Therapeutic Target Discovery Using High-Throughput Genetic Screens in Acute Myeloid Leukemia

Abstract
:1. Introduction
2. Epigenetic Regulators
2.1. Histone Writers
2.1.1. Males Absent on the First (MOF)
2.1.2. SET Domain Bifurcated 1 (SETDB1)
2.1.3. Lysine Acetyltransferase 2A (KAT2A)
2.1.4. Histone Acetyltransferase Binding to ORC1 (HBO1)
2.2. Histone Readers
2.2.1. Bromodomain-Containing Protein 4 (BRD4)
2.2.2. Eleven-Nineteen Leukemia (ENL)
2.3. Histone Erasers
2.3.1. Jumonji Domain Containing 1C (JMJD1C)
2.3.2. Sirtuin 1 (SIRT1)
3. Kinase Pathways
3.1. Glycogen Synthase Kinase 3 (GSK3)
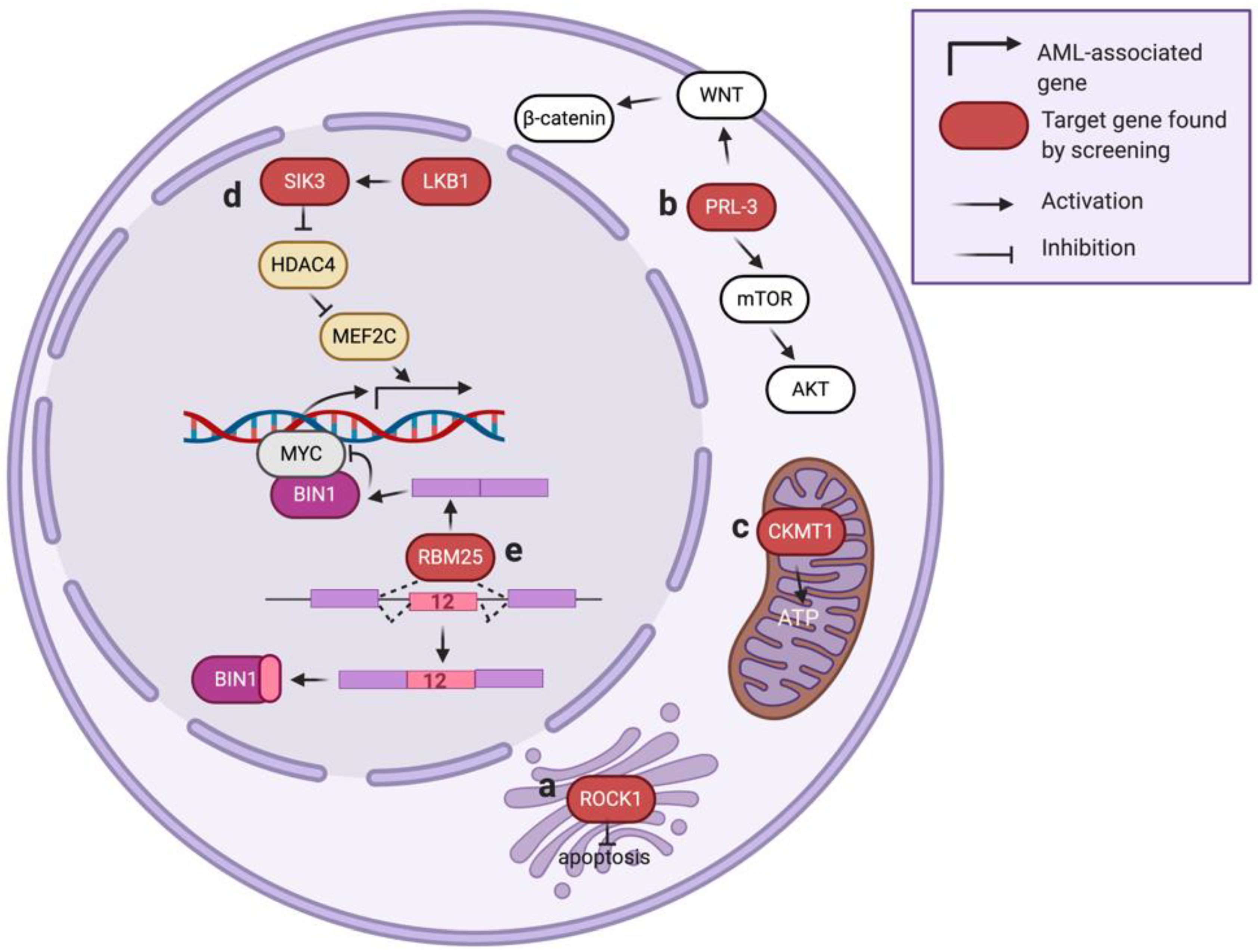
3.2. Rho-Associated Protein Kinase 1 (ROCK1)
3.3. Phosphatase of Regenerating Liver 3 (PRL-3)
3.4. Creatine Kinase, Mitochondrial 1 (CKMT1)
3.5. Liver Kinase B1 (LKB1)
4. Gene Expression Regulators
4.1. Zinc Finger E-Box-Binding Homeobox2 (ZEB2)
4.2. Zinc Finger Protein 64 (ZFP64)
4.3. RNA-Binding Protein 25 (RBM25)
4.4. RNA-Binding Protein 39 (RBM39)
5. Therapeutic Response Modulators
5.1. Cytarabine
5.1.1. Western Equine Encephalitis 1 (WEE1)
5.1.2. Deoxycytidine Kinase (DCK)
5.2. FLT3 Inhibitors
5.2.1. Protein Sprouty Homology 3 (SPRY3)
5.2.2. Ataxia Telangiectasia Mutated (ATM)
5.2.3. Glutaminase (GLS)
5.3. Venetoclax
5.3.1. Tumor Protein 53 (TP53)
5.3.2. Caseinolytic Peptidase b Protein Homolog (CLPB)
5.4. BET Inhibitors
5.4.1. Polycomb Repressive Complex 2 (PRC2)
5.4.2. Lysine-Specific Histone Demethylase 1A (KDM1A)
6. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stiller, C.; Marcos-gragera, R.; Angelis, R.D.; Mallone, S.; Tereanu, C.; Allemani, C.; Ricardi, U.; Schouten, H.C. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur. J. Cancer 2012, 48, 3257–3258. [Google Scholar] [CrossRef]
- Zjablovskaja, P.; Florian, M.C. Acute myeloid leukemia: Aging and epigenetics. Cancers 2020, 12, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antar, A.I.; Otrock, Z.K.; Jabbour, E.; Mohty, M.; Bazarbachi, A. FLT3 inhibitors in acute myeloid leukemia: Ten frequently asked questions. Leukemia 2020, 34, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Schmidts, I. The power and potential of integrated diagnostics in acute myeloid leukaemia. Br. J. Haematol. 2020, 188, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Dow, S.; Lucau-danila, A.; Anderson, K.; Arkin, A.P.; Astromoff, A.; Bakkoury, M.E.; Bangham, R.; Benito, R.; Brachat, S.; Andre, B.; et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 2002, 387–391. [Google Scholar] [CrossRef]
- Tijsterman, M.; Plasterk, R.H.A. Dicers at RISC: The mechanism of RNAi. Cell 2004, 117, 1–3. [Google Scholar] [CrossRef] [Green Version]
- McManus, M.T.; Sharp, P.A. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002, 3, 737–747. [Google Scholar] [CrossRef]
- Ni, W.; Qiao, J.; Hu, S.; Zhao, X.; Regouski, M.; Yang, M.; Polejaeva, I.A.; Chen, C. Efficient gene knockout in goats using CRISPR/Cas9 system. PLoS ONE 2014, 9, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Moffat, J.; Grueneberg, D.A.; Yang, X.; Kim, S.Y.; Kloepfer, A.M.; Hinkle, G.; Piqani, B.; Eisenhaure, T.M.; Luo, B.; Grenier, J.K.; et al. A Lentiviral RNAi Library for Human and Mouse Genes Applied to an Arrayed Viral High-Content Screen. Cell 2006, 124, 1283–1298. [Google Scholar] [CrossRef] [Green Version]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.S.; Zhang, F.; Irizarry, R.A.; Xiao, T.; Cong, L.; Love, M.I.; Li, W.; Xu, H.; Liu, J.S.; Brown, M. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 2014, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Valerio, D.G.; Xu, H.; Chen, C.W.; Hoshii, T.; Eisold, M.E.; Delaney, C.; Cusan, M.; Deshpande, A.J.; Huang, C.H.; Lujambio, A.; et al. Histone acetyltransferase activity of MOF is required for MLL-AF9 leukemogenesis. Cancer Res. 2017, 77, 1753–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuellar, L.; Herzner, A.M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SET DB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [Green Version]
- MacPherson, L.; Anokye, J.; Yeung, M.M.; Lam, E.Y.N.; Chan, Y.C.; Weng, C.F.; Yeh, P.; Knezevic, K.; Butler, M.S.; Hoegl, A.; et al. HBO1 is required for the maintenance of leukaemia stem cells. Nature 2020, 577, 266–270. [Google Scholar] [CrossRef]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [Green Version]
- Erb, M.A.; Scott, T.G.; Li, B.E.; Xie, H.; Paulk, J.; Seo, H.S.; Souza, A.; Roberts, J.M.; Dastjerdi, S.; Buckley, D.L.; et al. Transcription control by the ENL YEATS domain in acute leukaemia. Nature 2017, 543, 270–274. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Chen, M.; Eng, R.; DeJong, J.; Sinha, A.U.; Rahnamay, N.F.; Koche, R.; Al-Shahrour, F.; Minehart, J.C.; Chen, C.W.; et al. MLL-AF9-and HOXA9-mediated acute myeloid leukemia stem cell self-renewal requires JMJD1C. J. Clin. Investig. 2016, 126, 997–1011. [Google Scholar] [CrossRef]
- Chen, C.W.; Koche, R.P.; Sinha, A.U.; Deshpande, A.J.; Zhu, N.; Eng, R.; Doench, J.G.; Xu, H.; Chu, S.H.; Qi, J.; et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 2015, 21, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Banerji, V.; Frumm, S.M.; Ross, K.N.; Li, L.S.; Schinzel, A.C.; Hahn, C.K.; Kakoza, R.M.; Chow, K.T.; Ross, L.; Alexe, G.; et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Invest. 2012, 122, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wermke, M.; Camgoz, A.; Paszkowski-Rogacz, M.; Thieme, S.; Von Bonin, M.; Dahl, A.; Platzbecker, U.; Theis, M.; Ehninger, G.; Brenner, S.; et al. RNAi profiling of primary human AML cells identifies ROCK1 as a therapeutic target and nominates fasudil as an antileukemic drug. Blood 2015, 125, 3760–3768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Toh, S.H.M.; Chan, Z.L.; Quah, J.Y.; Chooi, J.Y.; Tan, T.Z.; Chong, P.S.Y.; Zeng, Q.; Chng, W.J. A loss-of-function genetic screening reveals synergistic targeting of AKT/mTOR and WTN/β-catenin pathways for treatment of AML with high PRL-3 phosphatase. J. Hematol. Oncol. 2018, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fenouille, N.; Bassil, C.F.; Ben-Sahra, I.; Benajiba, L.; Alexe, G.; Ramos, A.; Pikman, Y.; Conway, A.S.; Burgess, M.R.; Li, Q.; et al. The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat. Med. 2017, 23, 301–313.e6. [Google Scholar] [CrossRef]
- Tarumoto, Y.; Lu, B.; Somerville, T.D.D.; Huang, Y.H.; Milazzo, J.P.; Wu, X.S.; Klingbeil, O.; El Demerdash, O.; Shi, J.; Vakoc, C.R. LKB1, Salt-Inducible Kinases, and MEF2C Are Linked Dependencies in Acute Myeloid Leukemia. Mol. Cell 2018, 69, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Mar, B.G.; Zhang, H.; Puram, R.V.; Vazquez, F.; Weir, B.A.; Hahn, W.C.; Ebert, B.; Pellman, D. The EMT regulator ZEB2 is a novel dependency of human and murine acute myeloid leukemia. Blood 2017, 129, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Klingbeil, O.; Tarumoto, Y.; Somerville, T.D.D.; Huang, Y.H.; Wei, Y.; Wai, D.C.; Low, J.K.K.; Milazzo, J.P.; Wu, X.S.; et al. A Transcription Factor Addiction in Leukemia Imposed by the MLL Promoter Sequence. Cancer Cell 2018, 34, 970–981.e8. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Schuster, M.B.; Pundhir, S.; Rapin, N.; Bagger, F.O.; Sidiropoulos, N.; Hashem, N.; Porse, B.T. The splicing factor RBM25 controls MYC activity in acute myeloid leukemia. Nat. Commun. 2019, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-Binding Protein Network in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 369–384.e7. [Google Scholar] [CrossRef] [Green Version]
- Tibes, R.; Bogenberger, J.M.; Chaudhuri, L.; Hagelstrom, R.T.; Chow, D.; Buechel, M.E.; Gonzales, I.M.; Demuth, T.; Slack, J.; Mesa, R.A.; et al. RNAi screening of the kinome with cytarabine in leukemias. Blood 2012, 119, 2863–2872. [Google Scholar] [CrossRef] [Green Version]
- Kurata, M.; Rathe, S.K.; Bailey, N.J.; Aumann, N.K.; Jones, J.M.; Veldhuijzen, G.W.; Moriarity, B.S.; Largaespada, D.A. Using genome-wide CRISPR library screening with library resistant DCK to find new sources of Ara-C drug resistance in AML. Sci. Rep. 2016, 6, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Wu, C.; Wang, Y.; Qi, R.; Bhavanasi, D.; Zuo, Z.; Dos Santos, C.; Chen, S.; Chen, Y.; Zheng, H.; et al. A genome-wide CRISPR screen identifies genes critical for resistance to FLT3 inhibitor AC220. Cancer Res. 2017, 77, 4402–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallipoli, P.; Giotopoulos, G.; Tzelepis, K.; Costa, A.S.H.; Vohra, S.; Medina-Perez, P.; Basheer, F.; Marando, L.; Lisio, L.D.; Dias, J.M.L.; et al. Glutaminolysis is a metabolic dependency in FLT3 ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 2018, 131, 1639–1653. [Google Scholar] [CrossRef] [Green Version]
- Nechiporuk, T.; Kurtz, S.E.; Nikolova, O.; Liu, T.; Jones, C.L.; D’alessandro, A.; Culp-Hill, R.; D’almeida, A.; Joshi, S.K.; Rosenberg, M.; et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019, 9, 910–925. [Google Scholar] [CrossRef]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting mitochondrial structure sensitizes acute myeloid Leukemia to venetoclax treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef]
- Bell, C.C.; Fennell, K.A.; Chan, Y.C.; Rambow, F.; Yeung, M.M.; Vassiliadis, D.; Lara, L.; Yeh, P.; Martelotto, L.G.; Rogiers, A.; et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Lorch, Y.; Maier-Davis, B.; Kornberg, R.D. Mechanism of chromatin remodeling. Proc. Natl. Acad. Sci. USA 2010, 107, 3458–3462. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications-miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.F.; Chen, C.-W.; Armstrong, S.A. Drugging Chromatin in Cancer: Recent Advances and Novel Approaches. Mol. Cell 2015, 60, 561–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, M.; Eberl, H.C.; Matarese, F.; Marks, H.; Denissov, S.; Butter, F.; Lee, K.K.; Olsen, J.V.; Hyman, A.A.; Stunnenberg, H.G.; et al. Quantitative Interaction Proteomics and Genome-wide Profiling of Epigenetic Histone Marks and Their Readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef] [Green Version]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-Rearranged Leukemia Is Dependent on Aberrant H3K79 Methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Chan, A.K.N.; Chen, C.W. Rewiring the epigenetic networks in MLL-rearranged leukemias: Epigenetic dysregulation and pharmacological interventions. Front. Cell Dev. Biol. 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Dancy, B.M.; Cole, P.A. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015, 115, 2419–2452. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; McGirr, C.; Shakhbazov, K.; Barbier, V.; Krishnan, K.; Mukhopadhyay, P.; Hawthorne, P.; Trezise, A.; Ding, J.; Grimmond, S.M.; et al. Interaction of c-Myb with p300 is required for the induction of acute myeloid leukemia (AML) by human AML oncogenes. Blood 2014, 123, 2682–2690. [Google Scholar] [CrossRef] [Green Version]
- Neal, K.C.; Pannuti, A.; Smith, E.R.; Lucchesi, J.C. A new human member of the MYST family of histone acetyl transferases with high sequence similarity to Drosophila MOF. Biochim. Biophys. Acta -Gene Struct. Expr. 2000, 1490, 170–174. [Google Scholar] [CrossRef]
- Valerio, D.G.; Xu, H.; Eisold, M.E.; Woolthuis, C.M.; Pandita, T.K.; Armstrong, S.A. Histone acetyltransferase activity of MOF is required for adult but not early fetal hematopoiesis in mice. Blood 2017, 129, 48–59. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, J.; Zhao, L.; Liu, S.; Du, D.; Ding, H.; Chen, S.; Yue, L.; Liu, Y.C.; Zhang, C.; et al. Identification of novel inhibitors of histone acetyltransferase hMOF through high throughput screening. Eur. J. Med. Chem. 2018, 157, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, W.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Chatton, B.; Tempst, P.; Roeder, R.G.; Zhang, Y. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol. Cell 2003, 12, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Mazo, A.M.; Huang, D.H.; Mozer, B.A.; Dawid, I.B. The trithorax gene, a trans-acting regulator of the bithorax complex in Drosophila, encodes a protein with zinc-binding domains. Proc. Natl. Acad. Sci. USA 1990, 87, 2112–2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haubold, B.; Wiehe, T. How repetitive are genomes? BMC Bioinformatics 2006, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Kim, K.C. DZNep, inhibitor of S-adenosylhomocysteine hydrolase, down-regulates expression of SETDB1 H3K9me3 HMTase in human lung cancer cells. Biochem. Biophys. Res. Commun. 2013, 438, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Fournier, M.; Orpinell, M.; Grauffel, C.; Scheer, E.; Garnier, J.M.; Ye, T.; Chavant, V.; Joint, M.; Esashi, F.; Dejaegere, A.; et al. KAT2A/KAT2B-targeted acetylome reveals a role for PLK4 acetylation in preventing centrosome amplification. Nat. Commun. 2016, 7, 13227. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.H.; Li, X.J.; et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Vlassis, A.; Roques, C.; Lalonde, M.; González-Aguilera, C.; Lambert, J.; Lee, S.; Zhao, X.; Alabert, C.; Johansen, J.V.; et al. BRPF 3- HBO 1 regulates replication origin activation and histone H3K14 acetylation. EMBO J. 2016, 35, 176–192. [Google Scholar] [CrossRef]
- Mishima, Y.; Miyagi, S.; Saraya, A.; Negishi, M.; Endoh, M.; Endo, T.A.; Toyoda, T.; Shinga, J.; Katsumoto, T.; Chiba, T.; et al. The Hbo1-Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 2011, 118, 2443–2453. [Google Scholar] [CrossRef]
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wen, H.; Xi, Y.; Tanaka, K.; Wang, H.; Peng, D.; Ren, Y.; Jin, Q.; Dent, S.Y.R.; Li, W.; et al. AF9 YEATS domain links histone acetylation to DOT1L-mediated H3K79 methylation. Cell 2014, 159, 558–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Schreiner, S.A.; García-Cuéllar, M.P.; Fey, G.H.; Slany, R.K. The leukemogenic fusion of MLL with ENL creates a novel transcriptional transactivator. Leukemia 1999, 13, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. AFF4, a Component of the ELL/P-TEFb Elongation Complex and a Shared Subunit of MLL Chimeras, Can Link Transcription Elongation to Leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) family in transcriptional control. Nat. Rev. Mol. Cell Biol. 2012, 13, 543–547. [Google Scholar] [CrossRef]
- Wan, L.; Wen, H.; Li, Y.; Lyu, J.; Xi, Y.; Hoshii, T.; Joseph, J.K.; Wang, X.; Loh, Y.H.E.; Erb, M.A.; et al. ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 2017, 543, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Christott, T.; Bennett, J.; Coxon, C.; Monteiro, O.; Giroud, C.; Beke, V.; Felce, S.L.; Gamble, V.; Gileadi, C.; Poda, G.; et al. Discovery of a Selective Inhibitor for the YEATS Domains of ENL/AF9. SLAS Discov. 2019, 24, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Ma, H. Evolutionary history of histone demethylase families: Distinct evolutionary patterns suggest functional divergence. BMC Evol. Biol. 2008, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walport, L.J.; Hopkinson, R.J.; Schofield, C.J. Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 2012, 16, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Tan, L.; Nagata, Y.; Takemura, T.; Asahina, A.; Yokota, D.; Yagyu, T.; Shibata, K.; Fujisawa, S.; Ohnishi, K. JmjC-domain containing histone demethylase 1B-mediated p15Ink4b suppression promotes the proliferation of leukemic progenitor cells through modulation of cell cycle progression in acute myeloid leukemia. Mol. Carcinog. 2013, 52, 57–69. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Schemies, J.; Uciechowska, U.; Sippl, W.; Jung, M. NAD1(+)-Dependent Histone Deacetylases (Sirtuins) as Novel Therapeutic Targets. Med. Res. Rev. 2010, 30, 861–899. [Google Scholar] [CrossRef]
- Huang, Y.; Chen, J.; Lu, C.; Han, J.; Wang, G.; Song, C.; Zhu, S.; Wang, C.; Li, G.; Kang, J.; et al. HDAC1 and Klf4 interplay critically regulates human myeloid leukemia cell proliferation. Cell Death Dis. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Jia, M.-Y.; Fang, W.-Y.; Chen, X.-J.; Mu, L.-L.; Wang, Z.-Y.; Shen, Y.; Xiang, R.-F.; Wang, L.-N.; Wang, L.; et al. FLT3 inhibition upregulates HDAC8 via FOXO to inactivate p53 and promote maintenance of FLT3-ITD+ acute myeloid leukemia. Blood 2020, 135, 1472–1483. [Google Scholar] [CrossRef]
- Sroczynska, P.; Cruickshank, V.A.; Bukowski, J.P.; Miyagi, S.; Bagger, F.O.; Walfridsson, J.; Schuster, M.B.; Porse, B.; Helin, K. ShRNA screening identifies JMJD1C as being required for leukemia maintenance. Blood 2014, 123, 1870–1882. [Google Scholar] [CrossRef]
- Xu, X.; Wang, L.; Hu, L.; Dirks, W.G.; Zhao, Y.; Wei, Z.; Chen, D.; Li, Z.; Wang, Z.; Han, Y.; et al. Small molecular modulators of JMJD1C preferentially inhibit growth of leukemia cells. Int. J. Cancer 2020, 146, 400–412. [Google Scholar] [CrossRef]
- Yang, Y.; Hou, H.; Haller, E.M.; Nicosia, S.V.; Bai, W. Suppression of FOXO1 activity by FHL2 through SIRT1-mediated deacetylation. EMBO J. 2005, 24, 1021–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.Y.; Bhatia, R. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell 2012, 21, 266–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Dev. 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrington, T.P.; Johnson, G.L. Organization and regulation of mitogen-activated protein kinase signaling pathways. Curr. Opin. Cell Biol. 1999, 11, 211–218. [Google Scholar] [CrossRef]
- Tiong, I.S.; Wei, A.H. New drugs creating new challenges in acute myeloid leukemia. Genes Chromosom. Cancer 2019, 58, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Miano, M.; Micalizzi, C.; Calvillo, M.; Dufour, C. New targets in pediatric acute myeloid leukemia. Immunol. Lett. 2013, 155, 47–50. [Google Scholar] [CrossRef]
- Wang, Z.; Iwasaki, M.; Ficara, F.; Lin, C.; Matheny, C.; Wong, S.H.K.; Smith, K.S.; Cleary, M.L. GSK-3 Promotes Conditional Association of CREB and Its Coactivators with MEIS1 to Facilitate HOX-Mediated Transcription and Oncogenesis. Cancer Cell 2010, 17, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.E.; Culbert, A.A.; Chalmers, K.A.; Facci, L.; Skaper, S.D.; Reith, A.D. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J. Neurochem. 2001, 77, 94–102. [Google Scholar] [CrossRef]
- Maekawa, M.; Ishizaki, T.; Boku, S.; Watanabe, N.; Fujita, A.; Iwamatsu, A.; Obinata, T.; Ohashi, K.; Mizuno, K.; Narumiya, S. Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 1999, 285, 895–898. [Google Scholar] [CrossRef]
- Kroll, J.; Epting, D.; Kern, K.; Dietz, C.T.; Feng, Y.; Hammes, H.P.; Wieland, T.; Augustin, H.G. Inhibition of Rho-dependent kinases ROCK I/II activates VEGF-driven retinal neovascularization and sprouting angiogenesis. Am. J. Physiol. -Hear. Circ. Physiol. 2009, 296, 893–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.B.; Cheng, Q.; Liu, L.; Zhou, T.; Shan, C.Y.; Hu, X.Y.; Zhou, J.; Liu, E.H.; Li, P.; Gao, N. Mitochondrial translocation of cofilin is required for allyl isothiocyanate-mediated cell death via ROCK1/PTEN/PI3K signaling pathway. Cell Commun. Signal. 2013, 11, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Fu, R.; Liu, Y.; Li, J.; Zhang, H.; Hu, X.; Chen, Y.; Liu, X.; Li, Y.; Li, P.; et al. Dephosphorylation and mitochondrial translocation of cofilin sensitizes human leukemia cells to cerulenin-induced apoptosis via the ROCK1/Akt/JNK signaling pathway. Oncotarget 2016, 7, 20655–20668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerli, U.M.; Holt, R.U.; Holien, T.; Vaatsveen, T.K.; Zhan, F.; Egeberg, K.W.; Barlogie, B.; Waage, A.; Aarset, H.; Hong, Y.D.; et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood 2008, 111, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Abdollahi, P.; Vandsemb, E.N.; Hjort, M.A.; Misund, K.; Holien, T.; Sponaas, A.M.; Rø, T.B.; Slørdahl, T.S.; Børset, M. Src family kinases are regulated in multiple myeloma cells by phosphatase of regenerating liver-3. Mol. Cancer Res. 2017, 15, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharlow, E.R.; Wipf, P.; McQueeney, K.E.; Bakan, A.; Lazo, J.S. Investigational inhibitors of PTP4A3 phosphatase as antineoplastic agents. Expert Opin. Investig. Drugs 2014, 23, 661–673. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. the Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [Green Version]
- Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. Pim kinases and their relevance to the PI3K/AKT/mTOR pathway in the regulation of ovarian cancer. Biomolecules 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef] [Green Version]
- Esteve-Puig, R.; Gil, R.; González-Sánchez, E.; Bech-Serra, J.J.; Grueso, J.; Hernández-Losa, J.; Moliné, T.; Canals, F.; Ferrer, B.; Cortés, J.; et al. A Mouse Model Uncovers LKB1 as an UVB-Induced DNA Damage Sensor Mediating CDKN1A (p21WAF1/CIP1) Degradation. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef]
- Hou, X.; Liu, J.E.; Liu, W.; Liu, C.Y.; Liu, Z.Y.; Sun, Z.Y. A new role of NUAK1: Directly phosphorylating p53 and regulating cell proliferation. Oncogene 2011, 30, 2933–2942. [Google Scholar] [CrossRef] [PubMed]
- Csukasi, F.; Duran, I.; Barad, M.; Barta, T.; Gudernova, I.; Trantirek, L.; Martin, J.H.; Kuo, C.Y.; Woods, J.; Lee, H.; et al. The PTH/PTHrP-SIK3 pathway affects skeletogenesis through altered mTOR signaling. Sci. Transl. Med. 2018, 10, eaat9356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laszlo, G.S.; Alonzo, T.A.; Gudgeon, C.J.; Harrington, K.H.; Kentsis, A.; Gerbing, R.B.; Wang, Y.C.; Ries, R.E.; Raimondi, S.C.; Hirsch, B.A.; et al. High expression of myocyte enhancer factor 2C (MEF2C) is associated with adverse-risk features and poor outcome in pediatric acute myeloid leukemia: A report from the Children’s Oncology Group. J. Hematol. Oncol. 2015, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takei, H.; Kobayashi, S.S. Targeting transcription factors in acute myeloid leukemia. Int. J. Hematol. 2019, 109, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Ma, O.; Speck, N.A.; Friedman, A.D. Runx1 deletion or dominant inhibition reduces Cebpa transcription via conserved promoter and distal enhancer sites to favor monopoiesis over granulopoiesis. Blood 2012, 119, 4408–4418. [Google Scholar] [CrossRef] [Green Version]
- De Necochea-Campion, R.; Shouse, G.P.; Zhou, Q.; Mirshahidi, S.; Chen, C.S. Aberrant splicing and drug resistance in AML. J. Hematol. Oncol. 2016, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Zuo, D.; Park, M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J. Biol. Chem. 2005, 280, 35477–35489. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Tamamura, Y.; Katsube, K.I.; Yamaguchi, A. Zfp64 participates in Notch signaling and regulates differentiation in mesenchymal cells. J. Cell Sci. 2008, 121, 1613–1623. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Liu, X.; Liu, Y.; Zhang, Q.; Yao, Z.; Huang, B.; Zhang, P.; Li, N.; Cao, X. Zinc finger protein 64 promotes toll-like receptor-triggered proinflammatory and type I interferon production in macrophages by enhancing p65 subunit activation. J. Biol. Chem. 2013, 288, 24600–24608. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.; Ou, A.C.; Cho, A.; Benz, E.J.; Huang, S.-C. Novel Splicing Factor RBM25 Modulates Bcl-x Pre-mRNA 5′ Splice Site Selection. Mol. Cell. Biol. 2008, 28, 5924–5936. [Google Scholar] [CrossRef] [Green Version]
- Tari, M.; Manceau, V.; Matha Salone, J.; Kobayashi, A.; Pastré, D.; Maucuer, A. U2 AF 65 assemblies drive sequence-specific splice site recognition. EMBO Rep. 2019, 20, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, M.; Dipersio, J.F. Small molecule inhibitors in acute myeloid leukemia: From the bench to the clinic. Expert Rev. Hematol. 2014, 7, 439–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antar, A.; Otrock, Z.K.; El-Cheikh, J.; Kharfan-Dabaja, M.A.; Battipaglia, G.; Mahfouz, R.; Mohty, M.; Bazarbachi, A. Inhibition of FLT3 in AML: A focus on sorafenib. Bone Marrow Transplant. 2017, 52, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, B.C.; Gillet, J.P.; Patel, C.; Baer, M.R.; Bates, S.E.; Gottesman, M.M. Drug resistance: Still a daunting challenge to the successful treatment of AML. Drug Resist. Updat. 2012, 15, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Momparler, R.L. Optimization of cytarabine (ARA-C) therapy for acute myeloid leukemia. Exp. Hematol. Oncol. 2013, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Kellogg, D.R. Wee1-dependent mechanisms required for coordination of cell growth and cell division. J. Cell Sci. 2003, 116, 4883–4890. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.C.; Kim, J.; Fosmire, S.; Gearheart, C.M.; Van Linden, A.; Baturin, D.; Zaberezhnyy, V.; Patel, P.R.; Gao, D.; Tan, A.C.; et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia 2012, 26, 1266–1276. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, G.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.D.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Pratz, K.W.; Luger, S.M. Will FLT3 Inhibitors Fulfill Their Promise in AML? Curr Opin Hematol. 2014, 21, 72–78. [Google Scholar] [CrossRef]
- Celik-Selvi, B.E.; Stütz, A.; Mayer, C.-E.; Salhi, J.; Siegwart, G.; Sutterlüty, H. Sprouty3 and Sprouty4, Two Members of a Family Known to Inhibit FGF-Mediated Signaling, Exert Opposing Roles on Proliferation and Migration of Glioblastoma-Derived Cells. Cells 2019, 8, 808. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Chen, G.; Kuan, S.F.; Zhang, D.H.; Schlaepfer, D.D.; Hu, J. FAK/PYK2 promotes the Wnt/β-catenin pathway and intestinal tumorigenesis by phosphorylating GSK3β. Elife 2015, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bhoumik, A.; Takahashi, S.; Breitweiser, W.; Shiloh, Y.; Jones, N.; Ronai, Z. ATM-dependent phosphorylation of ATF2 is required for the DNA damage response. Mol. Cell 2005, 18, 577–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosentino, C.; Grieco, D.; Costanzo, V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. EMBO J. 2011, 30, 546–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumping, L.; Büttner, B.; Maier, O.; Rehmann, H.; Lequin, M.; Schlump, J.U.; Schmitt, B.; Schiebergen-Bronkhorst, B.; Prinsen, H.C.M.T.; Losa, M.; et al. Identification of a Loss-of-Function Mutation in the Context of Glutaminase Deficiency and Neonatal Epileptic Encephalopathy. JAMA Neurol. 2019, 76, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Juárez-Salcedo, L.M.; Desai, V.; Dalia, S. Venetoclax: Evidence to date and clinical potential. Drugs Context 2019, 8, 1–13. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Panuzzo, C.; Signorino, E.; Calabrese, C.; Ali, M.S.; Petiti, J.; Bracco, E.; Cilloni, D. Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia. J. Clin. Med. 2020, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- LeRoy, G.; Rickards, B.; Flint, S.J. The Double Bromodomain Proteins Brd2 and Brd3 Couple Histone Acetylation to Transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; He, N.; Zhou, Q. Brd4 Recruits P-TEFb to Chromosomes at Late Mitosis To Promote G1 Gene Expression and Cell Cycle Progression. Mol. Cell. Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, D.; Tontsch-Grunt, U.; Baum, A.; Popow, J.; Scharn, D.; Hofmann, M.H.; Engelhardt, H.; Kaya, O.; Beck, J.; Schweifer, N.; et al. The novel BET bromodomain inhibitor BI 894999 represses super-enhancer-associated transcription and synergizes with CDK9 inhibition in AML. Oncogene 2018, 37, 2687–2701. [Google Scholar] [CrossRef]
- Curie, I.; Paris, U. The Polycomb Complex PRC2 and its Mark in Life. Nature 2013, 469, 343–349. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Deb, G.; Wingelhofer, B.; Amaral, F.M.R.; Maiques-Diaz, A.; Chadwick, J.A.; Spencer, G.J.; Williams, E.L.; Leong, H.S.; Maes, T.; Somervaille, T.C.P. Pre-clinical activity of combined LSD1 and mTORC1 inhibition in MLL-translocated acute myeloid leukaemia. Leukemia 2019, 34, 1266–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Yu, J.S.L.; Tilgner, K.; Ong, S.H.; Koike-Yusa, H.; Yusa, K. Genome-wide CRISPR-KO Screen Uncovers mTORC1-Mediated Gsk3 Regulation in Naive Pluripotency Maintenance and Dissolution. Cell Rep. 2018, 24, 489–502. [Google Scholar] [CrossRef] [Green Version]
- Galeev, R.; Baudet, A.; Kumar, P.; Rundberg Nilsson, A.; Nilsson, B.; Soneji, S.; Törngren, T.; Borg, Å.; Kvist, A.; Larsson, J. Genome-wide RNAi Screen Identifies Cohesin Genes as Modifiers of Renewal and Differentiation in Human HSCs. Cell Rep. 2016, 14, 2988–3000. [Google Scholar] [CrossRef] [Green Version]
- Pusapati, G.V.; Kong, J.H.; Patel, B.B.; Krishnan, A.; Sagner, A.; Kinnebrew, M.; Briscoe, J.; Aravind, L.; Rohatgi, R. CRISPR Screens Uncover Genes that Regulate Target Cell Sensitivity to the Morphogen Sonic Hedgehog. Dev. Cell 2018, 44, 113–129.e8. [Google Scholar] [CrossRef] [Green Version]
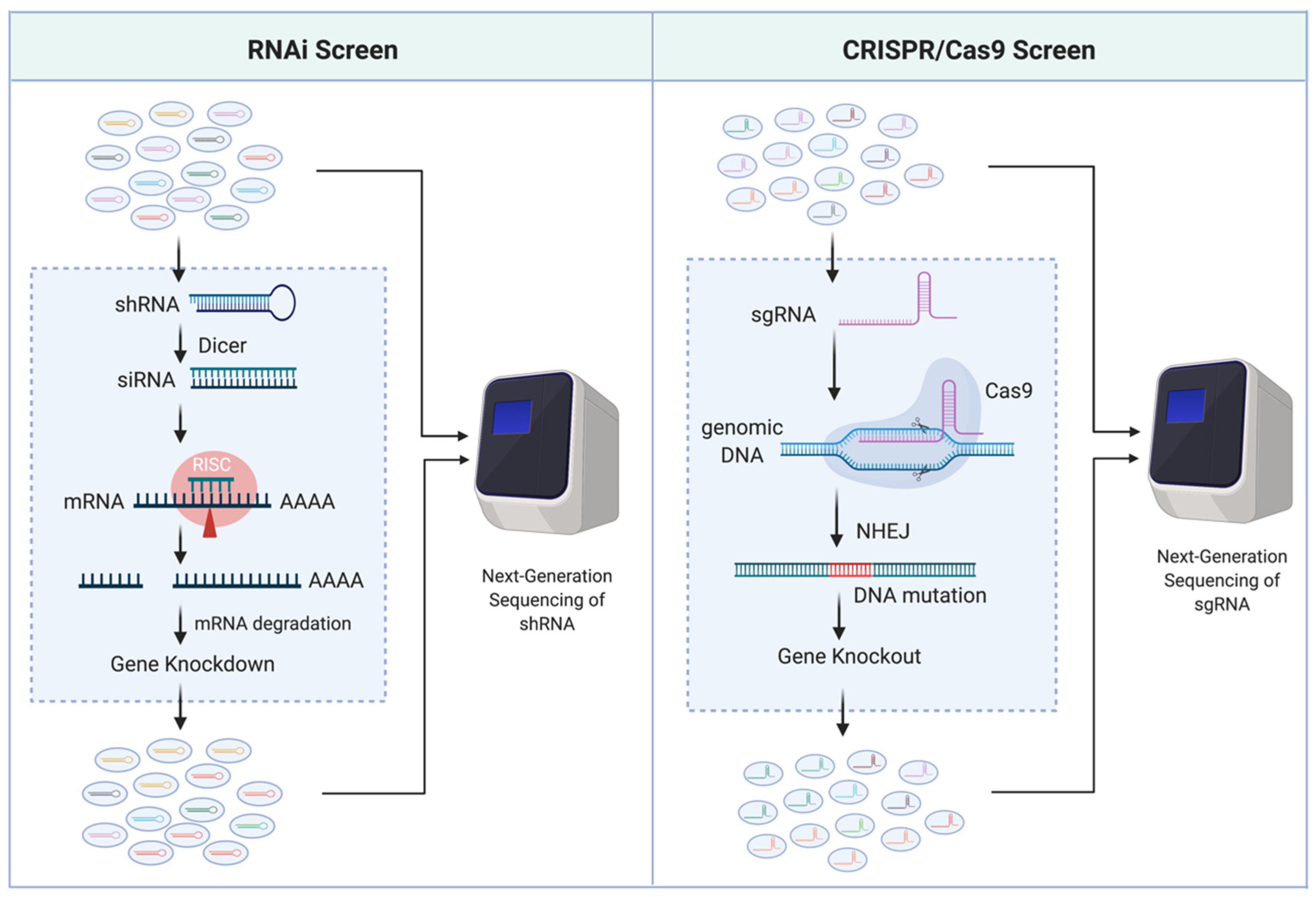
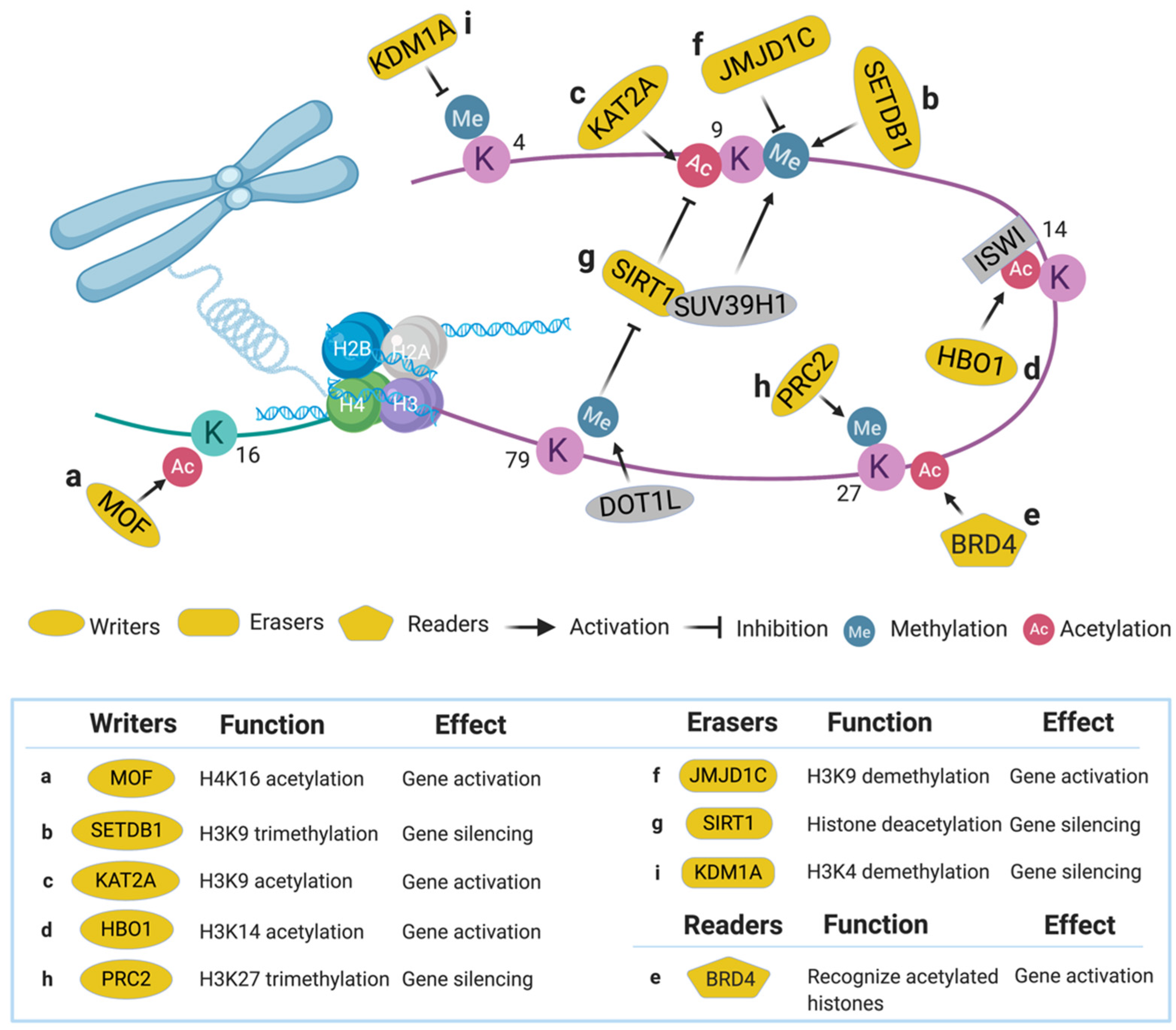


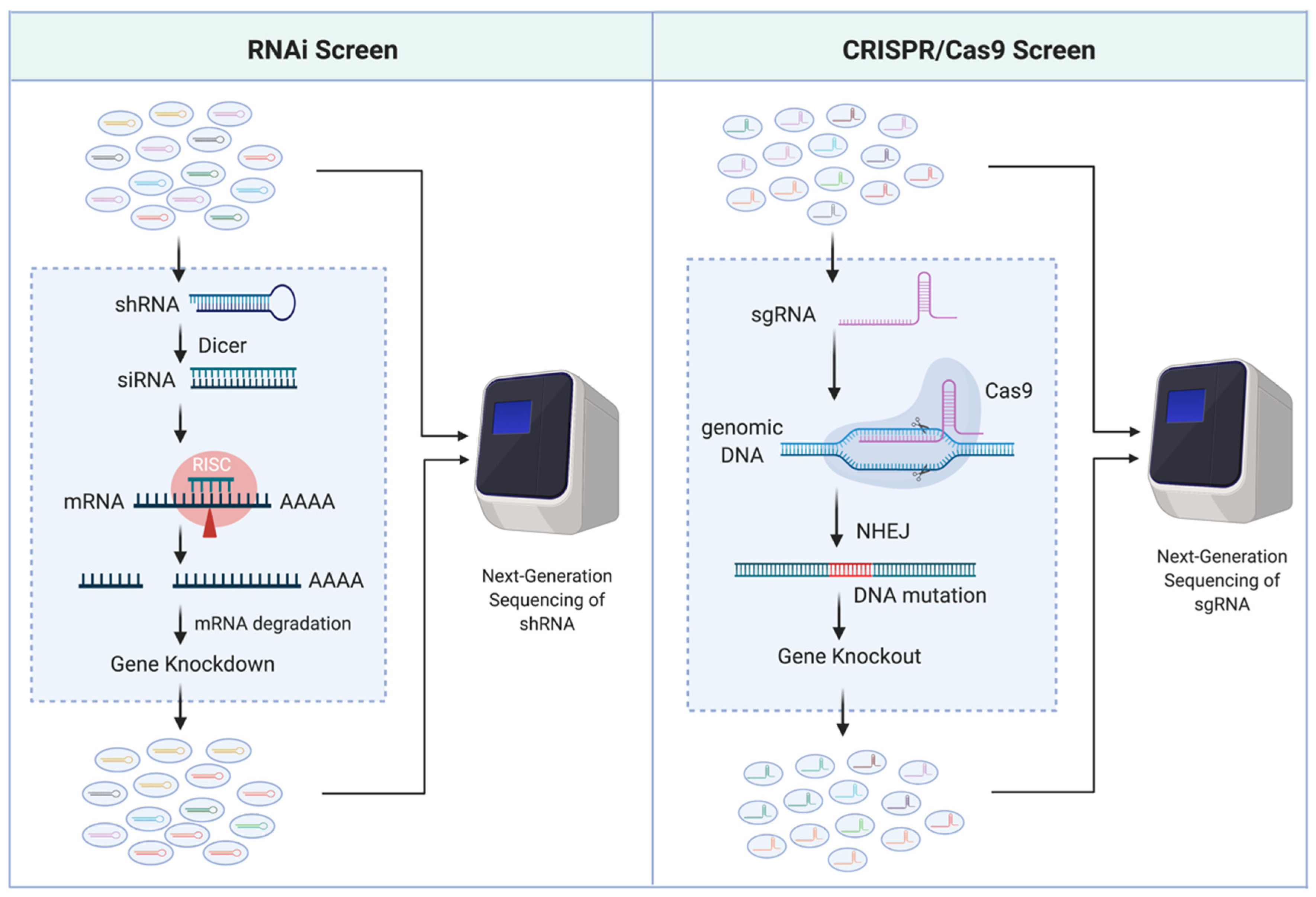{kind=link}
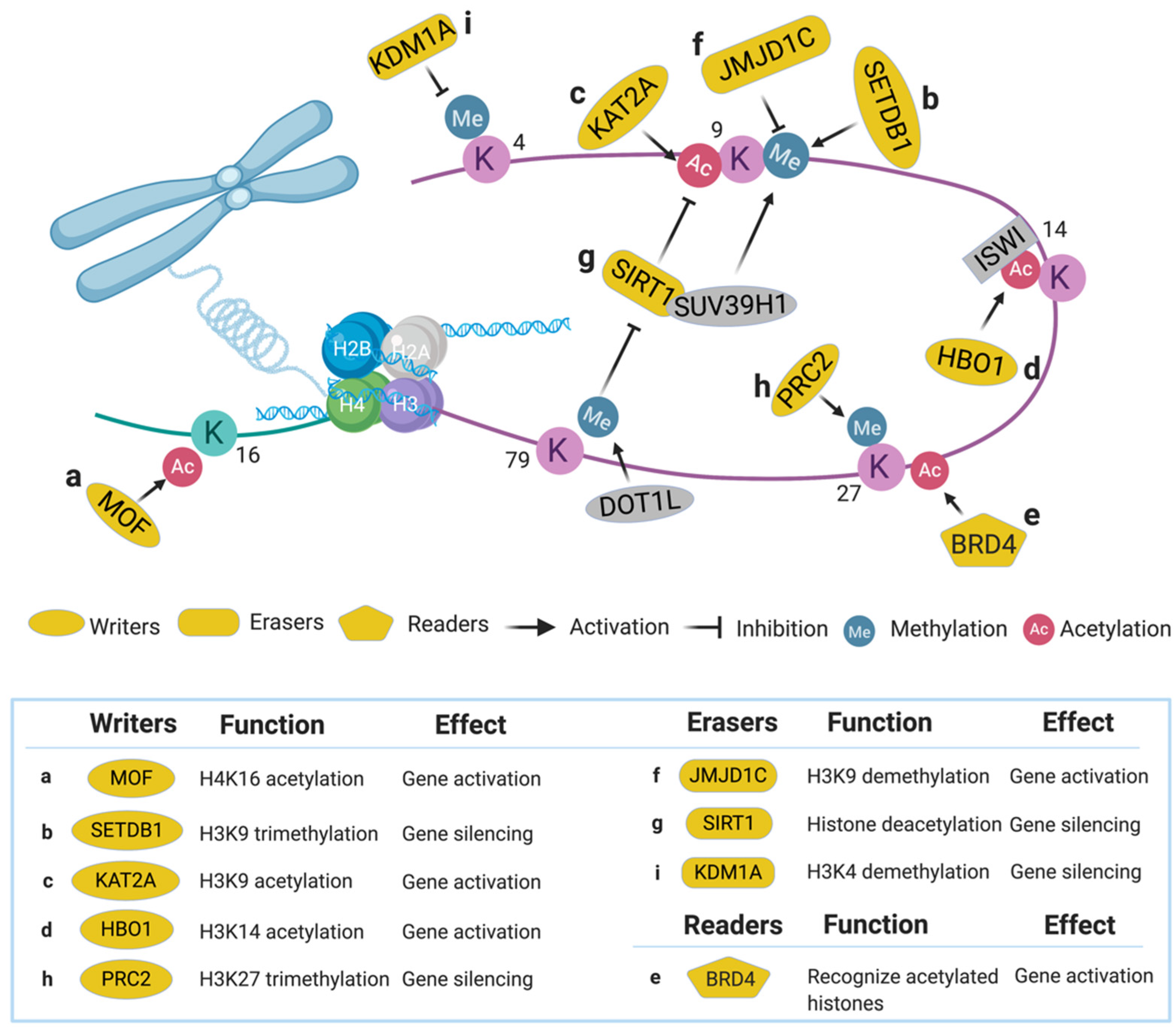{kind=link}
{kind=link}
| Gene Identified | Type of Screen | Gene # | Construct # | Report Year | Ref # | ||
|---|---|---|---|---|---|---|---|
| Epigenetic Regulators | Histone Writers | MOF SETDB1 KAT2A HBO1 | shRNA CRISPR/Cas9 CRISPR/Cas9 shRNA | 468 ~350 18,010 270 | 2252 ~15–25 per gene 90,709 1922 | 2017 2017 2016 2020 | [13] [14] [15] [16] |
| Histone Readers | BRD4 ENL | shRNA CRISPR/Cas9 | 243 18,080 | 1094 64,751 | 2011 2017 | [17] [18] | |
| Histone Erasers | JMJD1C SIRT1 | shRNA shRNA | 160 16,924 | 752 92,425 | 2016 2015 | [19] [20] | |
| Kinase Pathways | GSK3 ROCK1 PRL-3 CKMT1 LKB1 | shRNA shRNA shRNA shRNA CRISPR/Cas9 | ~1000 16,000 67 482 | ~5000 7709 80,000 361 6 sgRNAs per gene | 2012 2015 2018 2017 2018 | [21] [22] [23] [24] [25] | |
| Genes Expression Regulators | ZEB2 ZFP64 RBM25 RBM39 | shRNA CRISPR/Cas9 shRNA CRISPR/Cas9 | 11,194 1426 230 490 | 54,020 8658 613 2900 | 2017 2018 2019 2019 | [26] [27] [28] [29] | |
| Therapeutic Response Modulators | Cytarabine | WEE1 DCK | siRNA CRISPR/Cas9 | 572 18,080 | 2 siRNAs per gene 64,751 | 2012 2016 | [30] [31] |
| FLT3 inhibitors | SPRY3 ATM GLS | CRISPR/Cas9 shRNA CRISPR/Cas9 | 18,080 18,010 | 64,751 ~4–5 per genes 90,709 | 2017 2016 2018 | [32] [33] [34] | |
| Venetoclax | TP53 CLPB | CRISPR/Cas9 CRISPR/Cas9 | 18,010 18,675 | 90,709 110,257 | 2019 2019 | [35] [36] | |
| BET inhibitors | PRC2 KDM1A | shRNA CRISPR/Cas9 | 626 693 | 2917 ~1200 | 2015 2019 | [37] [38] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Garcia, M.; Wang, S.; Chen, C.-W. Therapeutic Target Discovery Using High-Throughput Genetic Screens in Acute Myeloid Leukemia. Cells 2020, 9, 1888. https://doi.org/10.3390/cells9081888
Liu Q, Garcia M, Wang S, Chen C-W. Therapeutic Target Discovery Using High-Throughput Genetic Screens in Acute Myeloid Leukemia. Cells. 2020; 9(8):1888. https://doi.org/10.3390/cells9081888
Chicago/Turabian StyleLiu, Qiao, Michelle Garcia, Shaoyuan Wang, and Chun-Wei Chen. 2020. "Therapeutic Target Discovery Using High-Throughput Genetic Screens in Acute Myeloid Leukemia" Cells 9, no. 8: 1888. https://doi.org/10.3390/cells9081888
APA StyleLiu, Q., Garcia, M., Wang, S., & Chen, C.-W. (2020). Therapeutic Target Discovery Using High-Throughput Genetic Screens in Acute Myeloid Leukemia. Cells, 9(8), 1888. https://doi.org/10.3390/cells9081888