RNA Recognition and Immunity—Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
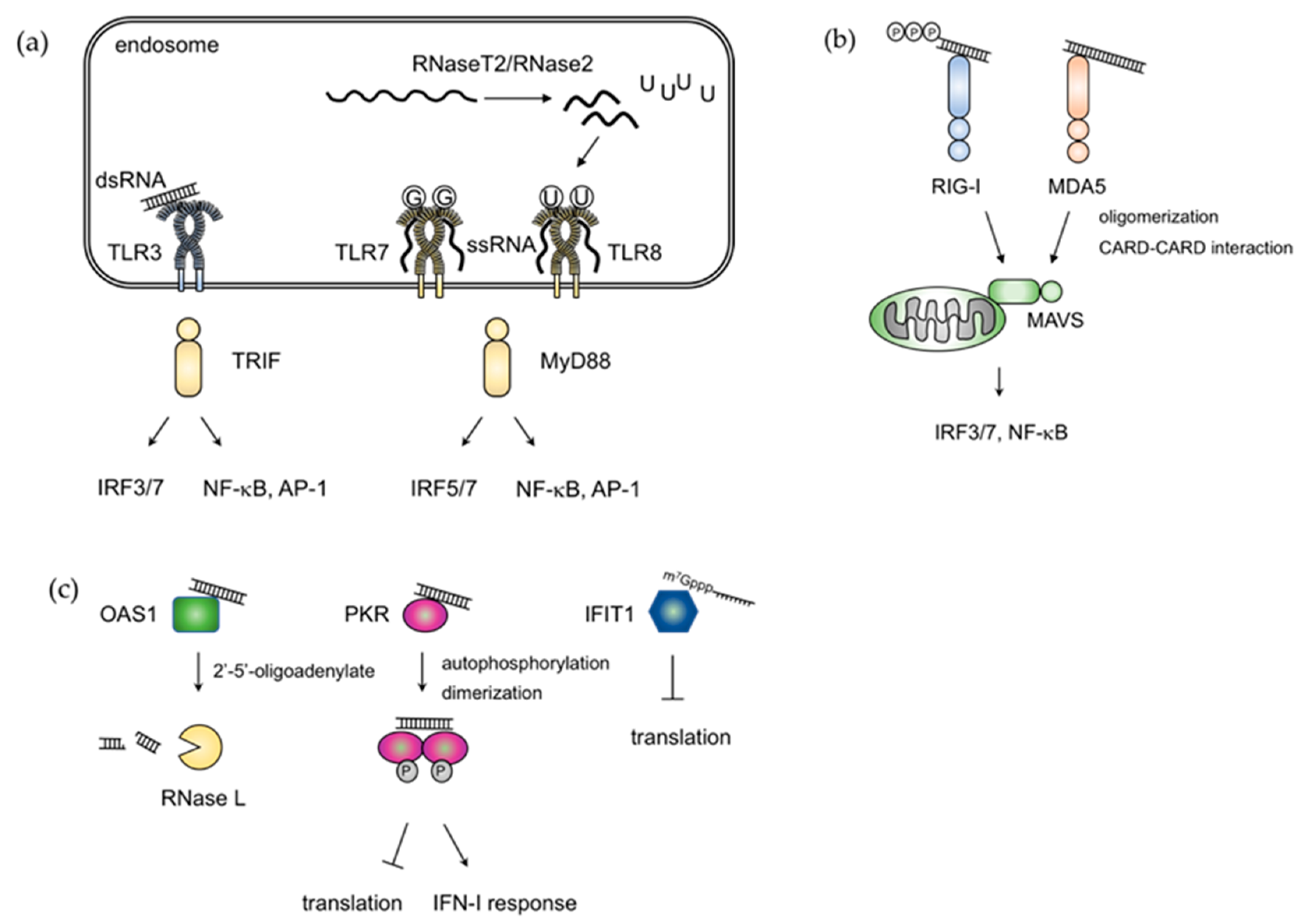
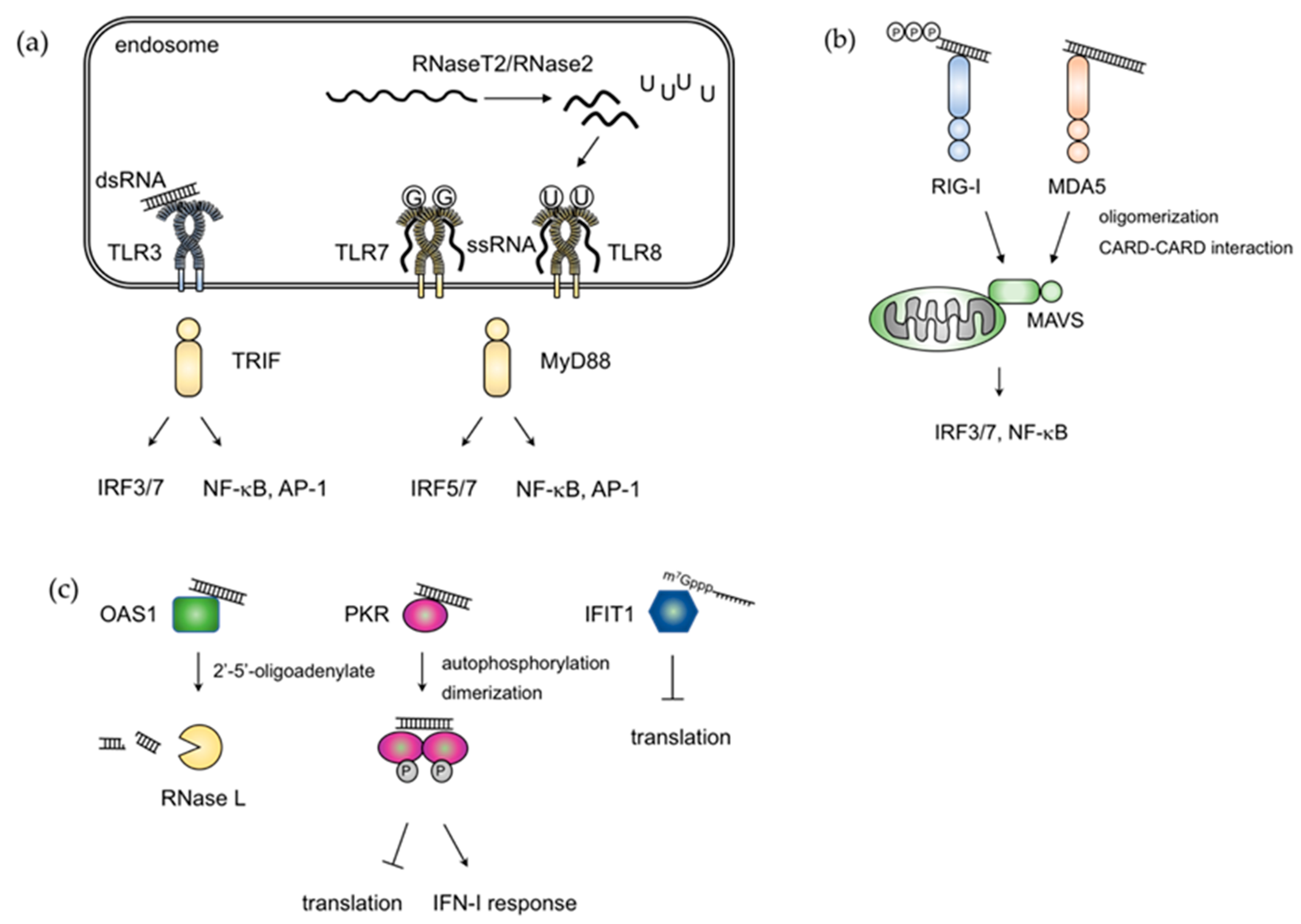
2. RNA Sensing Mechanisms
3. Elements for Discrimination between Self and Non-Self RNA
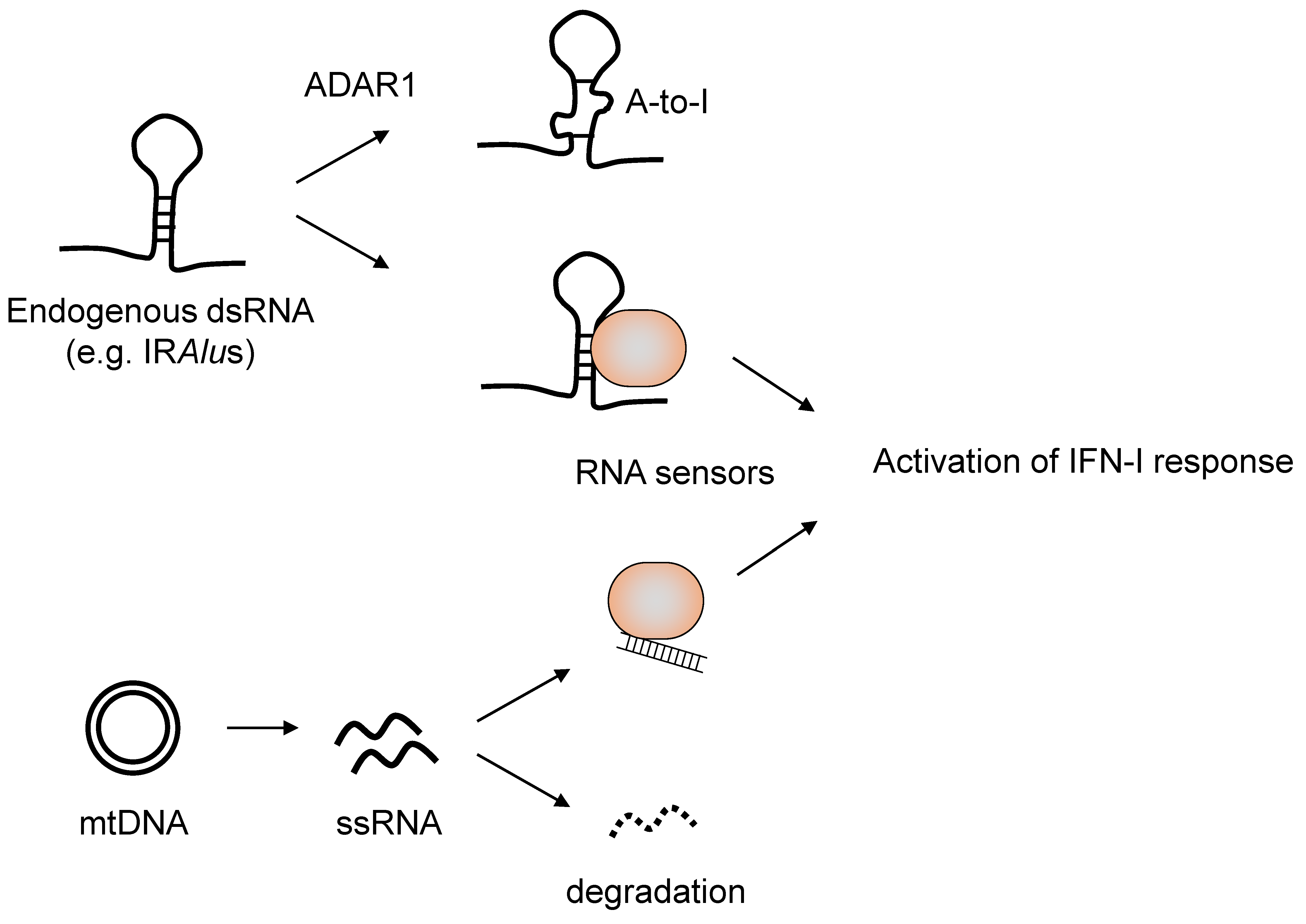
4. Recognition of Endogenous RNAs in Diseases
5. RBPs Directly Suppress Viral RNAs
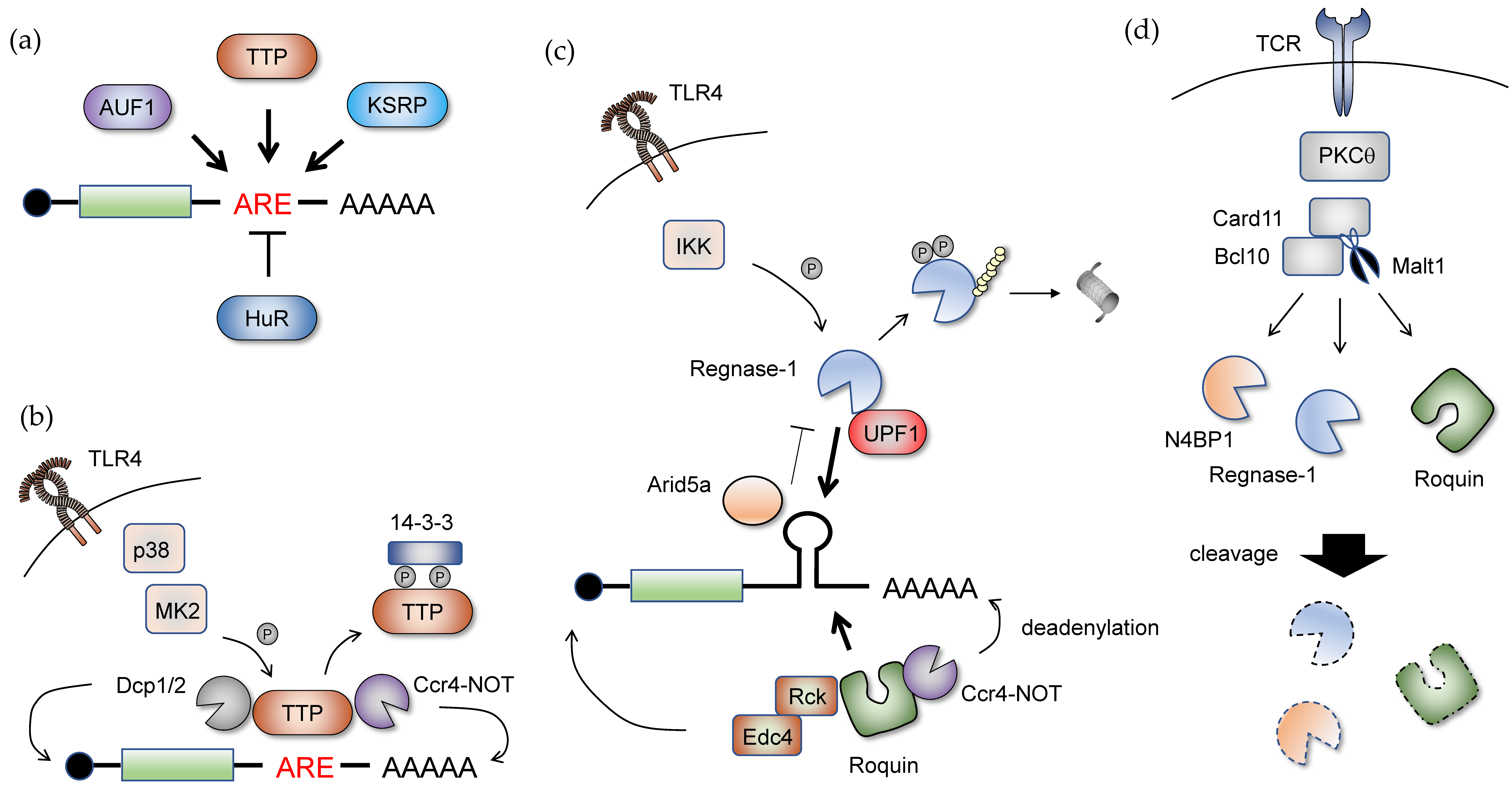
6. Posttranscriptional Mechanisms Regulating Host Inflammatory Gene Expression
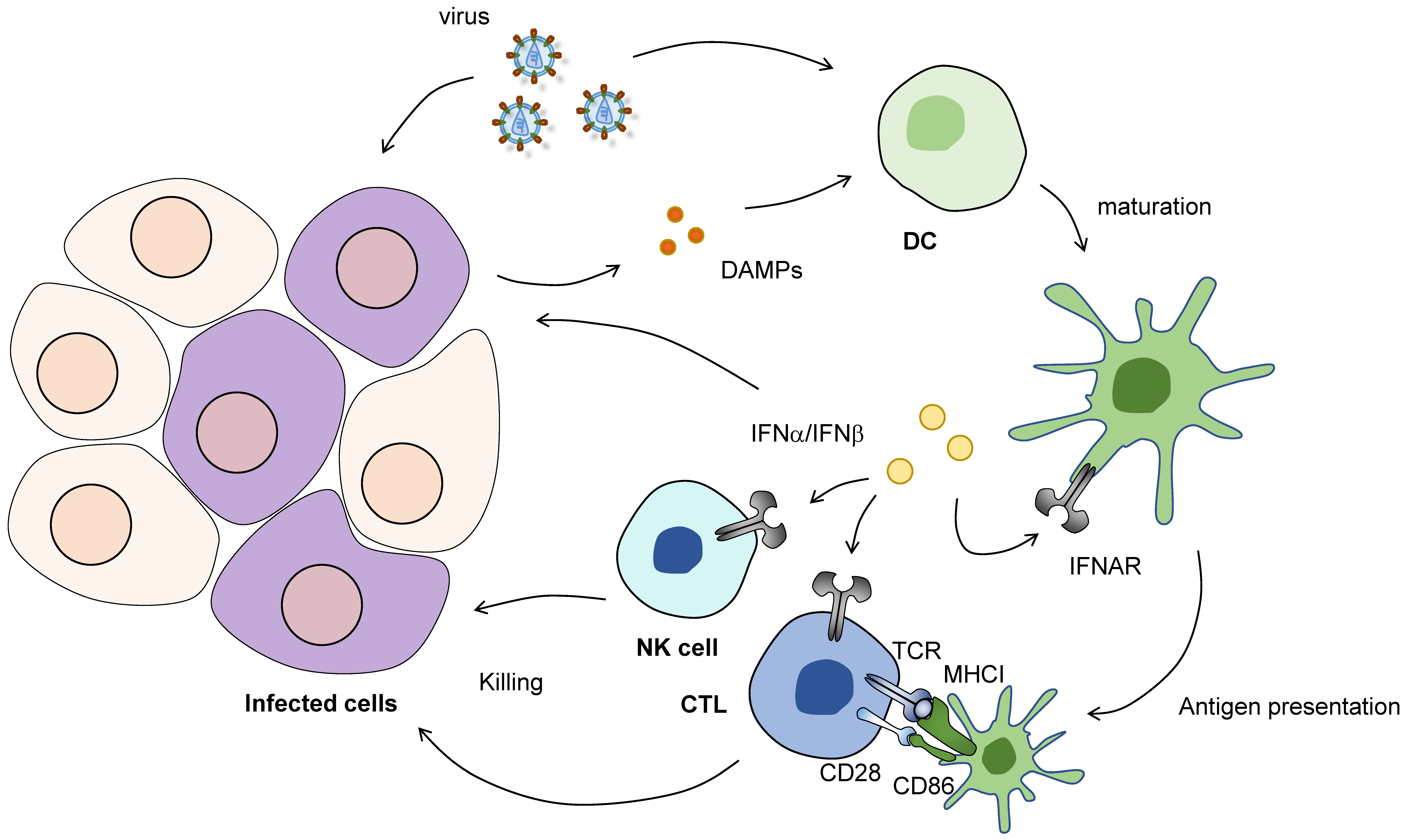
7. The Role of Posttranscriptional Regulation in Immune Homeostasis
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20303872 (accessed on 15 July 2020). [CrossRef] [PubMed] [Green Version]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. Available online: https://www.ncbi.nlm.nih.gov/pubmed/25789684 (accessed on 15 July 2020). [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. Available online: https://www.ncbi.nlm.nih.gov/pubmed/32164908 (accessed on 15 July 2020). [CrossRef] [PubMed]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. Damp-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. Available online: https://www.ncbi.nlm.nih.gov/pubmed/31558839 (accessed on 15 July 2020). [CrossRef] [PubMed]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Ten Oever, B.R. The Evolution of Antiviral Defense Systems. Cell Host Microbe 2016, 19, 142–149. [Google Scholar] [CrossRef]
- Takeda, K.; Kaisho, T.; Akira, S. Toll-like receptors. Annu. Rev. Immunol. 2003, 21, 335–376. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edelson, B.T.; Kc, W.; Juang, R.; Kohyama, M.; Benoit, L.A.; Klekotka, P.A.; Moon, C.; Albring, J.C.; Ise, W.; Michael, D.G.; et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J. Exp. Med. 2010, 207, 823–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune. Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; Cornen, S.; Daeron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kafasla, P.; Skliris, A.; Kontoyiannis, D.L. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat. Immunol. 2014, 15, 492–502. [Google Scholar] [CrossRef]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Cao, X. Long noncoding RNAs in innate immunity. Cell Mol. Immunol. 2016, 13, 138–147. [Google Scholar] [CrossRef]
- Chen, Y.G.; Satpathy, A.T.; Chang, H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017, 18, 962–972. [Google Scholar] [CrossRef]
- Hadjicharalambous, M.R.; Lindsay, M.A. Long Non-Coding RNAs and the Innate Immune Response. Noncoding RNA 2019, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.D.; Chen, Z.J. Sequence specific detection of bacterial 23S ribosomal RNA by TLR13. Elife 2012, 1, e00102. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, M.; Kruger, A.; Ferstl, R.; Kaufmann, A.; Nees, G.; Sigmund, A.; Bathke, B.; Lauterbach, H.; Suter, M.; Dreher, S.; et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 2012, 337, 1111–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Onomoto, K.; Jogi, M.; Akaboshi, T.; Fujita, T. Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 2015, 32, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Stark, G.R.; Darnell, J.E., Jr. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Ablasser, A.; Hertrich, C.; Wassermann, R.; Hornung, V. Nucleic acid driven sterile inflammation. Clin. Immunol. 2013, 147, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Jefferies, C. Role of DNA/RNA sensors and contribution to autoimmunity. Cytokine Growth Factor Rev. 2014, 25, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Tsomos, E.; Hammerstad, S.S.; Tomer, Y. Interferon alpha: The key trigger of type 1 diabetes. J. AutoImmun. 2018, 94, 7–15. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Leonard, J.N.; Ghirlando, R.; Askins, J.; Bell, J.K.; Margulies, D.H.; Davies, D.R.; Segal, D.M. The TLR3 signaling complex forms by cooperative receptor dimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlee, M.; Roth, A.; Hornung, V.; Hagmann, C.A.; Wimmenauer, V.; Barchet, W.; Coch, C.; Janke, M.; Mihailovic, A.; Wardle, G.; et al. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 2009, 31, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Schwerd, T.; Hamm, W.; Hellmuth, J.C.; Cui, S.; Wenzel, M.; Hoffmann, F.S.; Michallet, M.C.; Besch, R.; Hopfner, K.P.; et al. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. USA 2009, 106, 12067–12072. [Google Scholar] [CrossRef] [Green Version]
- Schuberth-Wagner, C.; Ludwig, J.; Bruder, A.K.; Herzner, A.M.; Zillinger, T.; Goldeck, M.; Schmidt, T.; Schmid-Burgk, J.L.; Kerber, R.; Wolter, S.; et al. A Conserved Histidine in the RNA Sensor RIG-I Controls Immune Tolerance to N1-2′O-Methylated Self RNA. Immunity 2015, 43, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Marq, J.B.; Kolakofsky, D.; Garcin, D. Unpaired 5′ ppp-nucleotides, as found in arenavirus double-stranded RNA panhandles, are not recognized by RIG-I. J. Biol. Chem. 2010, 285, 18208–18216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goubau, D.; Schlee, M.; Deddouche, S.; Pruijssers, A.J.; Zillinger, T.; Goldeck, M.; Schuberth, C.; Van der Veen, A.G.; Fujimura, T.; Rehwinkel, J.; et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 2014, 514, 372–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, X.; Linehan, M.M.; Iwasaki, A.; Pyle, A.M. RIG-I Selectively Discriminates against 5′-Monophosphate RNA. Cell Rep. 2019, 26, 2019–2027 e2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Peisley, A.; Lin, C.; Wu, B.; Orme-Johnson, M.; Liu, M.; Walz, T.; Hur, S. Cooperative assembly and dynamic disassembly of MDA5 filaments for viral dsRNA recognition. Proc. Natl. Acad. Sci. USA 2011, 108, 21010–21015. [Google Scholar] [CrossRef] [Green Version]
- Berke, I.C.; Modis, Y. MDA5 cooperatively forms dimers and ATP-sensitive filaments upon binding double-stranded RNA. EMBO J. 2012, 31, 1714–1726. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Peisley, A.; Richards, C.; Yao, H.; Zeng, X.; Lin, C.; Chu, F.; Walz, T.; Hur, S. Structural basis for dsRNA recognition, filament formation, and antiviral signal activation by MDA5. Cell 2013, 152, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Qu, K.; Modis, Y. Cryo-EM Structures of MDA5-dsRNA Filaments at Different Stages of ATP Hydrolysis. Mol. Cell 2018, 72, 999–1012 e1016. [Google Scholar] [CrossRef] [Green Version]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like receptor 8 senses degradation products of single-stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Sato, R.; Shukla, N.M.; David, S.A.; Isobe, T.; Miyake, K.; et al. Structural Analyses of Toll-like Receptor 7 Reveal Detailed RNA Sequence Specificity and Recognition Mechanism of Agonistic Ligands. Cell Rep. 2018, 25, 3371–3381 e3375. [Google Scholar] [CrossRef] [Green Version]
- Greulich, W.; Wagner, M.; Gaidt, M.M.; Stafford, C.; Cheng, Y.; Linder, A.; Carell, T.; Hornung, V. TLR8 Is a Sensor of RNase T2 Degradation Products. Cell 2019, 179, 1264–1275 e1213. [Google Scholar] [CrossRef]
- Ostendorf, T.; Zillinger, T.; Andryka, K.; Schlee-Guimaraes, T.M.; Schmitz, S.; Marx, S.; Bayrak, K.; Linke, R.; Salgert, S.; Wegner, J.; et al. Immune Sensing of Synthetic, Bacterial, and Protozoan RNA by Toll-like Receptor 8 Requires Coordinated Processing by RNase T2 and RNase 2. Immunity 2020, 52, 591–605. [Google Scholar] [CrossRef]
- Oda, H.; Nakagawa, K.; Abe, J.; Awaya, T.; Funabiki, M.; Hijikata, A.; Nishikomori, R.; Funatsuka, M.; Ohshima, Y.; Sugawara, Y.; et al. Aicardi-Goutieres syndrome is caused by IFIH1 mutations. Am. J. Hum. Genet. 2014, 95, 121–125. [Google Scholar] [CrossRef] [Green Version]
- Rice, G.I.; Del Toro Duany, Y.; Jenkinson, E.M.; Forte, G.M.; Anderson, B.H.; Ariaudo, G.; Bader-Meunier, B.; Baildam, E.M.; Battini, R.; Beresford, M.W.; et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014, 46, 503–509. [Google Scholar] [CrossRef]
- Jang, M.A.; Kim, E.K.; Now, H.; Nguyen, N.T.; Kim, W.J.; Yoo, J.Y.; Lee, J.; Jeong, Y.M.; Kim, C.H.; Kim, O.H.; et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Rutsch, F.; MacDougall, M.; Lu, C.; Buers, I.; Mamaeva, O.; Nitschke, Y.; Rice, G.I.; Erlandsen, H.; Kehl, H.G.; Thiele, H.; et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funabiki, M.; Kato, H.; Miyachi, Y.; Toki, H.; Motegi, H.; Inoue, M.; Minowa, O.; Yoshida, A.; Deguchi, K.; Sato, H.; et al. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 2014, 40, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartner, J.C.; Walkley, C.R.; Lu, J.; Orkin, S.H. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009, 10, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Liddicoat, B.J.; Piskol, R.; Chalk, A.M.; Ramaswami, G.; Higuchi, M.; Hartner, J.C.; Li, J.B.; Seeburg, P.H.; Walkley, C.R. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 2015, 349, 1115–1120. [Google Scholar] [CrossRef] [Green Version]
- Pestal, K.; Funk, C.C.; Snyder, J.M.; Price, N.D.; Treuting, P.M.; Stetson, D.B. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity 2015, 43, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Ahmad, S.; Mu, X.; Yang, F.; Greenwald, E.; Park, J.W.; Jacob, E.; Zhang, C.Z.; Hur, S. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 2018, 172, 797–810 e713. [Google Scholar] [CrossRef] [Green Version]
- Dhir, A.; Dhir, S.; Borowski, L.S.; Jimenez, L.; Teitell, M.; Rotig, A.; Crow, Y.J.; Rice, G.I.; Duffy, D.; Tamby, C.; et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018, 560, 238–242. [Google Scholar] [CrossRef]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, S.; Tus, K.; Li, Q.Z.; Wang, A.; Tian, X.H.; Zhou, J.; Liang, C.; Bartov, G.; McDaniel, L.D.; Zhou, X.J.; et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc. Natl. Acad. Sci. USA 2006, 103, 9970–9975. [Google Scholar] [CrossRef] [Green Version]
- Deane, J.A.; Pisitkun, P.; Barrett, R.S.; Feigenbaum, L.; Town, T.; Ward, J.M.; Flavell, R.A.; Bolland, S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007, 27, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.R.; Kashgarian, M.; Alexopoulou, L.; Flavell, R.A.; Akira, S.; Shlomchik, M.J. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J. Exp. Med. 2005, 202, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.W.; Lee, B.L.; Deguine, J.; John, S.; Shlomchik, M.J.; Barton, G.M. Tissue-Resident Macrophages Are Locally Programmed for Silent Clearance of Apoptotic Cells. Immunity 2017, 47, 913–927 e916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, R.; Saitoh, S.; Kanno, A.; Onji, M.; Shibata, T.; Ito, A.; Onji, M.; Matsumoto, M.; Akira, S.; Yoshida, N.; et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity 2011, 35, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majer, O.; Liu, B.; Kreuk, L.S.M.; Krogan, N.; Barton, G.M. UNC93B1 recruits syntenin-1 to dampen TLR7 signalling and prevent autoimmunity. Nature 2019, 575, 366–370. [Google Scholar] [CrossRef]
- Majer, O.; Liu, B.; Woo, B.J.; Kreuk, L.S.M.; Van Dis, E.; Barton, G.M. Release from UNC93B1 reinforces the compartmentalized activation of select TLRs. Nature 2019, 575, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Levin, D.; London, I.M. Regulation of protein synthesis: Activation by double-stranded RNA of a protein kinase that phosphorylates eukaryotic initiation factor 2. Proc. Natl. Acad. Sci. USA 1978, 75, 1121–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. MicroBiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [Green Version]
- Onomoto, K.; Jogi, M.; Yoo, J.S.; Narita, R.; Morimoto, S.; Takemura, A.; Sambhara, S.; Kawaguchi, A.; Osari, S.; Nagata, K.; et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PLoS ONE 2012, 7, e43031. [Google Scholar] [CrossRef]
- Liu, C.X.; Li, X.; Nan, F.; Jiang, S.; Gao, X.; Guo, S.K.; Xue, W.; Cui, Y.; Dong, K.; Ding, H.; et al. Structure and Degradation of Circular RNAs Regulate PKR Activation in Innate Immunity. Cell 2019, 177, 865–880 e821. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.A.; Gorna, M.W.; Baumann, C.L.; Burkard, T.R.; Burckstummer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Habjan, M.; Hubel, P.; Lacerda, L.; Benda, C.; Holze, C.; Eberl, C.H.; Mann, A.; Kindler, E.; Gil-Cruz, C.; Ziebuhr, J.; et al. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 2013, 9, e1003663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, S.; Martignano, F.; Torcia, M.G.; Mattiuz, G.; Conticello, S.G. Evidence for host-dependent RNA editing in the transcriptome of SARS-CoV-2. Sci. Adv. 2020, 6, eabb5813. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Banerjee, S.; Goldstein, S.A.; Dong, B.; Gaughan, C.; Rath, S.; Donovan, J.; Korennykh, A.; Silverman, R.H.; Weiss, S.R. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife 2017, 6. [Google Scholar] [CrossRef]
- Chung, H.; Calis, J.J.A.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Dao Thi, V.L.; Shilvock, A.R.; Hoffmann, H.H.; Rosenberg, B.R.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824 e814. [Google Scholar] [CrossRef] [Green Version]
- Silverman, R.H. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [Green Version]
- Yamasoba, D.; Sato, K.; Ichinose, T.; Imamura, T.; Koepke, L.; Joas, S.; Reith, E.; Hotter, D.; Misawa, N.; Akaki, K.; et al. N4BP1 restricts HIV-1 and its inactivation by MALT1 promotes viral reactivation. Nat. MicroBiol. 2019, 4, 1532–1544. [Google Scholar] [CrossRef] [Green Version]
- Fu, M.; Blackshear, P.J. RNA-binding proteins in immune regulation: A focus on CCCH zinc finger proteins. Nat. Rev. Immunol. 2017, 17, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Uehata, T.; Takeuchi, O. Regnase-1 Is an Endoribonuclease Essential for the Maintenance of Immune Homeostasis. J. Interferon Cytokine Res. 2017, 37, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Hao, S.; Baltimore, D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat. Immunol. 2009, 10, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau, N.L.; Wilusz, J.; Wilusz, C.J. The highways and byways of mRNA decay. Nat. Rev. Mol. Cell Biol. 2007, 8, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Barreau, C.; Paillard, L.; Osborne, H.B. AU-rich elements and associated factors: Are there unifying principles? Nucleic. Acids. Res. 2005, 33, 7138–7150. [Google Scholar] [CrossRef]
- Bakheet, T.; Hitti, E.; Khabar, K.S.A. ARED-Plus: An updated and expanded database of AU-rich element-containing mRNAs and pre-mRNAs. Nucleic. Acids. Res. 2018, 46, D218–D220. [Google Scholar] [CrossRef] [Green Version]
- Lykke-Andersen, J.; Wagner, E. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005, 19, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Sandler, H.; Kreth, J.; Timmers, H.T.; Stoecklin, G. Not1 mediates recruitment of the deadenylase Caf1 to mRNAs targeted for degradation by tristetraprolin. Nucleic. Acids. Res. 2011, 39, 4373–4386. [Google Scholar] [CrossRef]
- Fabian, M.R.; Frank, F.; Rouya, C.; Siddiqui, N.; Lai, W.S.; Karetnikov, A.; Blackshear, P.J.; Nagar, B.; Sonenberg, N. Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin. Nat. Struct. Mol. Biol. 2013, 20, 735–739. [Google Scholar] [CrossRef] [Green Version]
- Bulbrook, D.; Brazier, H.; Mahajan, P.; Kliszczak, M.; Fedorov, O.; Marchese, F.P.; Aubareda, A.; Chalk, R.; Picaud, S.; Strain-Damerell, C.; et al. Tryptophan-Mediated Interactions between Tristetraprolin and the CNOT9 Subunit Are Required for CCR4-NOT Deadenylase Complex Recruitment. J. Mol. Biol. 2018, 430, 722–736. [Google Scholar] [CrossRef]
- Sanduja, S.; Blanco, F.F.; Dixon, D.A. The roles of TTP and BRF proteins in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 2011, 2, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, F.F.; Jimbo, M.; Wulfkuhle, J.; Gallagher, I.; Deng, J.; Enyenihi, L.; Meisner-Kober, N.; Londin, E.; Rigoutsos, I.; Sawicki, J.A.; et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene 2016, 35, 2529–2541. [Google Scholar] [CrossRef]
- Guo, J.; Lv, J.; Chang, S.; Chen, Z.; Lu, W.; Xu, C.; Liu, M.; Pang, X. Inhibiting cytoplasmic accumulation of HuR synergizes genotoxic agents in urothelial carcinoma of the bladder. Oncotarget 2016, 7, 45249–45262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sueyoshi, T.; Kawasaki, T.; Kitai, Y.; Ori, D.; Akira, S.; Kawai, T. Hu Antigen R Regulates Antiviral Innate Immune Responses through the Stabilization of mRNA for Polo-like Kinase 2. J. Immunol. 2018, 200, 3814–3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppek, K.; Schott, J.; Reitter, S.; Poetz, F.; Hammond, M.C.; Stoecklin, G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 2013, 153, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mino, T.; Murakawa, Y.; Fukao, A.; Vandenbon, A.; Wessels, H.H.; Ori, D.; Uehata, T.; Tartey, S.; Akira, S.; Suzuki, Y.; et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell 2015, 161, 1058–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mino, T.; Iwai, N.; Endo, M.; Inoue, K.; Akaki, K.; Hia, F.; Uehata, T.; Emura, T.; Hidaka, K.; Suzuki, Y.; et al. Translation-dependent unwinding of stem-loops by UPF1 licenses Regnase-1 to degrade inflammatory mRNAs. Nucleic. Acids. Res. 2019, 47, 8838–8859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlundt, A.; Heinz, G.A.; Janowski, R.; Geerlof, A.; Stehle, R.; Heissmeyer, V.; Niessing, D.; Sattler, M. Structural basis for RNA recognition in roquin-mediated post-transcriptional gene regulation. Nat. Struct. Mol. Biol. 2014, 21, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Zhou, M.; Kiledjian, M.; Tong, L. The ROQ domain of Roquin recognizes mRNA constitutive-decay element and double-stranded RNA. Nat. Struct. Mol. Biol. 2014, 21, 679–685. [Google Scholar] [CrossRef] [Green Version]
- Glasmacher, E.; Hoefig, K.P.; Vogel, K.U.; Rath, N.; Du, L.; Wolf, C.; Kremmer, E.; Wang, X.; Heissmeyer, V. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat. Immunol. 2010, 11, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.A.; Carballo, E.; Lee, D.M.; Lai, W.S.; Thompson, M.J.; Patel, D.D.; Schenkman, D.I.; Gilkeson, G.S.; Broxmeyer, H.E.; Haynes, B.F.; et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996, 4, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Carballo, E.; Lai, W.S.; Blackshear, P.J. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 1998, 281, 1001–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: Implications for joint and gut-associated immunopathologies. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Carballo, E.; Lai, W.S.; Blackshear, P.J. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 2000, 95, 1891–1899. [Google Scholar] [CrossRef]
- Sauer, I.; Schaljo, B.; Vogl, C.; Gattermeier, I.; Kolbe, T.; Muller, M.; Blackshear, P.J.; Kovarik, P. Interferons limit inflammatory responses by induction of tristetraprolin. Blood 2006, 107, 4790–4797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogilvie, R.L.; Sternjohn, J.R.; Rattenbacher, B.; Vlasova, I.A.; Williams, D.A.; Hau, H.H.; Blackshear, P.J.; Bohjanen, P.R. Tristetraprolin mediates interferon-gamma mRNA decay. J. Biol. Chem. 2009, 284, 11216–11223. [Google Scholar] [CrossRef] [Green Version]
- Van Tubergen, E.; Vander Broek, R.; Lee, J.; Wolf, G.; Carey, T.; Bradford, C.; Prince, M.; Kirkwood, K.L.; D’Silva, N.J. Tristetraprolin regulates interleukin-6, which is correlated with tumor progression in patients with head and neck squamous cell carcinoma. Cancer 2011, 117, 2677–2689. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, M.; D’Silva, N.J.; Kirkwood, K.L. Tristetraprolin regulates interleukin-6 expression through p38 MAPK-dependent affinity changes with mRNA 3′ untranslated region. J. Interferon Cytokine Res. 2011, 31, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Gaba, A.; Grivennikov, S.I.; Do, M.V.; Stumpo, D.J.; Blackshear, P.J.; Karin, M. Cutting edge: IL-10-mediated tristetraprolin induction is part of a feedback loop that controls macrophage STAT3 activation and cytokine production. J. Immunol. 2012, 189, 2089–2093. [Google Scholar] [CrossRef] [Green Version]
- Mahtani, K.R.; Brook, M.; Dean, J.L.; Sully, G.; Saklatvala, J.; Clark, A.R. Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol. Cell Biol. 2001, 21, 6461–6469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrestensen, C.A.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F.; Pelo, J.W.; Worthington, M.T.; Sturgill, T.W. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J. Biol. Chem. 2004, 279, 10176–10184. [Google Scholar] [CrossRef] [Green Version]
- Stoecklin, G.; Stubbs, T.; Kedersha, N.; Wax, S.; Rigby, W.F.; Blackwell, T.K.; Anderson, P. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 2004, 23, 1313–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, E.A.; Smallie, T.; Ding, Q.; O’Neil, J.D.; Cunliffe, H.E.; Tang, T.; Rosner, D.R.; Klevernic, I.; Morrice, N.A.; Monaco, C.; et al. Dominant Suppression of Inflammation via Targeted Mutation of the mRNA Destabilizing Protein Tristetraprolin. J. Immunol. 2015, 195, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Stoecklin, G.; Van Way, S.; Hinkovska-Galcheva, V.; Guo, R.F.; Anderson, P.; Shanley, T.P. Tristetraprolin (TTP)-14-3-3 complex formation protects TTP from dephosphorylation by protein phosphatase 2a and stabilizes tumor necrosis factor-alpha mRNA. J. Biol. Chem. 2007, 282, 3766–3777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, W.H.; Lee, H.H.; Vo, M.T.; Kim, H.J.; Ko, M.S.; Im, Y.C.; Min, Y.J.; Lee, B.J.; Cho, W.J.; Park, J.W. Casein kinase 2 regulates the mRNA-destabilizing activity of tristetraprolin. J. Biol. Chem. 2011, 286, 21577–21587. [Google Scholar] [CrossRef] [Green Version]
- Hodson, D.J.; Janas, M.L.; Galloway, A.; Bell, S.E.; Andrews, S.; Li, C.M.; Pannell, R.; Siebel, C.W.; MacDonald, H.R.; De Keersmaecker, K.; et al. Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat. Immunol. 2010, 11, 717–724. [Google Scholar] [CrossRef]
- Galloway, A.; Saveliev, A.; Lukasiak, S.; Hodson, D.J.; Bolland, D.; Balmanno, K.; Ahlfors, H.; Monzon-Casanova, E.; Mannurita, S.C.; Bell, L.S.; et al. RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science 2016, 352, 453–459. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, L.D.; Wasserman, G.A.; Rah, Y.J.; Matsuura, K.Y.; Coleman, F.T.; Hilliard, K.L.; Pepper-Cunningham, Z.A.; Ieong, M.; Stumpo, D.J.; Blackshear, P.J.; et al. Myeloid ZFP36L1 does not regulate inflammation or host defense in mouse models of acute bacterial infection. PLoS ONE 2014, 9, e109072. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, B.; Xi, Q.; He, C.; Schneider, R.J. Selective degradation of AU-rich mRNAs promoted by the p37 AUF1 protein isoform. Mol. Cell Biol. 2003, 23, 6685–6693. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.Y.; Sadri, N.; Schneider, R.J. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev. 2006, 20, 3174–3184. [Google Scholar] [CrossRef] [Green Version]
- Sadri, N.; Schneider, R.J. Auf1/Hnrnpd-deficient mice develop pruritic inflammatory skin disease. J. Investig. Dermatol 2009, 129, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.J.; Zheng, X.; Lin, C.C.; Tsao, J.; Zhu, X.; Cody, J.J.; Coleman, J.M.; Gherzi, R.; Luo, M.; Townes, T.M.; et al. Posttranscriptional control of type I interferon genes by KSRP in the innate immune response against viral infection. Mol. Cell Biol. 2011, 31, 3196–3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.L.; Wait, R.; Mahtani, K.R.; Sully, G.; Clark, A.R.; Saklatvala, J. The 3′ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol. Cell Biol. 2001, 21, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Yiakouvaki, A.; Dimitriou, M.; Karakasiliotis, I.; Eftychi, C.; Theocharis, S.; Kontoyiannis, D.L. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J. Clin. Investig. 2012, 122, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Brennan, C.M.; Steitz, J.A. HuR and mRNA stability. Cell Mol. Life Sci. 2001, 58, 266–277. [Google Scholar] [CrossRef]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M., Jr.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikantan, S.; Tominaga, K.; Gorospe, M. Functional interplay between RNA-binding protein HuR and microRNAs. Curr. Protein Pept. Sci. 2012, 13, 372–379. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, K.; Ghoshal, B.; Ghosh, S.; Chakrabarty, Y.; Shwetha, S.; Das, S.; Bhattacharyya, S.N. Reversible HuR-microRNA binding controls extracellular export of miR-122 and augments stress response. EMBO Rep. 2016, 17, 1184–1203. [Google Scholar] [CrossRef]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef]
- Masuda, K.; Ripley, B.; Nishimura, R.; Mino, T.; Takeuchi, O.; Shioi, G.; Kiyonari, H.; Kishimoto, T. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, 9409–9414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatya, N.; Childs, E.E.; Cruz, J.A.; Aggor, F.E.Y.; Garg, A.V.; Berman, A.J.; Gudjonsson, J.E.; Atasoy, U.; Gaffen, S.L. IL-17 integrates multiple self-reinforcing, feed-forward mechanisms through the RNA binding protein Arid5a. Sci. Signal 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higa, M.; Oka, M.; Fujihara, Y.; Masuda, K.; Yoneda, Y.; Kishimoto, T. Regulation of inflammatory responses by dynamic subcellular localization of RNA-binding protein Arid5a. Proc. Natl. Acad. Sci. USA 2018, 115, E1214–E1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, H.; Takeuchi, O.; Teraguchi, S.; Matsushita, K.; Uehata, T.; Kuniyoshi, K.; Satoh, T.; Saitoh, T.; Matsushita, M.; Standley, D.M.; et al. The IkappaB kinase complex regulates the stability of cytokine-encoding mRNA induced by TLR-IL-1R by controlling degradation of regnase-1. Nat. Immunol. 2011, 12, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Arima, Y.; Kamimura, D.; Tanaka, Y.; Takahashi, N.; Uehata, T.; Maeda, K.; Satoh, T.; Murakami, M.; Akira, S. Phosphorylation-dependent Regnase-1 release from endoplasmic reticulum is critical in IL-17 response. J. Exp. Med. 2019, 216, 1431–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakiuchi, N.; Yoshida, K.; Uchino, M.; Kihara, T.; Akaki, K.; Inoue, Y.; Kawada, K.; Nagayama, S.; Yokoyama, A.; Yamamoto, S.; et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 2020, 577, 260–265. [Google Scholar] [CrossRef]
- Nanki, K.; Fujii, M.; Shimokawa, M.; Matano, M.; Nishikori, S.; Date, S.; Takano, A.; Toshimitsu, K.; Ohta, Y.; Takahashi, S.; et al. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 2020, 577, 254–259. [Google Scholar] [CrossRef]
- Uehata, T.; Iwasaki, H.; Vandenbon, A.; Matsushita, K.; Hernandez-Cuellar, E.; Kuniyoshi, K.; Satoh, T.; Mino, T.; Suzuki, Y.; Standley, D.M.; et al. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell 2013, 153, 1036–1049. [Google Scholar] [CrossRef] [Green Version]
- Gewies, A.; Gorka, O.; Bergmann, H.; Pechloff, K.; Petermann, F.; Jeltsch, K.M.; Rudelius, M.; Kriegsmann, M.; Weichert, W.; Horsch, M.; et al. Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell Rep. 2014, 9, 1292–1305. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, M.; Marsland, B.J.; Gehrig, J.; Held, W.; Favre, S.; Luther, S.A.; Perroud, M.; Golshayan, D.; Gaide, O.; Thome, M. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. 2014, 33, 2765–2781. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.W.; Hoffman, S.; Beal, A.M.; Dykon, A.; Ringenberg, M.A.; Hughes, A.C.; Dare, L.; Anderson, A.D.; Finger, J.; Kasparcova, V.; et al. MALT1 Protease Activity Is Required for Innate and Adaptive Immune Responses. PLoS ONE 2015, 10, e0127083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaworski, M.; Thome, M. The paracaspase MALT1: Biological function and potential for therapeutic inhibition. Cell Mol. Life Sci. 2016, 73, 459–473. [Google Scholar] [CrossRef] [Green Version]
- von Gamm, M.; Schaub, A.; Jones, A.N.; Wolf, C.; Behrens, G.; Lichti, J.; Essig, K.; Macht, A.; Pircher, J.; Ehrlich, A.; et al. Immune homeostasis and regulation of the interferon pathway require myeloid-derived Regnase-3. J. Exp. Med. 2019, 216, 1700–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minagawa, K.; Wakahashi, K.; Kawano, H.; Nishikawa, S.; Fukui, C.; Kawano, Y.; Asada, N.; Sato, M.; Sada, A.; Katayama, Y.; et al. Posttranscriptional modulation of cytokine production in T cells for the regulation of excessive inflammation by TFL. J. Immunol. 2014, 192, 1512–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Liu, S.; Fu, J.J.; Tony Wang, T.; Yao, X.; Kumar, A.; Liu, G.; Fu, M. Monocyte Chemotactic Protein-induced Protein 1 and 4 Form a Complex but Act Independently in Regulation of Interleukin-6 mRNA Degradation. J. Biol. Chem. 2015, 290, 20782–20792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinuesa, C.G.; Cook, M.C.; Angelucci, C.; Athanasopoulos, V.; Rui, L.; Hill, K.M.; Yu, D.; Domaschenz, H.; Whittle, B.; Lambe, T.; et al. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 2005, 435, 452–458. [Google Scholar] [CrossRef]
- Yu, D.; Tan, A.H.; Hu, X.; Athanasopoulos, V.; Simpson, N.; Silva, D.G.; Hutloff, A.; Giles, K.M.; Leedman, P.J.; Lam, K.P.; et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature 2007, 450, 299–303. [Google Scholar] [CrossRef]
- Linterman, M.A.; Rigby, R.J.; Wong, R.; Silva, D.; Withers, D.; Anderson, G.; Verma, N.K.; Brink, R.; Hutloff, A.; Goodnow, C.C.; et al. Roquin differentiates the specialized functions of duplicated T cell costimulatory receptor genes CD28 and ICOS. Immunity 2009, 30, 228–241. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Silva, D.G.; Martin, J.L.; Pratama, A.; Hu, X.; Chang, P.P.; Walters, G.; Vinuesa, C.G. Interferon-gamma excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity 2012, 37, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Vogel, K.U.; Edelmann, S.L.; Jeltsch, K.M.; Bertossi, A.; Heger, K.; Heinz, G.A.; Zoller, J.; Warth, S.C.; Hoefig, K.P.; Lohs, C.; et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity 2013, 38, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Jeltsch, K.M.; Hu, D.; Brenner, S.; Zoller, J.; Heinz, G.A.; Nagel, D.; Vogel, K.U.; Rehage, N.; Warth, S.C.; Edelmann, S.L.; et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat. Immunol. 2014, 15, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Pratama, A.; Ramiscal, R.R.; Silva, D.G.; Das, S.K.; Athanasopoulos, V.; Fitch, J.; Botelho, N.K.; Chang, P.P.; Hu, X.; Hogan, J.J.; et al. Roquin-2 shares functions with its paralog Roquin-1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity 2013, 38, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Lee, F.C.Y.; Ule, J. Advances in CLIP Technologies for Studies of Protein-RNA Interactions. Mol. Cell 2018, 69, 354–369. [Google Scholar] [CrossRef] [Green Version]
- Baltz, A.G.; Munschauer, M.; Schwanhausser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Queiroz, R.M.L.; Smith, T.; Villanueva, E.; Marti-Solano, M.; Monti, M.; Pizzinga, M.; Mirea, D.M.; Ramakrishna, M.; Harvey, R.F.; Dezi, V.; et al. Comprehensive identification of RNA-protein interactions in any organism using orthogonal organic phase separation (OOPS). Nat. Biotechnol. 2019, 37, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trendel, J.; Schwarzl, T.; Horos, R.; Prakash, A.; Bateman, A.; Hentze, M.W.; Krijgsveld, J. The Human RNA-Binding Proteome and Its Dynamics during Translational Arrest. Cell 2019, 176, 391–403 e319. [Google Scholar] [CrossRef] [Green Version]
- Urdaneta, E.C.; Vieira-Vieira, C.H.; Hick, T.; Wessels, H.H.; Figini, D.; Moschall, R.; Medenbach, J.; Ohler, U.; Granneman, S.; Selbach, M.; et al. Purification of cross-linked RNA-protein complexes by phenol-toluol extraction. Nat. Commun 2019, 10, 990. [Google Scholar] [CrossRef]
- Shulman, Z.; Stern-Ginossar, N. The RNA modification N(6)-methyladenosine as a novel regulator of the immune system. Nat. Immunol. 2020, 21, 501–512. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uehata, T.; Takeuchi, O. RNA Recognition and Immunity—Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms. Cells 2020, 9, 1701. https://doi.org/10.3390/cells9071701
Uehata T, Takeuchi O. RNA Recognition and Immunity—Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms. Cells. 2020; 9(7):1701. https://doi.org/10.3390/cells9071701
Chicago/Turabian StyleUehata, Takuya, and Osamu Takeuchi. 2020. "RNA Recognition and Immunity—Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms" Cells 9, no. 7: 1701. https://doi.org/10.3390/cells9071701
APA StyleUehata, T., & Takeuchi, O. (2020). RNA Recognition and Immunity—Innate Immune Sensing and Its Posttranscriptional Regulation Mechanisms. Cells, 9(7), 1701. https://doi.org/10.3390/cells9071701