Regulation of the Actin Cytoskeleton in Podocytes


Abstract
:1. Introduction
2. Structural Regulation of Actin Dynamics in Podocytes
2.1. Actin Dynamics
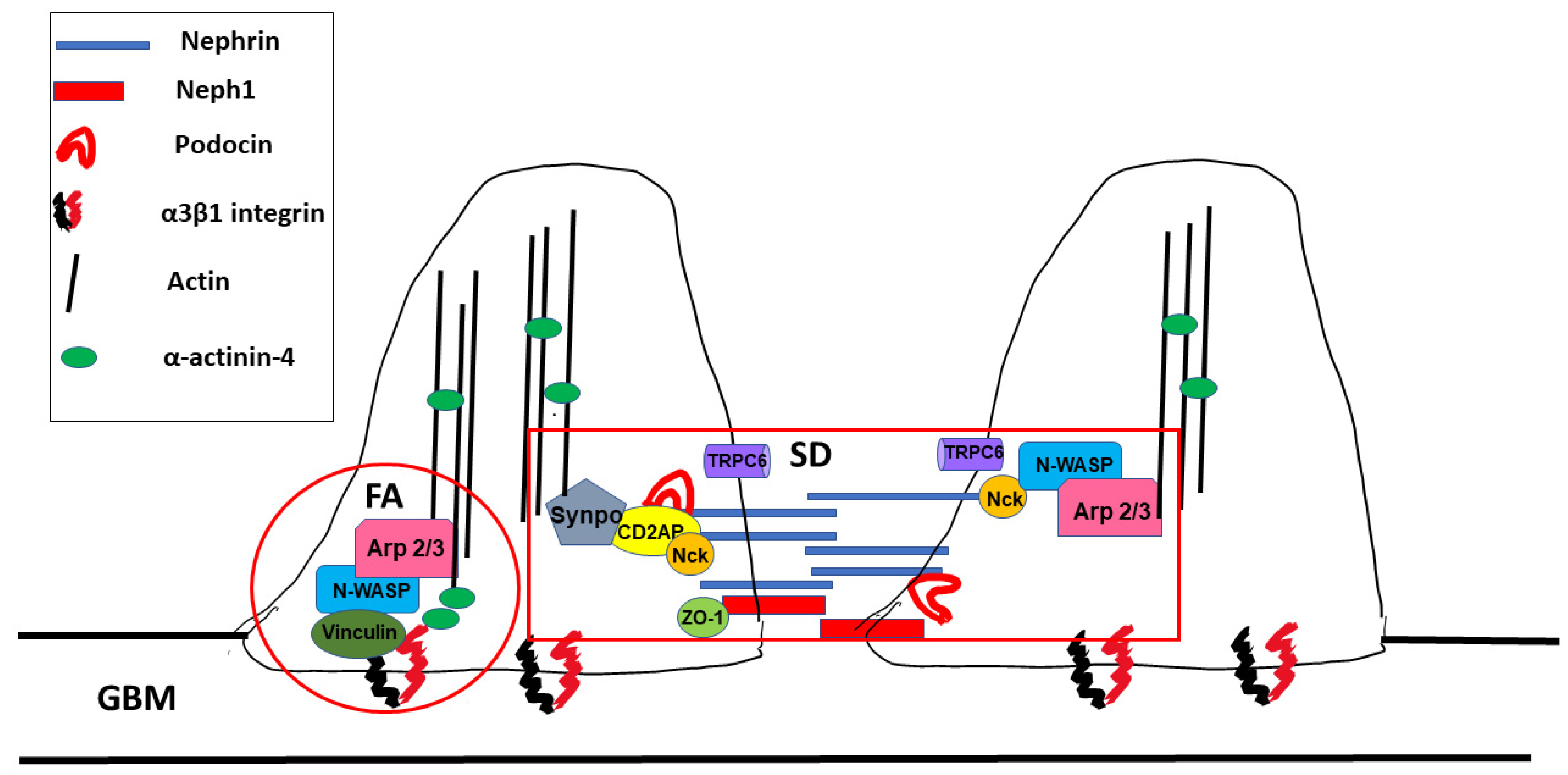
2.2. Podocyte Structure
2.3. The Slit Diaphragm
2.4. Signal Receptors
2.4.1. Nephrin
2.4.2. Nephrin Signaling
2.4.3. Neph1
2.4.4. Podocin
2.5. Adaptor Proteins/Signal Integrators
2.5.1. Adaptor Proteins
2.5.2. Rho/Small GTPases
2.5.3. Synaptopodin
2.5.4. α-actinin-4
2.5.5. TRPC6
2.6. Genetic Mutations Affecting Components of the Slit Diaphragm
2.6.1. Mutations Affecting Nephrin and Podocin
2.6.2. Mutations Affecting Adaptor Proteins/Signal Integrators
2.7. Focal Adhesions (FAs)
2.7.1. Integrins
2.7.2. GTPases
2.8. Genetic Mutations Affecting FA Proteins
2.9. Other Actin Associated Genetic Mutations
3. Other Mediators of Podocyte Actin Dynamics
3.1. Hemodynamic Factors
3.2. Cytokines/Hormones
3.2.1. TGF-ß1
3.2.2. IL-6
3.2.3. Angiotensin II (Ang II)
3.3. Drugs
3.3.1. Renin-Angiotensin System (RAS) Inhibitors
3.3.2. Glucocorticoids (GCs)
3.3.3. Cyclosporin A (CsA)
3.3.4. Rituximab
3.3.5. Abatacept
3.3.6. Dasatanib
4. Clinical Presentation of Genetic Mutations in Podocyte Actin
Clinical Presentation
5. Conclusions
Funding
Conflicts of Interest
References
- Srivastava, T.; Dai, H.; Heruth, D.P.; Alon, U.S.; Garola, R.E.; Zhou, J.; Duncan, R.S.; El-Meanawy, A.; McCarthy, E.T.; Sharma, R.; et al. Mechanotransduction signaling in podocytes from fluid flow shear stress. Am. J. Physiol. Ren. Physiol. 2018, 314, F22–F34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriz, W.; Lemley, K.V. A potential role for mechanical forces in the detachment of podocytes and the progression of CKD. J. Am. Soc. Nephrol. 2015, 26, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin dynamics, architecture, and mechanics in cell motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svitkina, T.M. Ultrastructure of the actin cytoskeleton. Curr. Opin. Cell Biol. 2018, 54, 1–8. [Google Scholar] [CrossRef]
- Svitkina, T. The Actin Cytoskeleton and Actin-Based Motility. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, R.; Holmes, K.C. Actin structure and function. Annu. Rev. Biophys. 2011, 40, 169–186. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Dominguez, R. Regulation of actin cytoskeleton dynamics in cells. Mol. Cells 2010, 29, 311–325. [Google Scholar] [CrossRef]
- Kriz, W.; Shirato, I.; Nagata, M.; LeHir, M.; Lemley, K.V. The podocyte’s response to stress: The enigma of foot process effacement. Am. J. Physiol. Ren. Physiol. 2013, 304, F333–F347. [Google Scholar] [CrossRef] [Green Version]
- Pavenstädt, H.; Kriz, W.; Kretzler, M. Cell biology of the glomerular podocyte. Physiol. Rev. 2003, 83, 253–307. [Google Scholar] [CrossRef] [Green Version]
- Andrews, P.M. Investigations of cytoplasmic contractile and cytoskeletal elements in the kidney glomerulus. Kidney Int. 1981, 20, 549–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schell, C.; Huber, T.B. The Evolving Complexity of the Podocyte Cytoskeleton. J. Am. Soc. Nephrol. 2017, 28, 3166–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, K.; Miyaki, T.; Kawasaki, Y.; Kinoshita, M.; Kakuta, S.; Sakai, T. Morphological Processes of Foot Process Effacement in Puromycin Aminonucleoside Nephrosis Revealed by FIB/SEM Tomography. J. Am. Soc. Nephrol. 2019, 30, 96–108. [Google Scholar] [CrossRef] [Green Version]
- Sever, S.; Schiffer, M. Actin dynamics at focal adhesions: A common endpoint and putative therapeutic target for proteinuric kidney diseases. Kidney Int. 2018, 93, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, K.; Kurihara, H.; Sakai, T. Actin filament organization of foot processes in vertebrate glomerular podocytes. Cell Tissue Res. 2007, 329, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Asanuma, K.; Yanagida-Asanuma, E.; Kim, K.; Mundel, P. Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007, 17, 428–437. [Google Scholar] [CrossRef]
- Perico, L.; Conti, S.; Benigni, A.; Remuzzi, G. Podocyte-actin dynamics in health and disease. Nat. Rev. Nephrol. 2016, 12, 692–710. [Google Scholar] [CrossRef] [PubMed]
- Suleiman, H.Y.; Roth, R.; Jain, S.; Heuser, J.E.; Shaw, A.S.; Miner, J.H. Injury-induced actin cytoskeleton reorganization in podocytes revealed by super-resolution microscopy. Jci Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, P. A Review of Podocyte Biology. Am. J. Nephrol. 2018, 47, 3–13. [Google Scholar] [CrossRef]
- Shirato, I. Podocyte process effacement in vivo. Microsc. Res. Tech. 2002, 57, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Shirato, I.; Sakai, T.; Kimura, K.; Tomino, Y.; Kriz, W. Cytoskeletal changes in podocytes associated with foot process effacement in Masugi nephritis. Am. J. Pathol. 1996, 148, 1283–1296. [Google Scholar] [PubMed]
- George, B.; Verma, R.; Soofi, A.A.; Garg, P.; Zhang, J.; Park, T.J.; Giardino, L.; Ryzhova, L.; Johnstone, D.B.; Wong, H.; et al. Crk1/2-dependent signaling is necessary for podocyte foot process spreading in mouse models of glomerular disease. J. Clin. Investig. 2012, 122, 674–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriz, W.; Lemley, K.V. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 2017, 91, 1283–1286. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.E.; Jones, N. Nephrin Signaling in the Podocyte: An Updated View of Signal Regulation at the Slit Diaphragm and Beyond. Front. Endocrinol. (Lausanne) 2018, 9, 302. [Google Scholar] [CrossRef]
- Succar, L.; Boadle, R.A.; Harris, D.C.; Rangan, G.K. Formation of tight junctions between neighboring podocytes is an early ultrastructural feature in experimental crescentic glomerulonephritis. Int. J. Nephrol. Renov. Dis. 2016, 9, 297–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, H.; Anderson, J.M.; Kerjaschki, D.; Farquhar, M.G. The altered glomerular filtration slits seen in puromycin aminonucleoside nephrosis and protamine sulfate-treated rats contain the tight junction protein ZO-1. Am. J. Pathol. 1992, 141, 805–816. [Google Scholar] [PubMed]
- Kestilä, M.; Lenkkeri, U.; Männikkö, M.; Lamerdin, J.; McCready, P.; Putaala, H.; Ruotsalainen, V.; Morita, T.; Nissinen, M.; Herva, R.; et al. Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol. Cell 1998, 1, 575–582. [Google Scholar] [CrossRef]
- Patrakka, J.; Tryggvason, K. Nephrin—A unique structural and signaling protein of the kidney filter. Trends Mol. Med. 2007, 13, 396–403. [Google Scholar] [CrossRef]
- Barletta, G.M.; Kovari, I.A.; Verma, R.K.; Kerjaschki, D.; Holzman, L.B. Nephrin and Neph1 co-localize at the podocyte foot process intercellular junction and form cis hetero-oligomers. J. Biol. Chem. 2003, 278, 19266–19271. [Google Scholar] [CrossRef] [Green Version]
- Gerke, P.; Huber, T.B.; Sellin, L.; Benzing, T.; Walz, G. Homodimerization and heterodimerization of the glomerular podocyte proteins nephrin and NEPH1. J. Am. Soc. Nephrol. 2003, 14, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Benzing, T. Signaling at the Slit Diaphragm. J. Am. Soc. Nephrol. 2004, 15, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Ruotsalainen, V.; Ljungberg, P.; Wartiovaara, J.; Lenkkeri, U.; Kestilä, M.; Jalanko, H.; Holmberg, C.; Tryggvason, K. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc. Natl. Acad. Sci. USA 1999, 96, 7962–7967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Sun, N.; Aoudjit, L.; Li, H.; Kawachi, H.; Lemay, S.; Takano, T. Nephrin mediates actin reorganization via phosphoinositide 3-kinase in podocytes. Kidney Int. 2008, 73, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Kovari, I.; Soofi, A.; Nihalani, D.; Patrie, K.; Holzman, L.B. Nephrin ectodomain engagement results in Src kinase activation, nephrin phosphorylation, Nck recruitment, and actin polymerization. J. Clin. Investig. 2006, 116, 1346–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- New, L.A.; Keyvani Chahi, A.; Jones, N. Direct regulation of nephrin tyrosine phosphorylation by Nck adaptor proteins. J. Biol. Chem. 2013, 288, 1500–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, N.; Blasutig, I.M.; Eremina, V.; Ruston, J.M.; Bladt, F.; Li, H.; Huang, H.; Larose, L.; Li, S.S.; Takano, T.; et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 2006, 440, 818–823. [Google Scholar] [CrossRef]
- Mullins, R.D. How WASP-family proteins and the Arp2/3 complex convert intracellular signals into cytoskeletal structures. Curr. Opin. Cell Biol. 2000, 12, 91–96. [Google Scholar] [CrossRef]
- Quack, I.; Woznowski, M.; Potthoff, S.A.; Palmer, R.; Königshausen, E.; Sivritas, S.; Schiffer, M.; Stegbauer, J.; Vonend, O.; Rump, L.C.; et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J. Biol. Chem. 2011, 286, 12959–12970. [Google Scholar] [CrossRef] [Green Version]
- Waters, A.M.; Wu, M.Y.; Huang, Y.W.; Liu, G.Y.; Holmyard, D.; Onay, T.; Jones, N.; Egan, S.E.; Robinson, L.A.; Piscione, T.D. Notch promotes dynamin-dependent endocytosis of nephrin. J. Am. Soc. Nephrol. 2012, 23, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tossidou, I.; Teng, B.; Menne, J.; Shushakova, N.; Park, J.K.; Becker, J.U.; Modde, F.; Leitges, M.; Haller, H.; Schiffer, M. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS ONE 2010, 5, e10185. [Google Scholar] [CrossRef] [Green Version]
- Geraldes, P. Protein phosphatases and podocyte function. Curr. Opin. Nephrol. Hypertens. 2018, 27, 49–55. [Google Scholar] [CrossRef]
- Denhez, B.; Lizotte, F.; Guimond, M.O.; Jones, N.; Takano, T.; Geraldes, P. Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes. J. Biol. Chem. 2015, 290, 350–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Koh, A.; Jeong, H.; Kim, E.; Ha, T.S.; Saleem, M.A.; Ryu, S.H. C1-Ten is a PTPase of nephrin, regulating podocyte hypertrophy through mTORC1 activation. Sci. Rep. 2017, 7, 12346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- New, L.A.; Martin, C.E.; Scott, R.P.; Platt, M.J.; Keyvani Chahi, A.; Stringer, C.D.; Lu, P.; Samborska, B.; Eremina, V.; Takano, T.; et al. Nephrin Tyrosine Phosphorylation Is Required to Stabilize and Restore Podocyte Foot Process Architecture. J. Am. Soc. Nephrol. 2016, 27, 2422–2435. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, S.; Zhao, F.; Lehtonen, E. CD2-associated protein directly interacts with the actin cytoskeleton. Am. J. Physiol. Ren. Physiol. 2002, 283, F734–F743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, D.K.; Winata, S.C.; Lyons, R.J.; Hughes, W.E.; Lehrbach, G.M.; Wasinger, V.; Corthals, G.; Cordwell, S.; Daly, R.J. A Cortactin-CD2-associated protein (CD2AP) complex provides a novel link between epidermal growth factor receptor endocytosis and the actin cytoskeleton. J. Biol. Chem. 2003, 278, 21805–21813. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, K.; Kim, K.; Oh, J.; Giardino, L.; Chabanis, S.; Faul, C.; Reiser, J.; Mundel, P. Synaptopodin regulates the actin-bundling activity of alpha-actinin in an isoform-specific manner. J. Clin. Investig. 2005, 115, 1188–1198. [Google Scholar] [CrossRef] [Green Version]
- Khoshnoodi, J.; Sigmundsson, K.; Ofverstedt, L.-G.; Skoglund, U.; Obrink, B.; Wartiovaara, J.; Tryggvason, K. Nephrin promotes cell-cell adhesion through homophilic interactions. Am. J. Pathol. 2003, 163, 2337–2346. [Google Scholar] [CrossRef] [Green Version]
- Garg, P.; Verma, R.; Nihalani, D.; Johnstone, D.B.; Holzman, L.B. Neph1 cooperates with nephrin to transduce a signal that induces actin polymerization. Mol. Cell. Biol. 2007, 27, 8698–8712. [Google Scholar] [CrossRef] [Green Version]
- Harita, Y.; Kurihara, H.; Kosako, H.; Tezuka, T.; Sekine, T.; Igarashi, T.; Hattori, S. Neph1, a component of the kidney slit diaphragm, is tyrosine-phosphorylated by the Src family tyrosine kinase and modulates intracellular signaling by binding to Grb2. J. Biol. Chem. 2008, 283, 9177–9186. [Google Scholar] [CrossRef] [Green Version]
- Roselli, S.; Gribouval, O.; Boute, N.; Sich, M.; Benessy, F.; Attié, T.; Gubler, M.C.; Antignac, C. Podocin localizes in the kidney to the slit diaphragm area. Am. J. Pathol. 2002, 160, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, K.; Simons, M.; Reiser, J.; Saleem, M.A.; Faul, C.; Kriz, W.; Shaw, A.S.; Holzman, L.B.; Mundel, P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J. Clin. Investig. 2001, 108, 1621–1629. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.B.; Simons, M.; Hartleben, B.; Sernetz, L.; Schmidts, M.; Gundlach, E.; Saleem, M.A.; Walz, G.; Benzing, T. Molecular basis of the functional podocin-nephrin complex: Mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 2003, 12, 3397–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shono, A.; Tsukaguchi, H.; Yaoita, E.; Nameta, M.; Kurihara, H.; Qin, X.S.; Yamamoto, T.; Doi, T. Podocin participates in the assembly of tight junctions between foot processes in nephrotic podocytes. J. Am. Soc. Nephrol. 2007, 18, 2525–2533. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.S. Roles of adaptor proteins in podocyte biology. World J. Nephrol. 2013, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tossidou, I.; Teng, B.; Worthmann, K.; Müller-Deile, J.; Jobst-Schwan, T.; Kardinal, C.; Schroder, P.; Bolanos-Palmieri, P.; Haller, H.; Willerding, J.; et al. Tyrosine Phosphorylation of CD2AP Affects Stability of the Slit Diaphragm Complex. J. Am. Soc. Nephrol. 2019, 30, 1220–1237. [Google Scholar] [CrossRef] [PubMed]
- Shih, N.Y.; Li, J.; Karpitskii, V.; Nguyen, A.; Dustin, M.L.; Kanagawa, O.; Miner, J.H.; Shaw, A.S. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science 1999, 286, 312–315. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.; New, L.A.; Fortino, M.A.; Eremina, V.; Ruston, J.; Blasutig, I.M.; Aoudjit, L.; Zou, Y.; Liu, X.; Yu, G.L.; et al. Nck proteins maintain the adult glomerular filtration barrier. J. Am. Soc. Nephrol. 2009, 20, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Huber, T.B.; Schmidts, M.; Gerke, P.; Schermer, B.; Zahn, A.; Hartleben, B.; Sellin, L.; Walz, G.; Benzing, T. The carboxyl terminus of Neph family members binds to the PDZ domain protein zonula occludens-1. J. Biol. Chem. 2003, 278, 13417–13421. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.C.; Rhodes, G.; Wang, E.; Pruthi, V.; Arif, E.; Saleem, M.A.; Wean, S.E.; Garg, P.; Verma, R.; Holzman, L.B.; et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J. Biol. Chem. 2008, 283, 35579–35589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattner, S.M.; Hodgin, J.B.; Nishio, M.; Wylie, S.A.; Saha, J.; Soofi, A.A.; Vining, C.; Randolph, A.; Herbach, N.; Wanke, R.; et al. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int. 2013, 84, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Mundel, P.; Reiser, J. Proteinuria: An enzymatic disease of the podocyte? Kidney Int. 2010, 77, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kistler, A.D.; Altintas, M.M.; Reiser, J. Podocyte GTPases regulate kidney filter dynamics. Kidney Int. 2012, 81, 1053–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Ellis, M.J.; Gomez, J.A.; Eisner, W.; Fennell, W.; Howell, D.N.; Ruiz, P.; Fields, T.A.; Spurney, R.F. Mechanisms of the proteinuria induced by Rho GTPases. Kidney Int. 2012, 81, 1075–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Jiang, R.; Aoudjit, L.; Jones, N.; Takano, T. Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 1621–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Ding, J.; Tsai, H.; Li, L.; Feng, Q.; Miao, J.; Fan, Q. Over-expressing transient receptor potential cation channel 6 in podocytes induces cytoskeleton rearrangement through increases of intracellular Ca2+ and RhoA activation. Exp. Biol. Med. 2011, 236, 184–193. [Google Scholar] [CrossRef]
- Mundel, P.; Gilbert, P.; Kriz, W. Podocytes in glomerulus of rat kidney express a characteristic 44 KD protein. J. Histochem. Cytochem. 1991, 39, 1047–1056. [Google Scholar] [CrossRef] [Green Version]
- Asanuma, K.; Yanagida-Asanuma, E.; Faul, C.; Tomino, Y.; Kim, K.; Mundel, P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat. Cell. Biol. 2006, 8, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Yanagida-Asanuma, E.; Asanuma, K.; Kim, K.; Donnelly, M.; Young Choi, H.; Hyung Chang, J.; Suetsugu, S.; Tomino, Y.; Takenawa, T.; Faul, C.; et al. Synaptopodin protects against proteinuria by disrupting Cdc42:IRSp53:Mena signaling complexes in kidney podocytes. Am. J. Pathol. 2007, 171, 415–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartram, M.P.; Habbig, S.; Pahmeyer, C.; Höhne, M.; Weber, L.T.; Thiele, H.; Altmüller, J.; Kottoor, N.; Wenzel, A.; Krueger, M.; et al. Three-layered proteomic characterization of a novel ACTN4 mutation unravels its pathogenic potential in FSGS. Hum. Mol. Genet. 2016, 25, 1152–1164. [Google Scholar] [CrossRef] [Green Version]
- Goode, N.P.; Shires, M.; Khan, T.N.; Mooney, A.F. Expression of alpha-actinin-4 in acquired human nephrotic syndrome: A quantitative immunoelectron microscopy study. Nephrol. Dial. Transplant. 2004, 19, 844–851. [Google Scholar] [CrossRef] [Green Version]
- Wagrowska-Danilewicz, M.; Stasikowska, O.; Danilewicz, M. Immunoexpression of podocyte-associated proteins in acquired human glomerulopathies with nephrotic syndrome. Pol. J. Pathol. 2006, 57, 17–21. [Google Scholar] [PubMed]
- Dandapani, S.V.; Sugimoto, H.; Matthews, B.D.; Kolb, R.J.; Sinha, S.; Gerszten, R.E.; Zhou, J.; Ingber, D.E.; Kalluri, R.; Pollak, M.R. Alpha-actinin-4 is required for normal podocyte adhesion. J. Biol. Chem. 2007, 282, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, A.; Gudermann, T. TRPC6. Handb. Exp. Pharm. 2007. [Google Scholar] [CrossRef]
- Dryer, S.E.; Reiser, J. TRPC6 channels and their binding partners in podocytes: Role in glomerular filtration and pathophysiology. Am. J. Physiol. Ren. Physiol. 2010, 299, F689–F701. [Google Scholar] [CrossRef] [Green Version]
- Reiser, J.; Polu, K.R.; Möller, C.C.; Kenlan, P.; Altintas, M.M.; Wei, C.; Faul, C.; Herbert, S.; Villegas, I.; Avila-Casado, C.; et al. TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function. Nat. Genet. 2005, 37, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Anderson, M.; Wilson, C.; Hagmann, H.; Benzing, T.; Dryer, S.E. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: Essential role of podocin in formation of this complex. Am. J. Physiol.—Cell Physiol. 2013, 305, C960–C971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dryer, S.E.; Roshanravan, H.; Kim, E.Y. TRPC channels: Regulation, dysregulation and contributions to chronic kidney disease. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 1041–1066. [Google Scholar] [CrossRef] [PubMed]
- Verheijden, K.A.T.; Sonneveld, R.; Bakker-van Bebber, M.; Wetzels, J.F.M.; van der Vlag, J.; Nijenhuis, T. The Calcium-Dependent Protease Calpain-1 Links TRPC6 Activity to Podocyte Injury. J. Am. Soc. Nephrol. 2018, 29, 2099–2109. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Jacobo, S.M.P.; Billing, D.; Rozkalne, A.; Gage, S.D.; Anagnostou, T.; Pavenstädt, H.; Hsu, H.-H.; Schlondorff, J.; Ramos, A.; et al. Antagonistic Regulation of Actin Dynamics and Cell Motility by TRPC5 and TRPC6 Channels. Sci. Signal. 2010, 3, ra77. [Google Scholar] [CrossRef] [Green Version]
- Ilatovskaya, D.V.; Palygin, O.; Chubinskiy-Nadezhdin, V.; Negulyaev, Y.A.; Ma, R.; Birnbaumer, L.; Staruschenko, A. Angiotensin II has acute effects on TRPC6 channels in podocytes of freshly isolated glomeruli. Kidney Int. 2014, 86, 506–514. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.; Roshanravan, H.; Khine, J.; Dryer, S.E. Angiotensin II Activation of TRPC6 Channels in Rat Podocytes Requires Generation of Reactive Oxygen Species. J. Cell. Physiol. 2014, 229, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, H.; Dryer, S.E. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: Role of podocin and reactive oxygen species. Am. J. Physiol. Ren. Physiol. 2014, 306, F1088–F1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farmer, L.K.; Rollason, R.; Whitcomb, D.J.; Ni, L.; Goodliff, A.; Lay, A.C.; Birnbaumer, L.; Heesom, K.J.; Xu, S.-Z.; Saleem, M.A.; et al. TRPC6 Binds to and Activates Calpain, Independent of Its Channel Activity, and Regulates Podocyte Cytoskeleton, Cell Adhesion, and Motility. J. Am. Soc. Nephrol. 2019, 30, 1910–1924. [Google Scholar] [CrossRef] [Green Version]
- Harita, Y.; Kurihara, H.; Kosako, H.; Tezuka, T.; Sekine, T.; Igarashi, T.; Ohsawa, I.; Ohta, S.; Hattori, S. Phosphorylation of Nephrin Triggers Ca2+ Signaling by Recruitment and Activation of Phospholipase C-{gamma}1. J. Biol. Chem. 2009, 284, 8951–8962. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, Q.; Wang, Y.; Chen, X.; Wang, Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol. Endocrinol. 2009, 23, 901–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canaud, G.; Bienaimé, F.; Viau, A.; Treins, C.; Baron, W.; Nguyen, C.; Burtin, M.; Berissi, S.; Giannakakis, K.; Muda, A.O.; et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat. Med. 2013, 19, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Brandt, D.T.; Grosse, R. Get to grips: Steering local actin dynamics with IQGAPs. Embo Rep. 2007, 8, 1019–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermle, T.; Schneider, R.; Schapiro, D.; Braun, D.A.; van der Ven, A.T.; Warejko, J.K.; Daga, A.; Widmeier, E.; Nakayama, M.; Jobst-Schwan, T.; et al. GAPVD1 and ANKFY1 Mutations Implicate RAB5 Regulation in Nephrotic Syndrome. J. Am. Soc. Nephrol. 2018, 29, 2123–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claverie-Martin, F.; Trindade, A.; Garcia-Gonzalez, N.C.; Callejon, A.C. Novel missense mutation affecting the LIM-A domain of LMX1B in a family with Nail-Patella syndrome. Intractable Rare Dis. Res. 2019, 8, 14–19. [Google Scholar] [CrossRef]
- Andeen, N.K.; Schleit, J.; Blosser, C.D.; Dorschner, M.O.; Hisama, F.M.; Smith, K.D. LMX1B-Associated Nephropathy With Type III Collagen Deposition in the Glomerular and Tubular Basement Membranes. Am. J. Kidney Dis. 2018, 72, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.; Lennon, R. Basement Membrane Defects in Genetic Kidney Diseases. Front. Pediatr. 2018, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boute, N.; Gribouval, O.; Roselli, S.; Benessy, F.; Lee, H.; Fuchshuber, A.; Dahan, K.; Gubler, M.C.; Niaudet, P.; Antignac, C. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 2000, 24, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Asharam, K.; Bhimma, R.; David, V.A.; Coovadia, H.M.; Qulu, W.P.; Naicker, T.; Gillies, C.E.; Vega-Warner, V.; Johnson, R.C.; Limou, S.; et al. NPHS2 V260E Is a Frequent Cause of Steroid-Resistant Nephrotic Syndrome in Black South African Children. Kidney Int. Rep. 2018, 3, 1354–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baylarov, R.; Senol, O.; Atan, M.; Berdeli, A. NPHS2 gene mutations in azerbaijani children with steroid-resistant nephrotic syndrome. Saudi J. Kidney Dis. Transplant. 2020, 31, 144–149. [Google Scholar] [CrossRef]
- Harita, Y.; Urae, S.; Akashio, R.; Isojima, T.; Miura, K.; Yamada, T.; Yamamoto, K.; Miyasaka, Y.; Furuyama, M.; Takemura, T.; et al. Clinical and genetic characterization of nephropathy in patients with nail-patella syndrome. Eur. J. Hum. Genet. 2020. [Google Scholar] [CrossRef]
- Lu, X.Y.; Liu, B.C.; Cao, Y.Z.; Song, C.; Su, H.; Chen, G.; Klein, J.D.; Zhang, H.X.; Wang, L.H.; Ma, H.P. High glucose reduces expression of podocin in cultured human podocytes by stimulating TRPC6. Am. J. Physiol. Ren. Physiol. 2019, 317, F1605–F1611. [Google Scholar] [CrossRef]
- Forst, A.L.; Olteanu, V.S.; Mollet, G.; Wlodkowski, T.; Schaefer, F.; Dietrich, A.; Reiser, J.; Gudermann, T.; Mederos y Schnitzler, M.; Storch, U. Podocyte Purinergic P2X4 Channels Are Mechanotransducers That Mediate Cytoskeletal Disorganization. J. Am. Soc. Nephrol. 2016, 27, 848–862. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.; Kim, E.Y.; Hagmann, H.; Benzing, T.; Dryer, S.E. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am. J. Physiol. Cell Physiol. 2013, 305, C276–C289. [Google Scholar] [CrossRef]
- Cormont, M.; Metón, I.; Mari, M.; Monzo, P.; Keslair, F.; Gaskin, C.; McGraw, T.E.; Le Marchand-Brustel, Y. CD2AP/CMS regulates endosome morphology and traffic to the degradative pathway through its interaction with Rab4 and c-Cbl. Traffic 2003, 4, 97–112. [Google Scholar] [CrossRef]
- Yaddanapudi, S.; Altintas, M.M.; Kistler, A.D.; Fernandez, I.; Möller, C.C.; Wei, C.; Peev, V.; Flesche, J.B.; Forst, A.L.; Li, J.; et al. CD2AP in mouse and human podocytes controls a proteolytic program that regulates cytoskeletal structure and cellular survival. J. Clin. Investig. 2011, 121, 3965–3980. [Google Scholar] [CrossRef] [Green Version]
- Löwik, M.M.; Groenen, P.J.T.A.; Pronk, I.; Lilien, M.R.; Goldschmeding, R.; Dijkman, H.B.; Levtchenko, E.N.; Monnens, L.A.; van den Heuvel, L.P. Focal segmental glomerulosclerosis in a patient homozygous for a CD2AP mutation. Kidney Int. 2007, 72, 1198–1203. [Google Scholar] [CrossRef] [PubMed]
- Takano, T.; Bareke, E.; Takeda, N.; Aoudjit, L.; Baldwin, C.; Pisano, P.; Matsuda, J.; El Andalousi, J.; Muhtadie, L.; Bernard, C.; et al. Recessive mutation in CD2AP causes focal segmental glomerulosclerosis in humans and mice. Kidney Int. 2019, 95, 57–61. [Google Scholar] [CrossRef] [PubMed]
- van Duijn, T.J.; Anthony, E.C.; Hensbergen, P.J.; Deelder, A.M.; Hordijk, P.L. Rac1 recruits the adapter protein CMS/CD2AP to cell-cell contacts. J. Biol. Chem. 2010, 285, 20137–20146. [Google Scholar] [CrossRef] [Green Version]
- Balbas, M.D.; Burgess, M.R.; Murali, R.; Wongvipat, J.; Skaggs, B.J.; Mundel, P.; Weins, A.; Sawyers, C.L. MAGI-2 scaffold protein is critical for kidney barrier function. Proc. Natl. Acad. Sci. USA 2014, 111, 14876–14881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, K.; Asanuma, K.; Fukuda, T.; Ohwada, S.; Yoshida, M.; Nishimori, K. MAGI-2 is critical for the formation and maintenance of the glomerular filtration barrier in mouse kidney. Am. J. Pathol. 2014, 184, 2699–2708. [Google Scholar] [CrossRef] [PubMed]
- Empitu, M.A.; Kadariswantiningsih, I.N.; Aizawa, M.; Asanuma, K. MAGI-2 and scaffold proteins in glomerulopathy. Am. J. Physiol. Ren. Physiol. 2018, 315, F1336–F1344. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, S.; Kudo, H.; Rao, J.; Kikuchi, A.; Widmeier, E.; Lawson, J.A.; Tan, W.; Hermle, T.; Warejko, J.K.; Shril, S.; et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat. Commun. 2018, 9, 1960. [Google Scholar] [CrossRef] [Green Version]
- Lehtonen, S.; Ryan, J.J.; Kudlicka, K.; Iino, N.; Zhou, H.; Farquhar, M.G. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc. Natl. Acad. Sci. USA 2005, 102, 9814–9819. [Google Scholar] [CrossRef] [Green Version]
- Kanai, M.; Jeon, H.; Ojima, M.; Nishino, T.; Usui, T.; Yadav, M.K.; Kulathunga, K.; Morito, N.; Takahashi, S.; Hamada, M. Phenotypic analysis of mice carrying human-type MAFB p.Leu239Pro mutation. Biochem. Biophys. Res. Commun. 2020, 523, 452–457. [Google Scholar] [CrossRef]
- Zankl, A.; Duncan, E.L.; Leo, P.J.; Clark, G.R.; Glazov, E.A.; Addor, M.-C.; Herlin, T.; Kim, C.A.; Leheup, B.P.; McGill, J.; et al. Multicentric Carpotarsal Osteolysis Is Caused by Mutations Clustering in the Amino-Terminal Transcriptional Activation Domain of MAFB. Am. J. Hum. Genet. 2012, 90, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Tsukaguchi, H.; Morita, H.; Higasa, K.; Tran, M.T.N.; Hamada, M.; Usui, T.; Morito, N.; Horita, S.; Hayashi, T.; et al. A mutation in transcription factor MAFB causes Focal Segmental Glomerulosclerosis with Duane Retraction Syndrome. Kidney Int. 2018, 94, 396–407. [Google Scholar] [CrossRef]
- Srivastava, T.; Garola, R.E.; Whiting, J.M.; Alon, U.S. Synaptopodin expression in idiopathic nephrotic syndrome of childhood. Kidney Int. 2001, 59, 118–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Kistler, A.; Faridi, M.H.; Meyer, J.O.; Tryniszewska, B.; Mehta, D.; Yue, L.; Dryer, S.; Reiser, J. Synaptopodin Limits TRPC6 Podocyte Surface Expression and Attenuates Proteinuria. J. Am. Soc. Nephrol. 2016, 27, 3308–3319. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, M.; Tsuruya, K.; Yotsueda, H.; Tokumoto, M.; Ikeda, H.; Katafuchi, R.; Fujimi, S.; Hirakata, H.; Iida, M. Expression of synaptopodin and GLEPP1 as markers of steroid responsiveness in primary focal segmental glomerulosclerosis. Life Sci. 2006, 79, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wang, Z.; Pan, X.; Wang, W.; Chen, X.; Ren, H.; Hao, C.; Han, B.; Chen, N. Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2010, 25, 824–835. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Ishibe, S. Targeting the podocyte cytoskeleton: From pathogenesis to therapy in proteinuric kidney disease. Nephrol. Dial. Transplant. 2016, 31, 1577–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, J.M.; Kim, S.H.; North, K.N.; Rennke, H.; Correia, L.A.; Tong, H.Q.; Mathis, B.J.; Rodríguez-Pérez, J.C.; Allen, P.G.; Beggs, A.H.; et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 2000, 24, 251–256. [Google Scholar] [CrossRef]
- Meng, L.; Cao, S.; Lin, N.; Zhao, J.; Cai, X.; Liang, Y.; Huang, K.; Lin, M.; Chen, X.; Li, D.; et al. Identification of a Novel ACTN4 Gene Mutation Which Is Resistant to Primary Nephrotic Syndrome Therapy. Biomed Res. Int. 2019, 2019, 5949485. [Google Scholar] [CrossRef]
- Henderson, J.M.; Al-Waheeb, S.; Weins, A.; Dandapani, S.V.; Pollak, M.R. Mice with altered alpha-actinin-4 expression have distinct morphologic patterns of glomerular disease. Kidney Int. 2008, 73, 741–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kos, C.H.; Le, T.C.; Sinha, S.; Henderson, J.M.; Kim, S.H.; Sugimoto, H.; Kalluri, R.; Gerszten, R.E.; Pollak, M.R. Mice deficient in alpha-actinin-4 have severe glomerular disease. J. Clin. Investig. 2003, 111, 1683–1690. [Google Scholar] [CrossRef] [Green Version]
- Gee, H.Y.; Saisawat, P.; Ashraf, S.; Hurd, T.W.; Vega-Warner, V.; Fang, H.; Beck, B.B.; Gribouval, O.; Zhou, W.; Diaz, K.A.; et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J. Clin. Investig. 2013, 123, 3243–3253. [Google Scholar] [CrossRef] [Green Version]
- Robins, R.; Baldwin, C.; Aoudjit, L.; Gupta, I.R.; Takano, T. Loss of Rho-GDIα sensitizes podocytes to lipopolysaccharide-mediated injury. Am. J. Physiol. Ren. Physiol. 2015, 308, F1207–F1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akilesh, S.; Suleiman, H.; Yu, H.; Stander, M.C.; Lavin, P.; Gbadegesin, R.; Antignac, C.; Pollak, M.; Kopp, J.B.; Winn, M.P.; et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J. Clin. Investig. 2011, 121, 4127–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, H.Y.; Zhang, F.; Ashraf, S.; Kohl, S.; Sadowski, C.E.; Vega-Warner, V.; Zhou, W.; Lovric, S.; Fang, H.; Nettleton, M.; et al. KANK deficiency leads to podocyte dysfunction and nephrotic syndrome. J. Clin. Investig. 2015, 125, 2375–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, H.Y.; Sadowski, C.E.; Aggarwal, P.K.; Porath, J.D.; Yakulov, T.A.; Schueler, M.; Lovric, S.; Ashraf, S.; Braun, D.A.; Halbritter, J.; et al. FAT1 mutations cause a glomerulotubular nephropathy. Nat. Commun. 2016, 7, 10822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goode, B.L.; Eck, M.J. Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 2007, 76, 593–627. [Google Scholar] [CrossRef] [PubMed]
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Cong, E.H.; Arrondel, C.; Tête, M.-J.; Montjean, R.; et al. INF2 Mutations in Charcot–Marie–Tooth Disease with Glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, B.; Chun, J.; Perez-Gill, C.; Yan, P.; Stillman, I.E.; Higgs, H.N.; Alper, S.L.; Schlöndorff, J.S.; Pollak, M.R. FSGS-Causing INF2 Mutation Impairs Cleaved INF2 N-Fragment Functions in Podocytes. J. Am. Soc. Nephrol. 2020, 31, 374–391. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Tonna, S.J.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat. Genet. 2010, 42, 72–76. [Google Scholar] [CrossRef]
- Gbadegesin, R.A.; Lavin, P.J.; Hall, G.; Bartkowiak, B.; Homstad, A.; Jiang, R.; Wu, G.; Byrd, A.; Lynn, K.; Wolfish, N.; et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int. 2012, 81, 94–99. [Google Scholar] [CrossRef] [Green Version]
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013, 83, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.S.; McNagny, K.M. The role of podocalyxin in health and disease. J. Am. Soc. Nephrol. 2009, 20, 1669–1676. [Google Scholar] [CrossRef] [Green Version]
- Doyonnas, R.; Kershaw, D.B.; Duhme, C.; Merkens, H.; Chelliah, S.; Graf, T.; McNagny, K.M. Anuria, omphalocele, and perinatal lethality in mice lacking the CD34-related protein podocalyxin. J. Exp. Med. 2001, 194, 13–27. [Google Scholar] [CrossRef]
- Kang, H.G.; Lee, M.; Lee, K.B.; Hughes, M.; Kwon, B.S.; Lee, S.; McNagny, K.M.; Ahn, Y.H.; Ko, J.M.; Ha, I.S.; et al. Loss of podocalyxin causes a novel syndromic type of congenital nephrotic syndrome. Exp. Mol. Med. 2017, 49, e414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.J.; Wu, R.M.; Ho, C.H.; Cheng, J.; Lin, H.Y.; Lin, C.H. Genetic analysis of PODXL gene in patients with familial and young-onset Parkinson’s disease in a Taiwanese population. Neurobiol. Aging 2019, 84, 235.e9–235.e10. [Google Scholar] [CrossRef] [PubMed]
- El-Ashmawy, H.M.; Selim, F.O.; Hosny, T.A.M.; Almassry, H.N. Association of serum podocalyxin levels with peripheral arterial disease in patients with type 2 diabetes. J. Diabetes Complicat. 2019, 33, 495–499. [Google Scholar] [CrossRef]
- Mansilla, M.; Wang, Y.; Hyett, J.; da Silva Costa, F.; Nie, G. Serum podocalyxin for early detection of preeclampsia at 11-13 weeks of gestation. Placenta 2018, 71, 13–15. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Wang, L.; Spurney, R.F. TRPC Channels in Proteinuric Kidney Diseases. Cells 2019, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.P.; Conlon, P.J.; Lynn, K.L.; Farrington, M.K.; Creazzo, T.; Hawkins, A.F.; Daskalakis, N.; Kwan, S.Y.; Ebersviller, S.; Burchette, J.L.; et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005, 308, 1801–1804. [Google Scholar] [CrossRef] [Green Version]
- Riehle, M.; Büscher, A.K.; Gohlke, B.-O.; Kaßmann, M.; Kolatsi-Joannou, M.; Bräsen, J.H.; Nagel, M.; Becker, J.U.; Winyard, P.; Hoyer, P.F.; et al. TRPC6 G757D Loss-of-Function Mutation Associates with FSGS. J. Am. Soc. Nephrol. 2016, 27, 2771–2783. [Google Scholar] [CrossRef] [Green Version]
- Santín, S.; Ars, E.; Rossetti, S.; Salido, E.; Silva, I.; García-Maset, R.; Giménez, I.; Ruíz, P.; Mendizábal, S.; Luciano Nieto, J.; et al. TRPC6 mutational analysis in a large cohort of patients with focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 3089–3096. [Google Scholar] [CrossRef] [Green Version]
- Heeringa, S.F.; Möller, C.C.; Du, J.; Yue, L.; Hinkes, B.; Chernin, G.; Vlangos, C.N.; Hoyer, P.F.; Reiser, J.; Hildebrandt, F. A novel TRPC6 mutation that causes childhood FSGS. PLoS ONE 2009, 4, e7771. [Google Scholar] [CrossRef]
- Zhu, B.; Chen, N.; Wang, Z.H.; Pan, X.X.; Ren, H.; Zhang, W.; Wang, W.M. Identification and functional analysis of a novel TRPC6 mutation associated with late onset familial focal segmental glomerulosclerosis in Chinese patients. Mutat. Res. 2009, 664, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jirka, G.; Rosenberg, P.B.; Buckley, A.F.; Gomez, J.A.; Fields, T.A.; Winn, M.P.; Spurney, R.F. Gq signaling causes glomerular injury by activating TRPC6. J. Clin. Investig. 2015, 125, 1913–1926. [Google Scholar] [CrossRef] [Green Version]
- Freichel, M.; Vennekens, R.; Olausson, J.; Stolz, S.; Philipp, S.E.; Weissgerber, P.; Flockerzi, V. Functional role of TRPC proteins in native systems: Implications from knockout and knock-down studies. J. Physiol. 2005, 567, 59–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinkes, B.; Wiggins, R.C.; Gbadegesin, R.; Vlangos, C.N.; Seelow, D.; Nürnberg, G.; Garg, P.; Verma, R.; Chaib, H.; Hoskins, B.E.; et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat. Genet. 2006, 38, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.; Ashraf, S.; Tan, W.; van der Ven, A.T.; Gee, H.Y.; Braun, D.A.; Fehér, K.; George, S.P.; Esmaeilniakooshkghazi, A.; Choi, W.I.; et al. Advillin acts upstream of phospholipase C ϵ1 in steroid-resistant nephrotic syndrome. J. Clin. Investig. 2017, 127, 4257–4269. [Google Scholar] [CrossRef]
- Yu, S.; Choi, W.I.; Choi, Y.J.; Kim, H.Y.; Hildebrandt, F.; Gee, H.Y. PLCE1 regulates the migration, proliferation, and differentiation of podocytes. Exp. Mol. Med. 2020, 52, 594–603. [Google Scholar] [CrossRef] [Green Version]
- Atchison, D.K.; O’Connor, C.L.; Menon, R.; Otto, E.A.; Ganesh, S.K.; Wiggins, R.C.; Smrcka, A.V.; Bitzer, M. Hypertension induces glomerulosclerosis in phospholipase C-ε1 deficiency. Am. J. Physiol. Ren. Physiol. 2020, 318, F1177–F1187. [Google Scholar] [CrossRef]
- Hashmi, J.A.; Safar, R.A.; Afzal, S.; Albalawi, A.M.; Abdu-Samad, F.; Iqbal, Z.; Basit, S. Whole exome sequencing identification of a novel insertion mutation in the phospholipase C ε-1 gene in a family with steroid resistant inherited nephrotic syndrome. Mol. Med. Rep. 2018, 18, 5095–5100. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Goyal, M.; Wharram, B.; Wiggins, J.; Kershaw, D.; Wiggins, R. GLEPP1 receptor tyrosine phosphatase (Ptpro) in rat PAN nephrosis. A marker of acute podocyte injury. Nephron 2002, 90, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Wharram, B.L.; Goyal, M.; Gillespie, P.J.; Wiggins, J.E.; Kershaw, D.B.; Holzman, L.B.; Dysko, R.C.; Saunders, T.L.; Samuelson, L.C.; Wiggins, R.C. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J. Clin. Investig. 2000, 106, 1281–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozaltin, F.; Ibsirlioglu, T.; Taskiran, E.Z.; Baydar, D.E.; Kaymaz, F.; Buyukcelik, M.; Kilic, B.D.; Balat, A.; Iatropoulos, P.; Asan, E.; et al. Disruption of PTPRO Causes Childhood-Onset Nephrotic Syndrome. Am. J. Hum. Genet. 2011, 89, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Goyal, M.; Kurnit, D.; Wharram, B.; Wiggins, J.; Holzman, L.; Kershaw, D.; Wiggins, R. Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat. Kidney Int. 2001, 60, 957–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, J.; Wang, H.P.; Mao, Y.Y.; Jin, J.; Chen, J.H. Reduced glomerular epithelial protein 1 expression and podocyte injury in immunoglobulin A nephropathy. J. Int. Med. Res. 2007, 35, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Sonnenberg, A. Cell-matrix adhesion of podocytes in physiology and disease. Nat. Rev. Nephrol. 2013, 9, 200–210. [Google Scholar] [CrossRef]
- Pozzi, A.; Jarad, G.; Moeckel, G.W.; Coffa, S.; Zhang, X.; Gewin, L.; Eremina, V.; Hudson, B.G.; Borza, D.B.; Harris, R.C.; et al. Beta1 integrin expression by podocytes is required to maintain glomerular structural integrity. Dev. Biol. 2008, 316, 288–301. [Google Scholar] [CrossRef] [Green Version]
- Has, C.; Spartà, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin α3 mutations with kidney, lung, and skin disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef] [Green Version]
- Verma, R.; Venkatareddy, M.; Kalinowski, A.; Patel, S.R.; Garg, P. Integrin Ligation Results in Nephrin Tyrosine Phosphorylation In Vitro. PLoS ONE 2016, 11, e0148906. [Google Scholar] [CrossRef] [Green Version]
- Schlaepfer, D.D.; Mitra, S.K.; Ilic, D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta (Bba) Mol. Cell Res. 2004, 1692, 77–102. [Google Scholar] [CrossRef]
- Schaller, M.D. Cellular functions of FAK kinases: Insight into molecular mechanisms and novel functions. J. Cell Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.D.; Kiosses, W.B.; Sieg, D.J.; Otey, C.A.; Schlaepfer, D.D.; Schwartz, M.A. Focal adhesion kinase suppresses Rho activity to promote focal adhesion turnover. J. Cell Sci. 2000, 113, 3673–3678. [Google Scholar] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef]
- Lausecker, F.; Tian, X.; Inoue, K.; Wang, Z.; Pedigo, C.E.; Hassan, H.; Liu, C.; Zimmer, M.; Jinno, S.; Huckle, A.L.; et al. Vinculin is required to maintain glomerular barrier integrity. Kidney Int. 2018, 93, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayek, S.S.; Koh, K.H.; Grams, M.E.; Wei, C.; Ko, Y.-A.; Li, J.; Samelko, B.; Lee, H.; Dande, R.R.; Lee, H.W.; et al. A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017, 23, 945–953. [Google Scholar] [CrossRef]
- Tian, X.; Kim, J.J.; Monkley, S.M.; Gotoh, N.; Nandez, R.; Soda, K.; Inoue, K.; Balkin, D.M.; Hassan, H.; Son, S.H.; et al. Podocyte-associated talin1 is critical for glomerular filtration barrier maintenance. J. Clin. Investig. 2014, 124, 1098–1113. [Google Scholar] [CrossRef] [Green Version]
- Shahbazi, R.; Baradaran, B.; Khordadmehr, M.; Safaei, S.; Baghbanzadeh, A.; Jigari, F.; Ezzati, H. Targeting ROCK signaling in health, malignant and non-malignant diseases. Immunol. Lett. 2020, 219, 15–26. [Google Scholar] [CrossRef]
- Ramachandran, R.; Schmid, S.L. The dynamin superfamily. Curr. Biol. 2018, 28, R411–R416. [Google Scholar] [CrossRef] [Green Version]
- Yamada, H.; Abe, T.; Satoh, A.; Okazaki, N.; Tago, S.; Kobayashi, K.; Yoshida, Y.; Oda, Y.; Watanabe, M.; Tomizawa, K.; et al. Stabilization of actin bundles by a dynamin 1/cortactin ring complex is necessary for growth cone filopodia. J. Neurosci. 2013, 33, 4514–4526. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Yaddanapudi, S.; Weins, A.; Osborn, T.; Reiser, J.; Pollak, M.; Hartwig, J.; Sever, S. Direct dynamin-actin interactions regulate the actin cytoskeleton. Embo J. 2010, 29, 3593–3606. [Google Scholar] [CrossRef] [Green Version]
- Schiffer, M.; Teng, B.; Gu, C.; Shchedrina, V.A.; Kasaikina, M.; Pham, V.A.; Hanke, N.; Rong, S.; Gueler, F.; Schroder, P.; et al. Pharmacological targeting of actin-dependent dynamin oligomerization ameliorates chronic kidney disease in diverse animal models. Nat. Med. 2015, 21, 601–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menon, M.; Schafer, D.A. Dynamin: Expanding its scope to the cytoskeleton. Int. Rev. Cell Mol. Biol. 2013, 302, 187–219. [Google Scholar] [CrossRef] [PubMed]
- Sever, S.; Chang, J.; Gu, C. Dynamin rings: Not just for fission. Traffic 2013, 14, 1194–1199. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Kitamura, S.; Truong, D.M.; Rieg, T.; Vallon, V.; Sakurai, H.; Bush, K.T.; Vera, D.R.; Ross, R.S.; Nigam, S.K. Beta1-integrin is required for kidney collecting duct morphogenesis and maintenance of renal function. Am. J. Physiol. Ren. Physiol. 2009, 297, F210–F217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karamatic Crew, V.; Burton, N.; Kagan, A.; Green, C.A.; Levene, C.; Flinter, F.; Brady, R.L.; Daniels, G.; Anstee, D.J. CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood 2004, 104, 2217–2223. [Google Scholar] [CrossRef] [PubMed]
- Donnan, M.D.; Scott, R.P.; Onay, T.; Tarjus, A.; Onay, U.V.; Quaggin, S.E. Genetic Deletion of Emp2 Does Not Cause Proteinuric Kidney Disease in Mice. Front. Med. (Lausanne) 2019, 6, 189. [Google Scholar] [CrossRef] [Green Version]
- Dorval, G.; Gribouval, O.; Martinez-Barquero, V.; Machuca, E.; Tête, M.J.; Baudouin, V.; Benoit, S.; Chabchoub, I.; Champion, G.; Chauveau, D.; et al. Clinical and genetic heterogeneity in familial steroid-sensitive nephrotic syndrome. Pediatr. Nephrol. 2018, 33, 473–483. [Google Scholar] [CrossRef]
- Wan, X.; Chen, Z.; Choi, W.-I.; Gee, H.Y.; Hildebrandt, F.; Zhou, W. Loss of Epithelial Membrane Protein 2 Aggravates Podocyte Injury via Upregulation of Caveolin-1. J. Am. Soc. Nephrol. Jasn 2016, 27, 1066–1075. [Google Scholar] [CrossRef] [Green Version]
- Hagel, M.; George, E.L.; Kim, A.; Tamimi, R.; Opitz, S.L.; Turner, C.E.; Imamoto, A.; Thomas, S.M. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol. Cell Biol. 2002, 22, 901–915. [Google Scholar] [CrossRef] [Green Version]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [Green Version]
- Chinthalapudi, K.; Rangarajan, E.S.; Izard, T. The interaction of talin with the cell membrane is essential for integrin activation and focal adhesion formation. Proc. Natl. Acad. Sci. USA 2018, 115, 10339–10344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Wu, C. ILK: A pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 2012, 24, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Bordoy, R.; Stanchi, F.; Moser, M.; Braun, A.; Kudlacek, O.; Wewer, U.M.; Yurchenco, P.D.; Fässler, R. PINCH1 regulates cell-matrix and cell-cell adhesions, cell polarity and cell survival during the peri-implantation stage. J. Cell Sci. 2005, 118, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Guo, C.; Ma, P.; Lai, Y.; Yang, F.; Cai, J.; Cheng, Z.; Zhang, K.; Liu, Z.; Tian, Y.; et al. Kindlin-2 Association with Rho GDP-Dissociation Inhibitor α Suppresses Rac1 Activation and Podocyte Injury. J. Am. Soc. Nephrol. 2017, 28, 3545–3562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mele, C.; Iatropoulos, P.; Donadelli, R.; Calabria, A.; Maranta, R.; Cassis, P.; Buelli, S.; Tomasoni, S.; Piras, R.; Krendel, M.; et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef]
- Tabibzadeh, N.; Fleury, D.; Labatut, D.; Bridoux, F.; Lionet, A.; Jourde-Chiche, N.; Vrtovsnik, F.; Schlegel, N.; Vanhille, P. MYH9-related disorders display heterogeneous kidney involvement and outcome. Clin. Kidney J. 2019, 12, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Gbadegesin, R.A.; Hall, G.; Adeyemo, A.; Hanke, N.; Tossidou, I.; Burchette, J.; Wu, G.; Homstad, A.; Sparks, M.A.; Gomez, J.; et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J. Am. Soc. Nephrol. 2014, 25, 1991–2002. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.; Lane, B.M.; Khan, K.; Pediaditakis, I.; Xiao, J.; Wu, G.; Wang, L.; Kovalik, M.E.; Chryst-Stangl, M.; Davis, E.E.; et al. The Human FSGS-Causing ANLN R431C Mutation Induces Dysregulated PI3K/AKT/mTOR/Rac1 Signaling in Podocytes. J. Am. Soc. Nephrol. 2018, 29, 2110–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endlich, K.; Kliewe, F.; Endlich, N. Stressed podocytes-mechanical forces, sensors, signaling and response. Pflug. Arch. 2017, 469, 937–949. [Google Scholar] [CrossRef] [PubMed]
- Kriz, W.; Lemley, K.V. Mechanical challenges to the glomerular filtration barrier: Adaptations and pathway to sclerosis. Pediatr. Nephrol. 2017, 32, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Hostetter, T.H. Hypertrophy and hyperfunction of the diabetic kidney. J. Clin. Investig. 2001, 107, 161–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hostetter, T.H.; Rennke, H.G.; Brenner, B.M. The case for intrarenal hypertension in the initiation and progression of diabetic and other glomerulopathies. Am. J. Med. 1982, 72, 375–380. [Google Scholar] [CrossRef]
- Lemley, K.V. Glomerular pathology and the progression of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2016, 310, F1385–F1388. [Google Scholar] [CrossRef] [Green Version]
- Caulfield, J.P.; Reid, J.J.; Farquhar, M.G. Alterations of the glomerular epithelium in acute aminonucleoside nephrosis. Evidence for formation of occluding junctions and epithelial cell detachment. Lab. Investig. 1976, 34, 43–59. [Google Scholar]
- Inokuchi, S.; Sakai, T.; Shirato, I.; Tomino, Y.; Koide, H. Ultrastructural changes in glomerular epithelial cells in acute puromycin aminonucleoside nephrosis: A study by high-resolution scanning electron microscopy. Virchows Arch. A Pathol. Anat. Histopathol. 1993, 423, 111–119. [Google Scholar] [CrossRef]
- Friedrich, C.; Endlich, N.; Kriz, W.; Endlich, K. Podocytes are sensitive to fluid shear stress in vitro. Am. J. Physiol. Ren. Physiol. 2006, 291, F856–F865. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Fukui, M.; Ebihara, I.; Osada, S.; Nagaoka, I.; Tomino, Y.; Koide, H. mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes 1993, 42, 450–456. [Google Scholar] [CrossRef]
- Iglesias-de la Cruz, M.C.; Ziyadeh, F.N.; Isono, M.; Kouahou, M.; Han, D.C.; Kalluri, R.; Mundel, P.; Chen, S. Effects of high glucose and TGF-beta1 on the expression of collagen IV and vascular endothelial growth factor in mouse podocytes. Kidney Int. 2002, 62, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Herman-Edelstein, M.; Thomas, M.C.; Thallas-Bonke, V.; Saleem, M.; Cooper, M.E.; Kantharidis, P. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-β: A model for diabetic podocytopathy. Diabetes 2011, 60, 1779–1788. [Google Scholar] [CrossRef] [Green Version]
- Nagayama, Y.; Braun, G.S.; Jakobs, C.M.; Maruta, Y.; van Roeyen, C.R.; Klinkhammer, B.M.; Boor, P.; Villa, L.; Raffetseder, U.; Trautwein, C.; et al. Gp130-dependent signaling in the podocyte. Am. J. Physiol. Ren. Physiol. 2014, 307, F346–F355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutabarrik, A.; Nakanishi, I.; Ishibashi, M. Interleukin-6 and interleukin-6 receptor are expressed by cultured glomerular epithelial cells. Scand. J. Immunol. 1994, 40, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Dummer, P.; Kopp, J.; Qiu, L.; Levi, M.; Faubel, S.; Blaine, J. Endocytosis of albumin by podocytes elicits an inflammatory response and induces apoptotic cell death. PLoS ONE 2013, 8, e54817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.-F.; Bao, D.; Su, H.; Wang, Y.-M.; Lei, C.-T.; Zhang, C.-Y.; Ye, C.; Tang, H.; Wan, C.; You, C.-Q.; et al. IL-6 increases podocyte motility via MLC-mediated focal adhesion impairment and cytoskeleton disassembly. J. Cell. Physiol. 2018, 233, 7173–7181. [Google Scholar] [CrossRef]
- Tonsawan, P.; Dylewski, J.; Lewis, L.; Blaine, J. Knockout of the neonatal Fc receptor in cultured podocytes alters IL-6 signaling and the actin cytoskeleton. Am. J. Physiol. Cell Physiol. 2019, 317, C1048–C1060. [Google Scholar] [CrossRef]
- Hsu, H.-H.; Hoffmann, S.; Endlich, N.; Velic, A.; Schwab, A.; Weide, T.; Schlatter, E.; Pavenstädt, H. Mechanisms of angiotensin II signaling on cytoskeleton of podocytes. J. Mol. Med. 2008, 86, 1379–1394. [Google Scholar] [CrossRef] [Green Version]
- Königshausen, E.; Zierhut, U.M.; Ruetze, M.; Potthoff, S.A.; Stegbauer, J.; Woznowski, M.; Quack, I.; Rump, L.C.; Sellin, L. Angiotensin II increases glomerular permeability by β-arrestin mediated nephrin endocytosis. Sci. Rep. 2016, 6, 39513. [Google Scholar] [CrossRef] [Green Version]
- Kriz, W. Podocytes as a target for treatment with ACE inhibitors and/or angiotensin-receptor blockers. Kidney Int. 2004, 65, 333–334. [Google Scholar] [CrossRef] [Green Version]
- Macconi, D.; Ghilardi, M.; Bonassi, M.E.; Mohamed, E.I.; Abbate, M.; Colombi, F.; Remuzzi, G.; Remuzzi, A. Effect of Angiotensin-Converting Enzyme Inhibition on Glomerular Basement Membrane Permeability and Distribution of Zonula Occludens-1 in MWF Rats. J. Am. Soc. Nephrol. 2000, 11, 477–489. [Google Scholar]
- Davis, B.J.; Cao, Z.; de Gasparo, M.; Kawachi, H.; Cooper, M.E.; Allen, T.J. Disparate effects of angiotensin II antagonists and calcium channel blockers on albuminuria in experimental diabetes and hypertension: Potential role of nephrin. J. Hypertens. 2003, 21, 209–216. [Google Scholar] [CrossRef]
- Guess, A.; Agrawal, S.; Wei, C.-C.; Ransom, R.F.; Benndorf, R.; Smoyer, W.E. Dose- and time-dependent glucocorticoid receptor signaling in podocytes. Am. J. Physiol. Ren. Physiol. 2010, 299, F845–F853. [Google Scholar] [CrossRef] [Green Version]
- Xing, C.Y.; Saleem, M.A.; Coward, R.J.; Ni, L.; Witherden, I.R.; Mathieson, P.W. Direct effects of dexamethasone on human podocytes. Kidney Int. 2006, 70, 1038–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schönenberger, E.; Ehrich, J.H.; Haller, H.; Schiffer, M. The podocyte as a direct target of immunosuppressive agents. Nephrol. Dial. Transplant. 2010, 26, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Fornoni, A.; Sageshima, J.; Wei, C.; Merscher-Gomez, S.; Aguillon-Prada, R.; Jauregui, A.N.; Li, J.; Mattiazzi, A.; Ciancio, G.; Chen, L.; et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci. Transl. Med. 2011, 3, 85ra46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fornoni, A.; Merscher, S.; Kopp, J.B. Lipid biology of the podocyte--new perspectives offer new opportunities. Nat. Rev. Nephrol. 2014, 10, 379–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiser, J.; von Gersdorff, G.; Loos, M.; Oh, J.; Asanuma, K.; Giardino, L.; Rastaldi, M.P.; Calvaresi, N.; Watanabe, H.; Schwarz, K.; et al. Induction of B7-1 in podocytes is associated with nephrotic syndrome. J. Clin. Investig. 2004, 113, 1390–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.C.; Fornoni, A.; Weins, A.; Hakroush, S.; Maiguel, D.; Sageshima, J.; Chen, L.; Ciancio, G.; Faridi, M.H.; Behr, D.; et al. Abatacept in B7-1-positive proteinuric kidney disease. N. Engl. J. Med. 2013, 369, 2416–2423. [Google Scholar] [CrossRef] [Green Version]
- Calizo, R.C.; Bhattacharya, S.; van Hasselt, J.G.C.; Wei, C.; Wong, J.S.; Wiener, R.J.; Ge, X.; Wong, N.J.; Lee, J.-J.; Cuttitta, C.M.; et al. Disruption of podocyte cytoskeletal biomechanics by dasatinib leads to nephrotoxicity. Nat. Commun. 2019, 10, 2061. [Google Scholar] [CrossRef] [Green Version]
- Dylewski, J.; Blaine, J. Focal Segmental Glomerulosclerosis and Its Pathophysiology. In Proteinuria: Basic Mechanisms, Pathophysiology and Clinical Relevance; Blaine, J., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 117–139. [Google Scholar]
- Kriz, W.; Hähnel, B.; Hosser, H.; Rösener, S.; Waldherr, R. Structural analysis of how podocytes detach from the glomerular basement membrane under hypertrophic stress. Front. Endocrinol. (Lausanne) 2014, 5, 207. [Google Scholar] [CrossRef] [Green Version]
- Eng, D.G.; Sunseri, M.W.; Kaverina, N.V.; Roeder, S.S.; Pippin, J.W.; Shankland, S.J. Glomerular parietal epithelial cells contribute to adult podocyte regeneration in experimental focal segmental glomerulosclerosis. Kidney Int. 2015, 88, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Kaverina, N.V.; Eng, D.G.; Schneider, R.R.; Pippin, J.W.; Shankland, S.J. Partial podocyte replenishment in experimental FSGS derives from nonpodocyte sources. Am. J. Physiol. Ren. Physiol. 2016, 310, F1397–F1413. [Google Scholar] [CrossRef] [Green Version]
- Miesen, L.; Steenbergen, E.; Smeets, B. Parietal cells-new perspectives in glomerular disease. Cell Tissue Res. 2017, 369, 237–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheong, H.I. Genetic tests in children with steroid-resistant nephrotic syndrome. Kidney Res. Clin. Pract. 2020, 39, 7–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landini, S.; Mazzinghi, B.; Becherucci, F.; Allinovi, M.; Provenzano, A.; Palazzo, V.; Ravaglia, F.; Artuso, R.; Bosi, E.; Stagi, S.; et al. Reverse Phenotyping after Whole-Exome Sequencing in Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2020, 15, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62. [Google Scholar] [CrossRef]
- Riedhammer, K.M.; Braunisch, M.C.; Günthner, R.; Wagner, M.; Hemmer, C.; Strom, T.M.; Schmaderer, C.; Renders, L.; Tasic, V.; Gucev, Z.; et al. Exome Sequencing and Identification of Phenocopies in Patients With Clinically Presumed Hereditary Nephropathies. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Protein | Function |
|---|---|---|
| Focal Adhesions | ||
| CD151 | CD151 | Transmembrane protein regulator |
| EMP2 | Epithelial membrane protein 2 | Cell adhesion and trafficking protein |
| ITGA3 | Integrin α3 | Anchoring protein |
| ITGB1 | Integrin β1 | Anchoring protein |
| Slit Diaphragm | ||
| ANKFY1 | Rabakyrin-5 | Nephrin trafficking |
| CD2AP | CD2-associated protein | Slit diaphragm linking protein to actin cytoskeleton |
| GAPVD1 | GTPase-activating protein and VPS9 domain-containing protein 1 | Nephrin trafficking |
| LMX1B | LIM homeobox transcription factor 1-β | Transcription regulator of podocin |
| MAFB | Transcription factor MafB | Transcription regulator of nephrin, podocin, CD2AP |
| MAGI2 | Membrane-associated guanylate kinase | Scaffolding protein for nephrin complex |
| NPHS1 | Nephrin | Slit diaphragm signaling protein |
| NPHS2 | Podocin | Mechanosensing protein linking plasma membrane to actin cytoskeleton |
| PLCε1 | Phospholipase Cε1 | Slit diaphragm signaling protein |
| TPRC6 | Transient receptor potential channel 6 | Regulates Ca2+ signaling for mechanosensation Activates RhoA and Rac1 |
| Motility/Actin Dynamics | ||
| ACTN4 | α-actinin-4 | Links focal adhesions to actin cytoskeleton |
| ANLN | Anillin | Scaffold protein linking RhoA with actin |
| ARHGDIA | Rho GDP-dissociation inhibitor α | Regulates RhoGTPase signaling |
| ARHGAP24 | Rho GTPase-activating protein 24 | Regulates RhoGTPase signaling |
| AVIL | Advillin | Ca2+ regulated actin-binding protein |
| CDK20 | Cyclin-dependent kinase 20 | Regulates RhoA/Rac through regulating DLC1 |
| DLC1 | Rho GTPase-activating protein 7 | Regulates RhoGTPase signaling |
| INF2 | Inverted formin 2 | Cuts actin filaments |
| ITSN1 ITSN2 | Guanin exchange factor proteins | Activates Cdc42 |
| FAT1 | Fat cadherin 1 | Connects slit diaphragm and actin cytoskeleton |
| KANK1 KANK2 KANK4 | Kidney ankyrin repeat-containing protein | Regulates actin polymerization |
| MAGI2 | Membrane-associated guanylate kinase | Scaffold protein |
| MYH9 | Heavy chain of non-muscle myosinIIA | Contractile protein |
| PODXL | Podocalyxin | Modulates actin cytoskeleton |
| PTPRO | Glomerular epithelial protein 1 (GLEPP1) | Glomerular pressure maintenance |
| SYNPO | Synaptopodin | Actin-associated protein for foot process motility |
| TNS2 | Tensin-2 | Motility regulating through MAGi2 interaction |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blaine, J.; Dylewski, J. Regulation of the Actin Cytoskeleton in Podocytes. Cells 2020, 9, 1700. https://doi.org/10.3390/cells9071700
Blaine J, Dylewski J. Regulation of the Actin Cytoskeleton in Podocytes. Cells. 2020; 9(7):1700. https://doi.org/10.3390/cells9071700
Chicago/Turabian StyleBlaine, Judith, and James Dylewski. 2020. "Regulation of the Actin Cytoskeleton in Podocytes" Cells 9, no. 7: 1700. https://doi.org/10.3390/cells9071700
APA StyleBlaine, J., & Dylewski, J. (2020). Regulation of the Actin Cytoskeleton in Podocytes. Cells, 9(7), 1700. https://doi.org/10.3390/cells9071700