2Y6 receptors. Cell Adh. Migr. 2007, 1, 131–132. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, W.; Liu, Y.; Xu, P.; Li, Z.; Wu, R.; Shi, X.; Tang, Y. P2Y6 Receptor-Mediated Microglial Phagocytosis in Radiation-Induced Brain Injury. Mol. Neurobiol. 2016, 53, 3552–3564. [Google Scholar] [CrossRef]
- Wendt, S.; Maricos, M.; Vana, N.; Meyer, N.; Guneykaya, D.; Semtner, M.; Kettenmann, H. Changes in phagocytosis and potassium channel activity in microglia of 5xFAD mice indicate alterations in purinergic signaling in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2017, 58, 41–53. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Ren, H.; Yue, M.; Huang, K.; Gu, H.; Liu, M.; Du, B.; Qian, M. P2Y(6) agonist uridine 5′-diphosphate promotes host defense against bacterial infection via monocyte chemoattractant protein-1-mediated monocytes/macrophages recruitment. J. Immunol. 2011, 186, 5376–5387. [Google Scholar] [CrossRef]
- Harry, G.J.; Kraft, A.D. Neuroinflammation and microglia: Considerations and approaches for neurotoxicity assessment. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.A.; Gomes, B.; Palmer, K.; Du, K.; Wiekowski, M.; Wilburn, B.; Petro, M.; Chou, C.-C.; Desquitado, C.; Schwarz, M.; et al. The pyrimidinergic P2Y6 receptor mediates a novel release of proinflammatory cytokines and chemokines in monocytic cells stimulated with UDP. Biochem. Biophys. Res. Commun. 2005, 330, 467–473. [Google Scholar] [CrossRef]
- Vieira, R.P.; Müller, T.; Grimm, M.; von Gernler, V.; Vetter, B.; Dürk, T.; Cicko, S.; Ayata, C.K.; Sorichter, S.; Robaye, B.; et al. Purinergic receptor type 6 contributes to airway inflammation and remodeling in experimental allergic airway inflammation. Am. J. Respir. Crit. Care Med. 2011, 184, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Warny, M.; Aboudola, S.; Robson, S.C.; Sévigny, J.; Communi, D.; Soltoff, S.P.; Kelly, C.P. P2Y(6) nucleotide receptor mediates monocyte interleukin-8 production in response to UDP or lipopolysaccharide. J. Biol. Chem. 2001, 276, 26051–26056. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Hornik, T.; Brown, G.C. Inhibition of UDP/P2Y6 purinergic signaling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 2014, 62, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Wen, R.-X.; Shen, H.; Huang, S.-X.; Wang, L.-P.; Li, Z.-W.; Peng, P.; Mamtilahun, M.; Tang, Y.-H.; Shen, F.-X.; Tian, H.-L.; et al. P2Y6 receptor inhibition aggravates ischemic brain injury by reducing microglial phagocytosis. CNS Neurosci. Ther. 2020, 26, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Madry, C.; Attwell, D. Receptors, ion channels, and signaling mechanisms underlying microglial dynamics. J. Biol. Chem. 2015, 290, 12443–12450. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Yamanaka, H.; Yanamoto, F.; Okubo, M.; Noguchi, K. Multiple P2Y subtypes in spinal microglia are involved in neuropathic pain after peripheral nerve injury. Glia 2012, 60, 1529–1539. [Google Scholar] [CrossRef]
- Bernier, L.P.; Ase, A.R.; Boué-Grabot, É.; Séguéla, P. Inhibition of P2 × 4 function by P2Y6 UDP receptors in microglia. Glia 2013, 61, 2038–2049. [Google Scholar] [CrossRef]
- Miller, A.M.; Stella, N. Microglial cell migration stimulated by ATP and C5a involve distinct molecular mechanisms: Quantification of migration by a novel near-infrared method. Glia 2009, 57, 875–883. [Google Scholar] [CrossRef]
- Ohsawa, K.; Irino, Y.; Nakamura, Y.; Akazawa, C.; Inoue, K.; Kohsaka, S. Involvement of P2 × 4 and P2Y12 receptors in ATP-induced microglial chemotaxis. Glia 2007, 55, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Navone, F.; Genevini, P.; Borgese, N.J.C. Autophagy and neurodegeneration: Insights from a cultured cell model of ALS. Cells 2015, 4, 354–386. [Google Scholar] [CrossRef]
- Vernon, P.J.; Tang, D. Eat-me: Autophagy, phagocytosis, and reactive oxygen species signaling. Antioxid. Redox Signal. 2013, 18, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Darios, F.; Stevanin, G. Impairment of Lysosome Function and Autophagy in Rare Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2714–2734. [Google Scholar] [CrossRef]
- Wirawan, E.; Vanden Berghe, T.; Lippens, S.; Agostinis, P.; Vandenabeele, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Eaten alive: A history of macroautophagy. Nat. Cell Biol. 2010, 12, 814–822. [Google Scholar] [CrossRef]
- Jacquel, A.; Benikhlef, N.; Paggetti, J.; Lalaoui, N.; Guery, L.; Dufour, E.K.; Ciudad, M.; Racoeur, C.; Micheau, O.; Delva, L.; et al. Colony-stimulating factor-1-induced oscillations in phosphatidylinositol-3 kinase/AKT are required for caspase activation in monocytes undergoing differentiation into macrophages. Blood 2009, 114, 3633–3641. [Google Scholar] [CrossRef]
- Jacquel, A.; Obba, S.; Boyer, L.; Dufies, M.; Robert, G.; Gounon, P.; Lemichez, E.; Luciano, F.; Solary, E.; Auberger, P. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood 2012, 119, 4527–4531. [Google Scholar] [CrossRef]
- Obba, S.; Hizir, Z.; Boyer, L.; Selimoglu-Buet, D.; Pfeifer, A.; Michel, G.; Hamouda, M.A.; Gonçalvès, D.; Cerezo, M.; Marchetti, S.; et al. The PRKAA1/AMPKα1 pathway triggers autophagy during CSF1-induced human monocyte differentiation and is a potential target in CMML. Autophagy 2015, 11, 1114–1129. [Google Scholar] [CrossRef]
- Kim, S.G.; Gao, Z.G.; Soltysiak, K.A.; Chang, T.S.; Brodie, C.; Jacobson, K.A. P2Y6 nucleotide receptor activates PKC to protect 1321N1 astrocytoma cells against tumor necrosis factor-induced apoptosis. Cell. Mol. Neurobiol. 2003, 23, 401–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-κB Signaling as a Therapeutic Target in Autoimmunity. J. Biomol. Screen. 2016, 21, 223–242. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in Cancer and Inflammatory Diseases and Their Therapeutic Approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [PubMed]
- Donkor, E.S. Stroke in the 21(st) Century: A Snapshot of the Burden, Epidemiology, and Quality of Life. Stroke Res. Treat. 2018, 2018, 3238165. [Google Scholar] [CrossRef]
- Weinstein, J.R.; Koerner, I.P.; Möller, T. Microglia in ischemic brain injury. Future Neurol. 2010, 5, 227–246. [Google Scholar] [CrossRef]
- Zhao, S.-C.; Ma, L.-S.; Chu, Z.-H.; Xu, H.; Wu, W.-Q.; Liu, F. Regulation of microglial activation in stroke. Acta Pharmacol. Sin. 2017, 38, 445–458. [Google Scholar] [CrossRef]
- da Fonseca, A.C.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R.S. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef]
- Guruswamy, R.; ElAli, A. Complex Roles of Microglial Cells in Ischemic Stroke Pathobiology: New Insights and Future Directions. Int. J. Mol. Sci. 2017, 18, 496. [Google Scholar] [CrossRef]
- Wang, M.-M.; Miao, D.; Cao, X.-P.; Tan, L.; Tan, L. Innate immune activation in Alzheimer’s disease. Ann. Transl. Med. 2018, 6, 177. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Cieślak, M.; Wojtczak, A. Role of purinergic receptors in the Alzheimer’s disease. Purinergic Signal. 2018, 14, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Fani Maleki, A.; Rivest, S. Innate Immune Cells: Monocytes, Monocyte-Derived Macrophages and Microglia as Therapeutic Targets for Alzheimer’s Disease and Multiple Sclerosis. Front. Cell. Neurosci. 2019, 13, 355. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Woods, L.T.; Ajit, D.; Camden, J.M.; Erb, L.; Weisman, G.A. Purinergic receptors as potential therapeutic targets in Alzheimer’s disease. Neuropharmacology 2016, 104, 169–179. [Google Scholar] [CrossRef]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s disease. Subcell Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Dunbar, G.L. Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl. Neurodegener. 2017, 6, 28. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat. Disord. 2004, 10 (Suppl. 1), S3–S7. [Google Scholar] [CrossRef] [PubMed]
- Tansey, M.G.; Goldberg, M.S. Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol. Dis. 2010, 37, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Arru, G.; Hosseini, S.; Niegowska, M.; Sechi, G.; Zarbo, I.R.; Sechi, L.A. Inflammation, Infectious Triggers, and Parkinson’s Disease. Front. Neurol. 2019, 10, 122. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Lou, Y.; Liu, G.; Wang, X.; Qian, Y.; Ding, J.; Chen, S.; Xiao, Q. Microglia P2Y6 receptor is related to Parkinson’s disease through neuroinflammatory process. J. Neuroinflamm. 2017, 14, 38. [Google Scholar] [CrossRef] [PubMed]
- Ampil, F.L.; Kim, D.D.; Ghali, G.E.; Baluna, R.G. How intensive should radiotherapy for head and neck cancer with synchronous distant metastases be? Review of cases. J. Oral Maxillofac. Surg. 2012, 70, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Welsh, L.C.; Dunlop, A.W.; McGovern, T.; McQuaid, D.; Dean, J.A.; Gulliford, S.L.; Bhide, S.A.; Harrington, K.J.; Nutting, C.M.; Newbold, K.L. Neurocognitive function after (chemo)-radiotherapy for head and neck cancer. Clin. Oncol. 2014, 26, 765–775. [Google Scholar] [CrossRef]
- Dietrich, J.; Monje, M.; Wefel, J.; Meyers, C. Clinical patterns and biological correlates of cognitive dysfunction associated with cancer therapy. Oncologist 2008, 13, 1285–1295. [Google Scholar] [CrossRef]
- Lumniczky, K.; Szatmári, T.; Sáfrány, G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517. [Google Scholar] [CrossRef]
- Greene-Schloesser, D.; Robbins, M.E.; Peiffer, A.M.; Shaw, E.G.; Wheeler, K.T.; Chan, M.D. Radiation-induced brain injury: A review. Front. Oncol. 2012, 2, 73. [Google Scholar] [CrossRef]
- Liu, B.; Hong, J.S. Role of microglia in inflammation-mediated neurodegenerative diseases: Mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003, 304, 1–7. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Colloca, L.; Ludman, T.; Bouhassira, D.; Baron, R.; Dickenson, A.H.; Yarnitsky, D.; Freeman, R.; Truini, A.; Attal, N.; Finnerup, N.B.; et al. Neuropathic pain. Nat. Rev. Dis. Primers 2017, 3, 17002. [Google Scholar] [CrossRef] [PubMed]
- Finnerup, N.B.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef]
- Ji, R.-R.; Nackley, A.; Huh, Y.; Terrando, N.; Maixner, W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology 2018, 129, 343–366. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef]
- Bian, J.; Zhang, Y.; Liu, Y.; Li, Q.; Tang, H.-B.; Liu, Q. P2Y6 Receptor-Mediated Spinal Microglial Activation in Neuropathic Pain. Pain Res. Manag. 2019, 2019, 2612534. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, W.; Li, Y.; Zhang, Q.; Ji, H.; Li, H.; Hu, Q. The role of P2Y(6)R in cardiovascular diseases and recent development of P2Y(6)R antagonists. Drug Discov. Today 2020, 25, 568–573. [Google Scholar] [CrossRef]
- Yu, W.; Hill, W.G. Lack of specificity shown by P2Y6 receptor antibodies. Naunyn Schmiedebergs Arch. Pharmacol. 2013, 386, 885–891. [Google Scholar] [CrossRef]
- Garcia, R.A.; Yan, M.; Search, D.; Zhang, R.; Carson, N.L.; Ryan, C.S.; Smith-Monroy, C.; Zheng, J.; Chen, J.; Kong, Y.; et al. P2Y6 receptor potentiates pro-inflammatory responses in macrophages and exhibits differential roles in atherosclerotic lesion development. PLoS ONE 2014, 9, e111385. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, Y.; Liu, Y. The Role f Nucleotides and Purinergic Signaling in Apoptotic Cell Clearance–Implications for Chronic Inflammatory Diseases. Front. Immunol. 2014, 5, 656. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anwar, S.; Pons, V.; Rivest, S. Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases. Cells 2020, 9, 1595. https://doi.org/10.3390/cells9071595
Anwar S, Pons V, Rivest S. Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases. Cells. 2020; 9(7):1595. https://doi.org/10.3390/cells9071595
Chicago/Turabian StyleAnwar, Shehata, Vincent Pons, and Serge Rivest. 2020. "Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases" Cells 9, no. 7: 1595. https://doi.org/10.3390/cells9071595
APA StyleAnwar, S., Pons, V., & Rivest, S. (2020). Microglia Purinoceptor P2Y6: An Emerging Therapeutic Target in CNS Diseases. Cells, 9(7), 1595. https://doi.org/10.3390/cells9071595