Regulation of Histone Ubiquitination in Response to DNA Double Strand Breaks


Abstract
1. Introduction
2. Histone Ubiquitination as a Response to DSBs
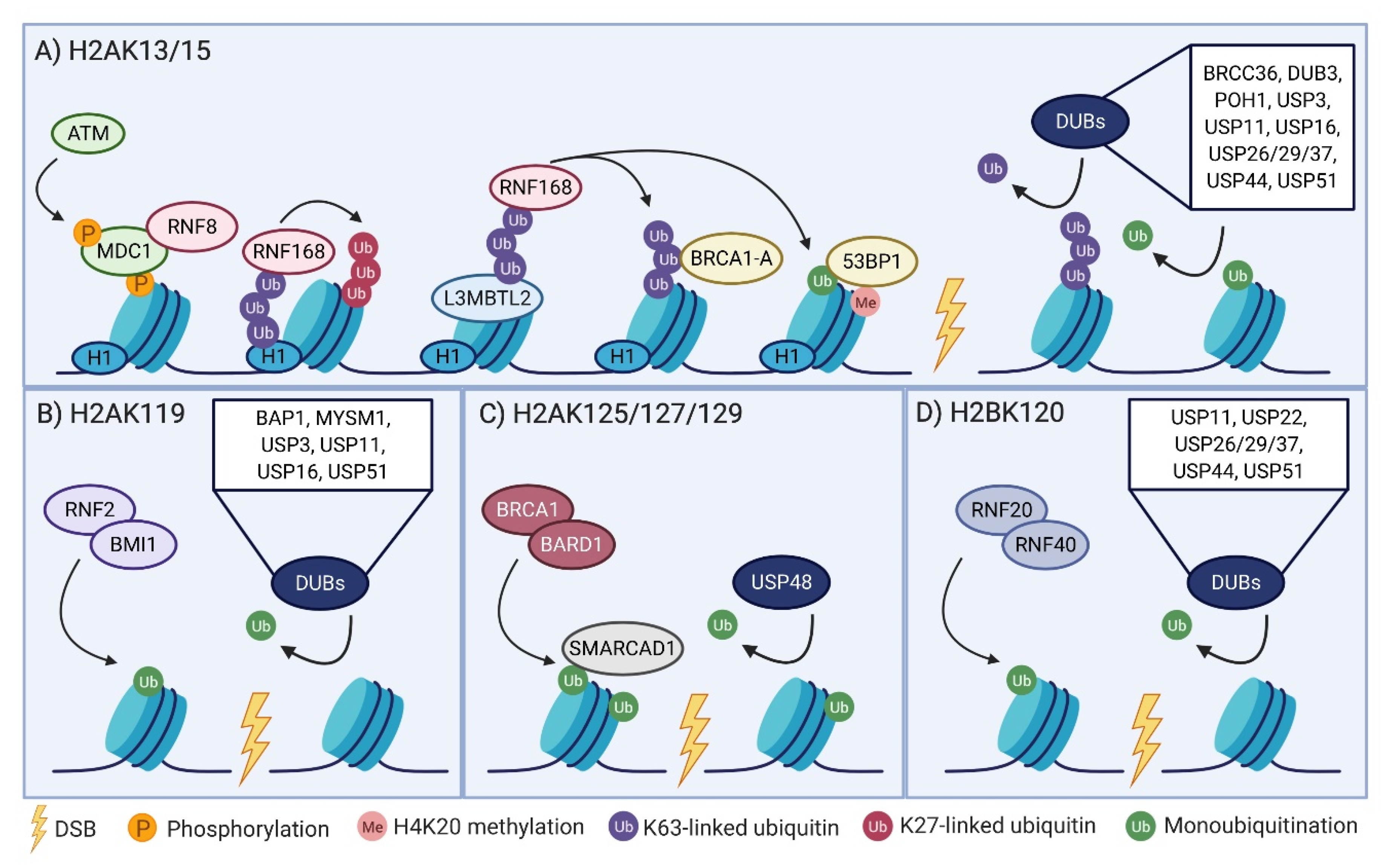
2.1. The RNF8-RNF168 Axis
2.2. H2AK119 Monoubiquitination Links Transcriptional Regulation to the DSB Response
2.3. BRCA1-BARD1 Regulation of Repair Pathway Choice Via H2AK125/127/129 Ubiquitination
2.4. A Non-Canonical Role of H2B Ubiquitination
2.5. The Potential Contribution of H3 and H4 Ubiquitination to DSB Repair
3. The Reversal of Histone Ubiquitination at DSBs
3.1. BAP1: Polycomb Activity at DSBs
3.2. BRCC36: Restricting End Resection through Removal of K63-Linked Polyubiquitin Chains
3.3. DUB3/USP17L2: Counteracting RNF8/RNF168-Mediated IRIF
3.4. MYSM1/2A-DUB: An Underexplored H2AK119ub1 Modifier
3.5. POH1/PSMD14: A Non-Degradative Function of the Proteasome
3.6. USP3: Antagonizing RNF8/RNF168 at H2A-Type Histones
3.7. USP11: Promoting HR through Deubiquitination of γH2AX
3.8. USP16/Ubp-M: Coordinating Transcriptional Repression at Sites Neighboring DSBs
3.9. USP22: Context-dependent Regulation of NHEJ by H2BK120ub1
3.10. USP26, USP29, USP37: Functionally Redundant DUB Activity towards H2A
3.11. USP44: Negative Regulation of Downstream Repair Machinery
3.12. USP48: Novel DUB Activity at H2AK125/127/129
3.13. USP51: Fine-Tuning the Repair of DSBs
4. Beyond Direct Histone Ubiquitination and Deubiquitination
5. DUBs as Therapeutic Targets
6. Concluding Remark
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia-telangiectasia mutated |
| ATXN3 | Ataxin 3 |
| BARD1 | BRCA1-associated RING domain protein 1 |
| BRISC | BRCC36 isopeptidase complex |
| CSR | Class switch recombination |
| DSB | Double strand break |
| DUB | Deubiquitinating enzyme/deubiquitinase |
| HR | Homologous recombination |
| IR | Ionizing radiation |
| IRIF | Ionizing radiation-induced foci |
| JAMM | Jab1/MPN domain-associated metalloisopeptidase |
| MINDY | Motif interacting with ubiquitin-containing novel DUB family |
| MJD | Machado-Josephin domain proteases |
| NCoR | Nuclear receptor corepressor |
| NHEJ | Non-homologous end joining |
| NuRD | Nucleosome remodeling deacetylase |
| OTU | Ovarian tumor protease |
| PcG | Polycomb-group protein |
| PR-DUB | Polycomb repressive deubiquitinase complex |
| PRC1 | Polycomb repressive complex 1 |
| PTM | Post-translational modification |
| SAGA | SPT-ADA-GCN5 acetyltransferase |
| SIM | SUMOylation-interacting motif |
| SUMO | Small ubiquitin-like modification |
| Ub | Ubiquitin |
| UIM | Ubiquitin interacting motif |
| UCH | Ubiquitin C-terminal hydrolase |
| USP | Ubiquitin specific protease |
| V(D)J recombination | Variable, diversity, and joining recombination |
| WT | Wild type |
| ZUFSPs | Zinc finger and UFSP domain proteins |
References
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Guo, R.; Xu, D. Non-homologous end joining: Advances and frontiers. Acta Biochim. Et. Biophys. Sin. 2016, 48, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed]
- Ranjha, L.; Howard, S.M.; Cejka, P. Main steps in DNA double-strand break repair: An introduction to homologous recombination and related processes. Chromosoma 2018, 127, 187–214. [Google Scholar] [CrossRef]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.-Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef]
- Hermanns, T.; Pichlo, C.; Woiwode, I.; Klopffleisch, K.; Witting, K.F.; Ovaa, H.; Baumann, U.; Hofmann, K. A family of unconventional deubiquitinases with modular chain specificity determinants. Nat. Commun. 2018, 9, 799. [Google Scholar] [CrossRef]
- Draker, R.; Sarcinella, E.; Cheung, P. USP10 deubiquitylates the histone variant H2A.Z and both are required for androgen receptor-mediated gene activation. Nucleic Acids Res. 2011, 39, 3529–3542. [Google Scholar] [CrossRef]
- Nakagawa, T.; Kajitani, T.; Togo, S.; Masuko, N.; Ohdan, H.; Hishikawa, Y.; Koji, T.; Matsuyama, T.; Ikura, T.; Muramatsu, M.; et al. Deubiquitylation of histone H2A activates transcriptional initiation via trans-histone cross-talk with H3K4 di- and trimethylation. Genes Dev. 2008, 22, 37–49. [Google Scholar] [CrossRef]
- Buszczak, M.; Paterno, S.; Spradling, A.C. Drosophila stem cells share a common requirement for the histone H2B ubiquitin protease scrawny. Science 2009, 323, 248–251. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Rogakou, E.P.; Yamazaki, V.; Kirchgessner, C.U.; Gellert, M.; Bonner, W.M. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 2000, 10, 886–895. [Google Scholar] [CrossRef]
- Huen, M.S.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 2007, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef]
- Kolas, N.K.; Chapman, J.R.; Nakada, S.; Ylanko, J.; Chahwan, R.; Sweeney, F.D.; Panier, S.; Mendez, M.; Wildenhain, J.; Thomson, T.M.; et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 2007, 318, 1637–1640. [Google Scholar] [CrossRef]
- Wang, B.; Elledge, S.J. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 20759–20763. [Google Scholar] [CrossRef]
- Doil, C.; Mailand, N.; Bekker-Jensen, S.; Menard, P.; Larsen, D.H.; Pepperkok, R.; Ellenberg, J.; Panier, S.; Durocher, D.; Bartek, J.; et al. RNF168 Binds and Amplifies Ubiquitin Conjugates on Damaged Chromosomes to Allow Accumulation of Repair Proteins. Cell 2009, 136, 435–446. [Google Scholar] [CrossRef]
- Stewart, G.S.; Panier, S.; Townsend, K.; Al-Hakim, A.K.; Kolas, N.K.; Miller, E.S.; Nakada, S.; Ylanko, J.; Olivarius, S.; Mendez, M.; et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009, 136, 420–434. [Google Scholar] [CrossRef]
- Campbell, S.J.; Edwards, R.A.; Leung, C.C.; Neculai, D.; Hodge, C.D.; Dhe-Paganon, S.; Glover, J.N. Molecular insights into the function of RING finger (RNF)-containing proteins hRNF8 and hRNF168 in Ubc13/Mms2-dependent ubiquitylation. J. Biol. Chem. 2012, 287, 23900–23910. [Google Scholar] [CrossRef]
- Mattiroli, F.; Vissers, J.H.; van Dijk, W.J.; Ikpa, P.; Citterio, E.; Vermeulen, W.; Marteijn, J.A.; Sixma, T.K. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell 2012, 150, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Thorslund, T.; Ripplinger, A.; Hoffmann, S.; Wild, T.; Uckelmann, M.; Villumsen, B.; Narita, T.; Sixma, T.K.; Choudhary, C.; Bekker-Jensen, S.; et al. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature 2015, 527, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Deng, M.; Lou, Z. Ubiquitin and the DNA double-strand break repair pathway. Genome Instab. Dis. 2020, 1, 69–80. [Google Scholar] [CrossRef]
- Nowsheen, S.; Aziz, K.; Aziz, A.; Deng, M.; Qin, B.; Luo, K.; Jeganathan, K.B.; Zhang, H.; Liu, T.; Yu, J.; et al. L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nat. Cell Biol. 2018, 20, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huen, M.S.; Lu, L.Y.; Ye, L.; Dou, Y.; Ljungman, M.; Chen, J.; Yu, X. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol. Cell Biol. 2009, 29, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.N.; Oehler, J.; Torrecilla, I.; Kilgas, S.; Li, S.; Vaz, B.; Guérillon, C.; Fielden, J.; Hernandez-Carralero, E.; Cabrera, E.; et al. The p97-Ataxin 3 complex regulates homeostasis of the DNA damage response E3 ubiquitin ligase RNF8. EMBO J. 2019, 38, e102361. [Google Scholar] [CrossRef]
- Zhong, X.; Pittman, R.N. Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum. Mol. Genet. 2006, 15, 2409–2420. [Google Scholar] [CrossRef]
- Winborn, B.J.; Travis, S.M.; Todi, S.V.; Scaglione, K.M.; Xu, P.; Williams, A.J.; Cohen, R.E.; Peng, J.; Paulson, H.L. The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 2008, 283, 26436–26443. [Google Scholar] [CrossRef]
- Ackermann, L.; Schell, M.; Pokrzywa, W.; Kevei, É.; Gartner, A.; Schumacher, B.; Hoppe, T. E4 ligase-specific ubiquitination hubs coordinate DNA double-strand-break repair and apoptosis. Nat. Struct. Mol. Biol. 2016, 23, 995–1002. [Google Scholar] [CrossRef]
- Pfeiffer, A.; Luijsterburg, M.S.; Acs, K.; Wiegant, W.W.; Helfricht, A.; Herzog, L.K.; Minoia, M.; Böttcher, C.; Salomons, F.A.; van Attikum, H.; et al. Ataxin-3 consolidates the MDC1-dependent DNA double-strand break response by counteracting the SUMO-targeted ubiquitin ligase RNF4. EMBO J. 2017, 36, 1066–1083. [Google Scholar] [CrossRef]
- Gatti, M.; Pinato, S.; Maiolica, A.; Rocchio, F.; Prato, M.G.; Aebersold, R.; Penengo, L. RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep. 2015, 10, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Mallette, F.A.; Mattiroli, F.; Cui, G.; Young, L.C.; Hendzel, M.J.; Mer, G.; Sixma, T.K.; Richard, S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012, 31, 1865–1878. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Liu, K.; Mao, Z.; Luo, J.; Gu, W.; Zhao, W. USP11 Is a Negative Regulator to gammaH2AX Ubiquitylation by RNF8/RNF168. J. Biol. Chem. 2016, 291, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Ichijima, Y.; Fradet-Turcotte, A.; Leung, C.C.; Kaustov, L.; Arrowsmith, C.H.; Durocher, D. Tandem protein interaction modules organize the ubiquitin-dependent response to DNA double-strand breaks. Mol. Cell 2012, 47, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Sobhian, B.; Shao, G.; Lilli, D.R.; Culhane, A.C.; Moreau, L.A.; Xia, B.; Livingston, D.M.; Greenberg, R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 2007, 316, 1198–1202. [Google Scholar] [CrossRef]
- Hu, X.; Paul, A.; Wang, B. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J. Biol. Chem. 2012, 287, 25510–25519. [Google Scholar] [CrossRef]
- Guzzo, C.M.; Berndsen, C.E.; Zhu, J.; Gupta, V.; Datta, A.; Greenberg, R.A.; Wolberger, C.; Matunis, M.J. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci. Signal. 2012, 5, ra88. [Google Scholar] [CrossRef]
- Hu, Y.; Scully, R.; Sobhian, B.; Xie, A.; Shestakova, E.; Livingston, D.M. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011, 25, 685–700. [Google Scholar] [CrossRef]
- Savage, K.I.; Harkin, D.P. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef]
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680. [Google Scholar] [CrossRef]
- Fradet-Turcotte, A.; Canny, M.D.; Escribano-Diaz, C.; Orthwein, A.; Leung, C.C.; Huang, H.; Landry, M.C.; Kitevski-LeBlanc, J.; Noordermeer, S.M.; Sicheri, F.; et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 2013, 499, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, I.; Oehler, J.; Ramadan, K. The role of ubiquitin-dependent segregase p97 (VCP or Cdc48) in chromatin dynamics after DNA double strand breaks. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2017, 372, 20160282. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, K. p97/VCP- and Lys48-linked polyubiquitination form a new signaling pathway in DNA damage response. Cell Cycle 2012, 11, 1062–1069. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Meerang, M.; Ritz, D.; Paliwal, S.; Garajova, Z.; Bosshard, M.; Mailand, N.; Janscak, P.; Hübscher, U.; Meyer, H.; Ramadan, K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double-strand breaks. Nat. Cell Biol. 2011, 13, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Acs, K.; Luijsterburg, M.S.; Ackermann, L.; Salomons, F.A.; Hoppe, T.; Dantuma, N.P. The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol. 2011, 18, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aroumougame, A.; Lobrich, M.; Li, Y.; Chen, D.; Chen, J.; Gong, Z. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014, 28, 2693–2698. [Google Scholar] [CrossRef]
- Panier, S.; Boulton, S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014, 15, 7–18. [Google Scholar] [CrossRef]
- Zimmermann, M.; de Lange, T. 53BP1: Pro choice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef]
- Goldknopf, I.L.; Taylor, C.W.; Baum, R.M.; Yeoman, L.C.; Olson, M.O.; Prestayko, A.W.; Busch, H. Isolation and characterization of protein A24, a “histone-like” non-histone chromosomal protein. J. Biol. Chem. 1975, 250, 7182–7187. [Google Scholar]
- Goldknopf, I.L.; Busch, H. Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate-protein A24. Proc. Natl. Acad. Sci. USA 1977, 74, 864–868. [Google Scholar] [CrossRef] [PubMed]
- Nickel, B.E.; Davie, J.R. Structure of polyubiquitinated histone H2A. Biochemistry 1989, 28, 964–968. [Google Scholar] [CrossRef] [PubMed]
- De Napoles, M.; Mermoud, J.E.; Wakao, R.; Tang, Y.A.; Endoh, M.; Appanah, R.; Nesterova, T.B.; Silva, J.; Otte, A.P.; Vidal, M.; et al. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 2004, 7, 663–676. [Google Scholar] [CrossRef]
- Fang, J.; Chen, T.; Chadwick, B.; Li, E.; Zhang, Y. Ring1b-mediated H2A ubiquitination associates with inactive X chromosomes and is involved in initiation of X inactivation. J. Biol. Chem. 2004, 279, 52812–52815. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Tsukada, Y.; Zhang, Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell 2005, 20, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Saurin, A.J.; Shiels, C.; Williamson, J.; Satijn, D.P.; Otte, A.P.; Sheer, D.; Freemont, P.S. The human polycomb group complex associates with pericentromeric heterochromatin to form a novel nuclear domain. J. Cell Biol. 1998, 142, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Munoz, I.; Taghavi, P.; Kuijl, C.; Neefjes, J.; van Lohuizen, M. Association of BMI1 with polycomb bodies is dynamic and requires PRC2/EZH2 and the maintenance DNA methyltransferase DNMT1. Mol. Cell Biol. 2005, 25, 11047–11058. [Google Scholar] [CrossRef]
- Kruhlak, M.; Crouch, E.E.; Orlov, M.; Montano, C.; Gorski, S.A.; Nussenzweig, A.; Misteli, T.; Phair, R.D.; Casellas, R. The ATM repair pathway inhibits RNA polymerase I transcription in response to chromosome breaks. Nature 2007, 447, 730–734. [Google Scholar] [CrossRef]
- Ismail, I.H.; Andrin, C.; McDonald, D.; Hendzel, M.J. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 2010, 191, 45–60. [Google Scholar] [CrossRef]
- Facchino, S.; Abdouh, M.; Chatoo, W.; Bernier, G. BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery. J. Neurosci. 2010, 30, 10096–10111. [Google Scholar] [CrossRef]
- Ginjala, V.; Nacerddine, K.; Kulkarni, A.; Oza, J.; Hill, S.J.; Yao, M.; Citterio, E.; van Lohuizen, M.; Ganesan, S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol. Cell Biol. 2011, 31, 1972–1982. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.R.; Peng, G.; Hung, W.C.; Lin, S.Y. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J. Biol. Chem. 2011, 286, 28599–28607. [Google Scholar] [CrossRef] [PubMed]
- Kakarougkas, A.; Ismail, A.; Chambers, A.L.; Riballo, E.; Herbert, A.D.; Kunzel, J.; Lobrich, M.; Jeggo, P.A.; Downs, J.A. Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 2014, 55, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, N.M.; Rafalska-Metcalf, I.U.; Balane-Bolivar, C.; Janicki, S.M.; Greenberg, R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010, 141, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.K.; Giadrossi, S.; Casanova, M.; Brookes, E.; Vidal, M.; Koseki, H.; Brockdorff, N.; Fisher, A.G.; Pombo, A. Ring1-mediated ubiquitination of H2A restrains poised RNA polymerase II at bivalent genes in mouse ES cells. Nat. Cell Biol. 2007, 9, 1428–1435. [Google Scholar] [CrossRef]
- Zhou, W.; Zhu, P.; Wang, J.; Pascual, G.; Ohgi, K.A.; Lozach, J.; Glass, C.K.; Rosenfeld, M.G. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol. Cell 2008, 29, 69–80. [Google Scholar] [CrossRef]
- Chou, D.M.; Adamson, B.; Dephoure, N.E.; Tan, X.; Nottke, A.C.; Hurov, K.E.; Gygi, S.P.; Colaiacovo, M.P.; Elledge, S.J. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc. Natl. Acad. Sci. USA 2010, 107, 18475–18480. [Google Scholar] [CrossRef]
- Ui, A.; Nagaura, Y.; Yasui, A. Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol. Cell 2015, 58, 468–482. [Google Scholar] [CrossRef]
- Chandler, H.; Patel, H.; Palermo, R.; Brookes, S.; Matthews, N.; Peters, G. Role of polycomb group proteins in the DNA damage response--a reassessment. PLoS ONE 2014, 9, e102968. [Google Scholar] [CrossRef]
- Ismail, I.H.; Davidson, R.; Gagne, J.P.; Xu, Z.Z.; Poirier, G.G.; Hendzel, M.J. Germline mutations in BAP1 impair its function in DNA double-strand break repair. Cancer Res. 2014, 74, 4282–4294. [Google Scholar] [CrossRef]
- Wu, C.Y.; Kang, H.Y.; Yang, W.L.; Wu, J.; Jeong, Y.S.; Wang, J.; Chan, C.H.; Lee, S.W.; Zhang, X.; Lamothe, B.; et al. Critical role of monoubiquitination of histone H2AX protein in histone H2AX phosphorylation and DNA damage response. J. Biol. Chem. 2011, 286, 30806–30815. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Westermark, U.K.; Reyngold, M.; Olshen, A.B.; Baer, R.; Jasin, M.; Moynahan, M.E. BARD1 participates with BRCA1 in homology-directed repair of chromosome breaks. Mol. Cell. Biol. 2003, 23, 7926–7936. [Google Scholar] [CrossRef]
- Morris, J.R.; Solomon, E. BRCA1: BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum. Mol Genet. 2004, 13, 807–817. [Google Scholar] [CrossRef]
- Wu-Baer, F.; Lagrazon, K.; Yuan, W.; Baer, R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003, 278, 34743–34746. [Google Scholar] [CrossRef]
- Reid, L.J.; Shakya, R.; Modi, A.P.; Lokshin, M.; Cheng, J.T.; Jasin, M.; Baer, R.; Ludwig, T. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl. Acad. Sci. USA 2008, 105, 20876–20881. [Google Scholar] [CrossRef]
- Kalb, R.; Mallery, D.L.; Larkin, C.; Huang, J.T.; Hiom, K. BRCA1 is a histone-H2A-specific ubiquitin ligase. Cell Rep. 2014, 8, 999–1005. [Google Scholar] [CrossRef]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Kumaraswamy, E.; Shiekhattar, R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol. Cell Biol. 2007, 27, 6733–6741. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, X.; Chini, C.C.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef]
- Minsky, N.; Shema, E.; Field, Y.; Schuster, M.; Segal, E.; Oren, M. Monoubiquitinated H2B is associated with the transcribed region of highly expressed genes in human cells. Nat. Cell Biol. 2008, 10, 483–488. [Google Scholar] [CrossRef]
- Pavri, R.; Zhu, B.; Li, G.; Trojer, P.; Mandal, S.; Shilatifard, A.; Reinberg, D. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 2006, 125, 703–717. [Google Scholar] [CrossRef]
- Kao, C.-F.; Hillyer, C.; Tsukuda, T.; Henry, K.; Berger, S.; Osley, M.A. Rad6 plays a role in transcriptional activation through ubiquitylation of histone H2B. Genes Dev. 2004, 18, 184–195. [Google Scholar] [CrossRef]
- Thorne, A.W.; Sautiere, P.; Briand, G.; Crane-Robinson, C. The structure of ubiquitinated histone H2B. EMBO J. 1987, 6, 1005–1010. [Google Scholar] [CrossRef]
- Zhu, B.; Zheng, Y.; Pham, A.D.; Mandal, S.S.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Monoubiquitination of human histone H2B: The factors involved and their roles in HOX gene regulation. Mol. Cell 2005, 20, 601–611. [Google Scholar] [CrossRef]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- So, C.C.; Ramachandran, S.; Martin, A. E3 Ubiquitin Ligases RNF20 and RNF40 Are Required for Double-Stranded Break (DSB) Repair: Evidence for Monoubiquitination of Histone H2B Lysine 120 as a Novel Axis of DSB Signaling and Repair. Mol. Cell Biol. 2019, 39. [Google Scholar] [CrossRef]
- Game, J.C.; Williamson, M.S.; Spicakova, T.; Brown, J.M. The RAD6/BRE1 histone modification pathway in Saccharomyces confers radiation resistance through a RAD51-dependent process that is independent of RAD18. Genetics 2006, 173, 1951–1968. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Li, D.; Lu, Z.; Liu, G.; Wang, M.; Xing, P.; Wang, M.; Dong, Y.; Wang, X.; Li, J.; et al. Bre1-dependent H2B ubiquitination promotes homologous recombination by stimulating histone eviction at DNA breaks. Nucleic Acids Res. 2018, 46, 11326–11339. [Google Scholar] [CrossRef]
- Chernikova, S.B.; Dorth, J.A.; Razorenova, O.V.; Game, J.C.; Brown, J.M. Deficiency in Bre1 impairs homologous recombination repair and cell cycle checkpoint response to radiation damage in mammalian cells. Radiat. Res. 2010, 174, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Shema, E.; Tirosh, I.; Aylon, Y.; Huang, J.; Ye, C.; Moskovits, N.; Raver-Shapira, N.; Minsky, N.; Pirngruber, J.; Tarcic, G.; et al. The histone H2B-specific ubiquitin ligase RNF20/hBRE1 acts as a putative tumor suppressor through selective regulation of gene expression. Genes Dev. 2008, 22, 2664–2676. [Google Scholar] [CrossRef]
- Fierz, B.; Chatterjee, C.; McGinty, R.K.; Bar-Dagan, M.; Raleigh, D.P.; Muir, T.W. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol 2011, 7, 113–119. [Google Scholar] [CrossRef]
- Fleming, A.B.; Kao, C.F.; Hillyer, C.; Pikaart, M.; Osley, M.A. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol. Cell 2008, 31, 57–66. [Google Scholar] [CrossRef]
- Chandrasekharan, M.B.; Huang, F.; Sun, Z.W. Ubiquitination of histone H2B regulates chromatin dynamics by enhancing nucleosome stability. Proc. Natl. Acad. Sci. USA 2009, 106, 16686–16691. [Google Scholar] [CrossRef]
- Chen, H.Y.; Sun, J.M.; Zhang, Y.; Davie, J.R.; Meistrich, M.L. Ubiquitination of histone H3 in elongating spermatids of rat testes. J. Biol. Chem. 1998, 273, 13165–13169. [Google Scholar] [CrossRef]
- Wang, H.; Zhai, L.; Xu, J.; Joo, H.Y.; Jackson, S.; Erdjument-Bromage, H.; Tempst, P.; Xiong, Y.; Zhang, Y. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol. Cell 2006, 22, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Li, T.; Yi, X.; MacCoss, M.J.; Moon, R.T.; Zheng, N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature 2006, 443, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Tinline-Purvis, H.; Walker, C.A.; Folkes, L.K.; Stratford, M.R.; Hayles, J.; Hoe, K.-L.; Kim, D.-U.; Park, H.-O.; Kearsey, S.E.; et al. Break-induced ATR and Ddb1-Cul4(Cdt)2 ubiquitin ligase-dependent nucleotide synthesis promotes homologous recombination repair in fission yeast. Genes Dev. 2010, 24, 2705–2716. [Google Scholar] [CrossRef]
- Yan, Q.; Dutt, S.; Xu, R.; Graves, K.; Juszczynski, P.; Manis, J.P.; Shipp, M.A. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol. Cell 2009, 36, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Ai, X.; Eugeni, E.E.; Zhang, L.; Carpenter, L.R.; Jelinek, M.A.; Freitas, M.A.; Parthun, M.R. Histone H4 lysine 91 acetylation a core domain modification associated with chromatin assembly. Mol. Cell 2005, 18, 123–130. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hebert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar] [CrossRef]
- Nishi, R.; Wijnhoven, P.; le Sage, C.; Tjeertes, J.; Galanty, Y.; Forment, J.V.; Clague, M.J.; Urbe, S.; Jackson, S.P. Systematic characterization of deubiquitylating enzymes for roles in maintaining genome integrity. Nat. Cell Biol. 2014, 16, 1016–1026. [Google Scholar] [CrossRef]
- Mosbech, A.; Lukas, C.; Bekker-Jensen, S.; Mailand, N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J. Biol. Chem. 2013, 288, 16579–16587. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; de Ayala Alonso, A.G.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Muller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Sahtoe, D.D.; van Dijk, W.J.; Ekkebus, R.; Ovaa, H.; Sixma, T.K. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 2016, 7, 10292. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Wang, J.; Chen, J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J. Biol. Chem. 2010, 285, 30982–30988. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Arciero, C.A.; Wang, C.; Broccoli, D.; Godwin, A.K. BRCC36 is essential for ionizing radiation-induced BRCA1 phosphorylation and nuclear foci formation. Cancer Res. 2006, 66, 5039–5046. [Google Scholar] [CrossRef]
- Shao, G.; Lilli, D.R.; Patterson-Fortin, J.; Coleman, K.A.; Morrissey, D.E.; Greenberg, R.A. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc. Natl. Acad. Sci. USA 2009, 106, 3166–3171. [Google Scholar] [CrossRef]
- Ng, H.M.; Wei, L.; Lan, L.; Huen, M.S. The Lys63-deubiquitylating Enzyme BRCC36 Limits DNA Break Processing and Repair. J. Biol. Chem. 2016, 291, 16197–16207. [Google Scholar] [CrossRef]
- Cooper, E.M.; Cutcliffe, C.; Kristiansen, T.Z.; Pandey, A.; Pickart, C.M.; Cohen, R.E. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009, 28, 621–631. [Google Scholar] [CrossRef]
- Hu, X.; Kim, J.A.; Castillo, A.; Huang, M.; Liu, J.; Wang, B. NBA1/MERIT40 and BRE interaction is required for the integrity of two distinct deubiquitinating enzyme BRCC36-containing complexes. J. Biol. Chem. 2011, 286, 11734–11745. [Google Scholar] [CrossRef]
- Dong, Y.; Hakimi, M.A.; Chen, X.; Kumaraswamy, E.; Cooch, N.S.; Godwin, A.K.; Shiekhattar, R. Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair. Mol. Cell 2003, 12, 1087–1099. [Google Scholar] [CrossRef]
- Kim, H.; Chen, J.; Yu, X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science 2007, 316, 1202–1205. [Google Scholar] [CrossRef]
- Delgado-Diaz, M.R.; Martin, Y.; Berg, A.; Freire, R.; Smits, V.A. Dub3 controls DNA damage signalling by direct deubiquitination of H2AX. Mol. Oncol. 2014, 8, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Boyer, L.A.; Latek, R.R.; Peterson, C.L. The SANT domain: A unique histone-tail-binding module? Nat. Rev. Mol. Cell Biol. 2004, 5, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Yoneyama, M.; Tochio, N.; Umehara, T.; Koshiba, S.; Inoue, M.; Yabuki, T.; Aoki, M.; Seki, E.; Matsuda, T.; Watanabe, S.; et al. Structural and functional differences of SWIRM domain subtypes. J. Mol. Biol. 2007, 369, 222–238. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhou, W.; Wang, J.; Puc, J.; Ohgi, K.A.; Erdjument-Bromage, H.; Tempst, P.; Glass, C.K.; Rosenfeld, M.G. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol. Cell 2007, 27, 609–621. [Google Scholar] [CrossRef]
- Won, H.; Nandakumar, V.; Yates, P.; Sanchez, S.; Jones, L.; Huang, X.F.; Chen, S.Y. Epigenetic control of dendritic cell development and fate determination of common myeloid progenitor by Mysm1. Blood 2014, 124, 2647–2656. [Google Scholar] [CrossRef]
- Wang, T.; Nandakumar, V.; Jiang, X.X.; Jones, L.; Yang, A.G.; Huang, X.F.; Chen, S.Y. The control of hematopoietic stem cell maintenance, self-renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef]
- Yao, T.; Cohen, R.E. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature 2002, 419, 403–407. [Google Scholar] [CrossRef]
- Verma, R.; Aravind, L.; Oania, R.; McDonald, W.H.; Yates, J.R., 3rd; Koonin, E.V.; Deshaies, R.J. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science 2002, 298, 611–615. [Google Scholar] [CrossRef]
- Butler, L.R.; Densham, R.M.; Jia, J.; Garvin, A.J.; Stone, H.R.; Shah, V.; Weekes, D.; Festy, F.; Beesley, J.; Morris, J.R. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012, 31, 3918–3934. [Google Scholar] [CrossRef]
- Krogan, N.J.; Lam, M.H.; Fillingham, J.; Keogh, M.C.; Gebbia, M.; Li, J.; Datta, N.; Cagney, G.; Buratowski, S.; Emili, A.; et al. Proteasome involvement in the repair of DNA double-strand breaks. Mol. Cell 2004, 16, 1027–1034. [Google Scholar] [CrossRef]
- Kakarougkas, A.; Ismail, A.; Katsuki, Y.; Freire, R.; Shibata, A.; Jeggo, P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013, 41, 10298–10311. [Google Scholar] [CrossRef]
- Lancini, C.; van den Berk, P.C.; Vissers, J.H.; Gargiulo, G.; Song, J.Y.; Hulsman, D.; Serresi, M.; Tanger, E.; Blom, M.; Vens, C.; et al. Tight regulation of ubiquitin-mediated DNA damage response by USP3 preserves the functional integrity of hematopoietic stem cells. J. Exp. Med. 2014, 211, 1759–1777. [Google Scholar] [CrossRef]
- Sloper-Mould, K.E.; Eyre, H.J.; Wang, X.W.; Sutherland, G.R.; Baker, R.T. Characterization and chromosomal localization of USP3, a novel human ubiquitin-specific protease. J. Biol. Chem. 1999, 274, 26878–26884. [Google Scholar] [CrossRef] [PubMed]
- Nicassio, F.; Corrado, N.; Vissers, J.H.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Zhu, Q.; Wani, G.; He, J.; Wang, Q.E.; Wani, A.A. USP3 counteracts RNF168 via deubiquitinating H2A and gammaH2AX at lysine 13 and 15. Cell Cycle 2014, 13, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, H.; Liu, J.; Cheruiyot, A.; Lee, J.H.; Ordog, T.; Lou, Z.; You, Z.; Zhang, Z. USP51 deubiquitylates H2AK13,15ub and regulates DNA damage response. Genes Dev. 2016, 30, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Ting, X.; Xia, L.; Yang, J.; He, L.; Si, W.; Shang, Y.; Sun, L. USP11 acts as a histone deubiquitinase functioning in chromatin reorganization during DNA repair. Nucleic Acids Res. 2019, 47, 9721–9740. [Google Scholar] [CrossRef]
- Wiltshire, T.D.; Lovejoy, C.A.; Wang, T.; Xia, F.; O’Connor, M.J.; Cortez, D. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J. Biol. Chem. 2010, 285, 14565–14571. [Google Scholar] [CrossRef]
- Joo, H.Y.; Zhai, L.; Yang, C.; Nie, S.; Erdjument-Bromage, H.; Tempst, P.; Chang, C.; Wang, H. Regulation of cell cycle progression and gene expression by H2A deubiquitination. Nature 2007, 449, 1068–1072. [Google Scholar] [CrossRef]
- Bekker-Jensen, S.; Rendtlew Danielsen, J.; Fugger, K.; Gromova, I.; Nerstedt, A.; Lukas, C.; Bartek, J.; Lukas, J.; Mailand, N. HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell Biol. 2010, 12, 80–86. [Google Scholar] [CrossRef]
- Sowa, M.E.; Bennett, E.J.; Gygi, S.P.; Harper, J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell 2009, 138, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, H.; Wang, H. The histone H2A deubiquitinase USP16 interacts with HERC2 and fine-tunes cellular response to DNA damage. J. Biol. Chem. 2014, 289, 32883–32894. [Google Scholar] [CrossRef] [PubMed]
- Sen Nkwe, N.; Daou, S.; Uriarte, M.; Gagnon, J.; Iannantuono, N.V.; Barbour, H.; Yu, H.; Masclef, L.; Fernandez, E.; Zamorano Cuervo, N.; et al. A potent nuclear export mechanism imposes USP16 cytoplasmic localization during interphase. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Haddad, D.; Li, C.; Le, M.X.; Ling, A.K.; So, C.C.; Nepal, R.M.; Gommerman, J.L.; Yu, K.; Ketela, T.; et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated gammaH2AX Formation. Cell Rep. 2016, 15, 1554–1565. [Google Scholar] [CrossRef]
- Li, C.; Irrazabal, T.; So, C.C.; Berru, M.; Du, L.; Lam, E.; Ling, A.K.; Gommerman, J.L.; Pan-Hammarstrom, Q.; Martin, A. The H2B deubiquitinase Usp22 promotes antibody class switch recombination by facilitating non-homologous end joining. Nat. Commun. 2018, 9, 1006. [Google Scholar] [CrossRef]
- Atanassov, B.S.; Mohan, R.D.; Lan, X.; Kuang, X.; Lu, Y.; Lin, K.; McIvor, E.; Li, W.; Zhang, Y.; Florens, L.; et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol. Cell 2016, 62, 558–571. [Google Scholar] [CrossRef]
- Koutelou, E.; Wang, L.; Schibler, A.C.; Chao, H.P.; Kuang, X.; Lin, K.; Lu, Y.; Shen, J.; Jeter, C.R.; Salinger, A.; et al. USP22 controls multiple signaling pathways that are essential for vasculature formation in the mouse placenta. Development 2019, 146. [Google Scholar] [CrossRef]
- Kosinsky, R.L.; Wegwitz, F.; Hellbach, N.; Dobbelstein, M.; Mansouri, A.; Vogel, T.; Begus-Nahrmann, Y.; Johnsen, S.A. Usp22 deficiency impairs intestinal epithelial lineage specification in vivo. Oncotarget 2015, 6, 37906–37918. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Gao, B.; Sun, Y.; Cao, L.; Genardi, S.M.; Wang, C.R.; Li, H.; Sun, Z.; Yang, Y.; et al. USP22 controls iNKT immunity through MED1 suppression of histone H2A monoubiquitination. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Pfeiffer, H.K.; Thorne, A.W.; McMahon, S.B. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 2008, 7, 1522–1524. [Google Scholar] [CrossRef]
- Paduch, D.A.; Mielnik, A.; Schlegel, P.N. Novel mutations in testis-specific ubiquitin protease 26 gene may cause male infertility and hypogonadism. Reprod. Biomed. Online 2005, 10, 747–754. [Google Scholar] [CrossRef]
- Wang, P.J.; McCarrey, J.R.; Yang, F.; Page, D.C. An abundance of X-linked genes expressed in spermatogonia. Nat. Genet. 2001, 27, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Pinto, S.M.; Getnet, D.; Nirujogi, R.S.; Manda, S.S.; Chaerkady, R.; Madugundu, A.K.; Kelkar, D.S.; Isserlin, R.; Jain, S.; et al. A draft map of the human proteome. Nature 2014, 509, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Typas, D.; Luijsterburg, M.S.; Wiegant, W.W.; Diakatou, M.; Helfricht, A.; Thijssen, P.E.; van den Broek, B.; Mullenders, L.H.; van Attikum, H. The de-ubiquitylating enzymes USP26 and USP37 regulate homologous recombination by counteracting RAP80. Nucleic Acids Res. 2015, 43, 6919–6933. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Fuchs, G.; Shema, E.; Vesterman, R.; Kotler, E.; Wolchinsky, Z.; Wilder, S.; Golomb, L.; Pribluda, A.; Zhang, F.; Haj-Yahya, M.; et al. RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol. Cell 2012, 46, 662–673. [Google Scholar] [CrossRef]
- Lan, X.; Atanassov, B.S.; Li, W.; Zhang, Y.; Florens, L.; Mohan, R.D.; Galardy, P.J.; Washburn, M.P.; Workman, J.L.; Dent, S.Y.R. USP44 Is an Integral Component of N-CoR that Contributes to Gene Repression by Deubiquitinating Histone H2B. Cell Rep. 2016, 17, 2382–2393. [Google Scholar] [CrossRef]
- Uckelmann, M.; Densham, R.M.; Baas, R.; Winterwerp, H.H.K.; Fish, A.; Sixma, T.K.; Morris, J.R. USP48 restrains resection by site-specific cleavage of the BRCA1 ubiquitin mark from H2A. Nat. Commun. 2018, 9, 229. [Google Scholar] [CrossRef]
- Zong, D.; Callen, E.; Pegoraro, G.; Lukas, C.; Lukas, J.; Nussenzweig, A. Ectopic expression of RNF168 and 53BP1 increases mutagenic but not physiological non-homologous end joining. Nucleic Acids Res. 2015, 43, 4950–4961. [Google Scholar] [CrossRef]
- Ai, H.; Guo, Y.; Sun, D.; Liu, S.; Qi, Y.; Guo, J.; Qu, Q.; Gong, Q.; Zhao, S.; Li, J.; et al. Examination of the Deubiquitylation Site Selectivity of USP51 by Using Chemically Synthesized Ubiquitylated Histones. Chembiochem 2019, 20, 221–229. [Google Scholar] [CrossRef]
- Jensen, D.E.; Proctor, M.; Marquis, S.T.; Gardner, H.P.; Ha, S.I.; Chodosh, L.A.; Ishov, A.M.; Tommerup, N.; Vissing, H.; Sekido, Y.; et al. BAP1: A novel ubiquitin hydrolase which binds to the BRCA1 RING finger and enhances BRCA1-mediated cell growth suppression. Oncogene 1998, 16, 1097–1112. [Google Scholar] [CrossRef] [PubMed]
- Campagne, A.; Lee, M.K.; Zielinski, D.; Michaud, A.; Le Corre, S.; Dingli, F.; Chen, H.; Shahidian, L.Z.; Vassilev, I.; Servant, N.; et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat. Commun. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Jiang, J.; Lan, L.; Nakajima, S.; Kanno, S.-i.; Koseki, H.; Yasui, A. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic Acids Res. 2008, 36, 2939–2947. [Google Scholar] [CrossRef]
- Campbell, S.; Ismail, I.H.; Young, L.C.; Poirier, G.G.; Hendzel, M.J. Polycomb repressive complex 2 contributes to DNA double-strand break repair. Cell Cycle 2013, 12, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Huang, J.; Chen, J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009, 23, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.X.; Nguyen, Q.; Chou, Y.; Wang, T.; Nandakumar, V.; Yates, P.; Jones, L.; Wang, L.; Won, H.; Lee, H.R.; et al. Control of B cell development by the histone H2A deubiquitinase MYSM1. Immunity 2011, 35, 883–896. [Google Scholar] [CrossRef]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.; Muller, C.W.; Vermeulen, M.; Muller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Zhu, Q.; Sharma, N.; He, J.; Wani, G.; Wani, A.A. USP7 deubiquitinase promotes ubiquitin-dependent DNA damage signaling by stabilizing RNF168*. Cell Cycle 2015, 14, 1413–1425. [Google Scholar] [CrossRef]
- Wang, Q.; Ma, S.; Song, N.; Li, X.; Liu, L.; Yang, S.; Ding, X.; Shan, L.; Zhou, X.; Su, D.; et al. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Investig. 2016, 126, 2205–2220. [Google Scholar] [CrossRef]
- Van der Knaap, J.A.; Kumar, B.R.; Moshkin, Y.M.; Langenberg, K.; Krijgsveld, J.; Heck, A.J.; Karch, F.; Verrijzer, C.P. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol. Cell 2005, 17, 695–707. [Google Scholar] [CrossRef]
- Luo, M.; Zhou, J.; Leu, N.A.; Abreu, C.M.; Wang, J.; Anguera, M.C.; de Rooij, D.G.; Jasin, M.; Wang, P.J. Polycomb protein SCML2 associates with USP7 and counteracts histone H2A ubiquitination in the XY chromatin during male meiosis. PLoS Genet. 2015, 11, e1004954. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Wang, X.; Tian, Q.; Hu, Z.; Peng, C.; Jiang, P.; Wang, T.; Guo, W.; Chen, Y.; et al. The Deubiquitylating Enzyme USP4 Cooperates with CtIP in DNA Double-Strand Break End Resection. Cell Rep. 2015, 13, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kruswick, A.; Dang, H.; Tran, A.D.; Kwon, S.M.; Wang, X.W.; Oberdoerffer, P. Ubiquitin-specific protease 21 stabilizes BRCA2 to control DNA repair and tumor growth. Nat. Commun. 2017, 8, 137. [Google Scholar] [CrossRef]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef]
- Pasupala, N.; Morrow, M.E.; Que, L.T.; Malynn, B.A.; Ma, A.; Wolberger, C. OTUB1 non-catalytically stabilizes the E2 ubiquitin-conjugating enzyme UBE2E1 by preventing its autoubiquitination. J. Biol. Chem. 2018, 293, 18285–18295. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Chen, Y.; Lu, L.-Y.; Wu, Y.; Paulsen, M.T.; Ljungman, M.; Ferguson, D.O.; Yu, X. Chfr and RNF8 synergistically regulate ATM activation. Nat. Struct. Mol. Biol. 2011, 18, 761–768. [Google Scholar] [CrossRef]
- Xu, Y.; Sun, Y.; Jiang, X.; Ayrapetov, M.K.; Moskwa, P.; Yang, S.; Weinstock, D.M.; Price, B.D. The p400 ATPase regulates nucleosome stability and chromatin ubiquitination during DNA repair. J. Cell Biol. 2010, 191, 31–43. [Google Scholar] [CrossRef]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell Biol. 2010, 190, 741–749. [Google Scholar] [CrossRef]
- Zhao, Y.; Brickner, J.R.; Majid, M.C.; Mosammaparast, N. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair. Trends Cell Biol. 2014, 24, 426–434. [Google Scholar] [CrossRef]
- Dantuma, N.P.; van Attikum, H. Spatiotemporal regulation of posttranslational modifications in the DNA damage response. EMBO J. 2016, 35, 6–23. [Google Scholar] [CrossRef]
- Adorno, M.; Sikandar, S.; Mitra, S.S.; Kuo, A.; Nicolis di Robilant, B.; Haro-Acosta, V.; Ouadah, Y.; Quarta, M.; Rodriguez, J.; Qian, D.; et al. Usp16 contributes to somatic stem-cell defects in Down’s syndrome. Nature 2013, 501, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, P.; Han, L.; Guo, Z.; Wang, X.; Chen, Z.; Nie, J.; Yin, S.; Piccioni, M.; Tsun, A.; et al. Cutting edge: Ubiquitin-specific protease 4 promotes Th17 cell function under inflammation by deubiquitinating and stabilizing RORγt. J. Immunol. 2015, 194, 4094–4097. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, B.; Suvarnapunya, A.E.; Venkatesan, M.; Edelmann, M.J. Deubiquitinating enzymes as promising drug targets for infectious diseases. Curr. Pharm. Des. 2013, 19, 3234–3247. [Google Scholar] [CrossRef] [PubMed]
- Masoomian, B.; Shields, J.A.; Shields, C.L. Overview of BAP1 cancer predisposition syndrome and the relationship to uveal melanoma. J. Curr. Ophthalmol. 2018, 30, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, Z.Y.; Du, H.; Li, S.Z.; Tu, R.; Jia, Y.F.; Zheng, Z.; Song, X.M.; Du, R.L.; Zhang, X.D. DUB3 deubiquitinates and stabilizes NRF2 in chemotherapy resistance of colorectal cancer. Cell Death Differ. 2019, 26, 2300–2313. [Google Scholar] [CrossRef]
- Wu, X.; Liu, M.; Zhu, H.; Wang, J.; Dai, W.; Li, J.; Zhu, D.; Tang, W.; Xiao, Y.; Lin, J.; et al. Ubiquitin-specific protease 3 promotes cell migration and invasion by interacting with and deubiquitinating SUZ12 in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 277. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-H.; Lu, S.-X.; Liu, L.-L.; Li, Y.; Yang, X.; He, Y.-F.; Chen, S.-L.; Cai, S.-H.; Wang, H.; Yun, J.-P. POH1 Knockdown Induces Cancer Cell Apoptosis via p53 and Bim. Neoplasia 2018, 20, 411–424. [Google Scholar] [CrossRef]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef]
- Song, Y.; Ray, A.; Das, D.S.; Samur, M.K.; Carrasco, R.D.; Munshi, N.C.; Chauhan, D.; Anderson, K.C. Deubiquitylating Enzyme Rpn11/POH1/PSMD14 As Therapeutic Target in Multiple Myeloma. Blood 2016, 128, 4469. [Google Scholar] [CrossRef]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.G.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.P.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell 2012, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Gavory, G.; O’Dowd, C.R.; Helm, M.D.; Flasz, J.; Arkoudis, E.; Dossang, A.; Hughes, C.; Cassidy, E.; McClelland, K.; Odrzywol, E.; et al. Discovery and characterization of highly potent and selective allosteric USP7 inhibitors. Nat. Chem. Biol. 2018, 14, 118–125. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| DUB | DUB Family | Associated Adaptor Proteins/ Complexes | Histone Substrates | Functions in DSB Repair | Refs. |
|---|---|---|---|---|---|
| BAP1 | UCH | ASXL1/2/3 (PR-DUB complex) | H2A(X)K119ub1 | Promotes HR, transcriptional regulation #, chromatin accessibility # | [71,106,107,108,109,110,111,112] |
| BRCC36 | JAMM | Abraxas, RAP80, BRCA1 (BRCA1-A complex) KIAA0517, BRCC45, MERIT40 (BRISC complex) | H2A(X) K63-linked chains | Counteracts RNF8/RNF168, restricts end resection | [17,40,65,113,114,115,116,117,118,119,120] |
| DUB3/USP17L2 | USP | H2A(X) | Antagonizes RNF8/RNF168, promotes HR | [33,107,121] | |
| MYSM1/2A-DUB | JAMM | H2AK119ub1 | Promotes HR #, transcriptional regulation #, chromatin accessibility # | [109,110,122,123,124,125,126] | |
| POH1/PSMD14 | JAMM | 26S proteasomal subunits | H2A K63-linked chains | Regulates 53BP1, promotes HR | [109,117,127,128,129,130,131] |
| USP3 | USP | H2A(X)K13/15 H2AX119ub1 | Protection from spontaneous DNA damage, antagonizes RNF168 | [18,109,110,132,133,134,135,136,137] | |
| USP11 | USP | γH2AX H2AK119ub1 H2BK120ub1 | Promotes HR, transcriptional regulation #, chromatin accessibility # | [33,109,137,138] | |
| USP16/Ubp-M | USP | H2AK119ub1 H2AK13/15 * | Transcriptional regulation, antagonizes RNF8/RNF168 # | [65,110,136,139,140,141,142,143] | |
| USP22 | USP | ATXN7, ATXN7L3, ENY2 | H2BK120ub1 | Promotes NHEJ, transcriptional regulation # | [91,110,144,145,146,147,148,149,150] |
| USP26, USP29, USP37 | USP | H2AK13/15 K63-linked chains * H2BK120ub1 * | Promotes HR, antagonizes RNF8/RNF168, antagonizes BRCA1-A complex formation | [110,151,152,153,154,155] | |
| USP44 | USP | TBL1X, TBL1XR1, HDAC3, NCOR1, NCOR2 (NCoR complex) | H2AK13/15 γH2AX H2BK120ub1 * | Counteracts RNF8/RNF168 to regulate IRIF assembly | [109,110,135,156,157] |
| USP48 | USP | H2AK125/127/129 | Counteracts BRCA1-BARD1 to restrict 53BP1 repositioning and end resection | [158] | |
| USP51 | USP | ATXN7L3, ENY2 | H2AK13/15 H2AK119ub1 * K27-linked chains * H2BK120ub1 * | Termination of DSB repair, transcriptional regulation #, chromatin accessibility # | [136,146,159,160] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aquila, L.; Atanassov, B.S. Regulation of Histone Ubiquitination in Response to DNA Double Strand Breaks. Cells 2020, 9, 1699. https://doi.org/10.3390/cells9071699
Aquila L, Atanassov BS. Regulation of Histone Ubiquitination in Response to DNA Double Strand Breaks. Cells. 2020; 9(7):1699. https://doi.org/10.3390/cells9071699
Chicago/Turabian StyleAquila, Lanni, and Boyko S. Atanassov. 2020. "Regulation of Histone Ubiquitination in Response to DNA Double Strand Breaks" Cells 9, no. 7: 1699. https://doi.org/10.3390/cells9071699
APA StyleAquila, L., & Atanassov, B. S. (2020). Regulation of Histone Ubiquitination in Response to DNA Double Strand Breaks. Cells, 9(7), 1699. https://doi.org/10.3390/cells9071699