Role of EGFR in the Nervous System



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
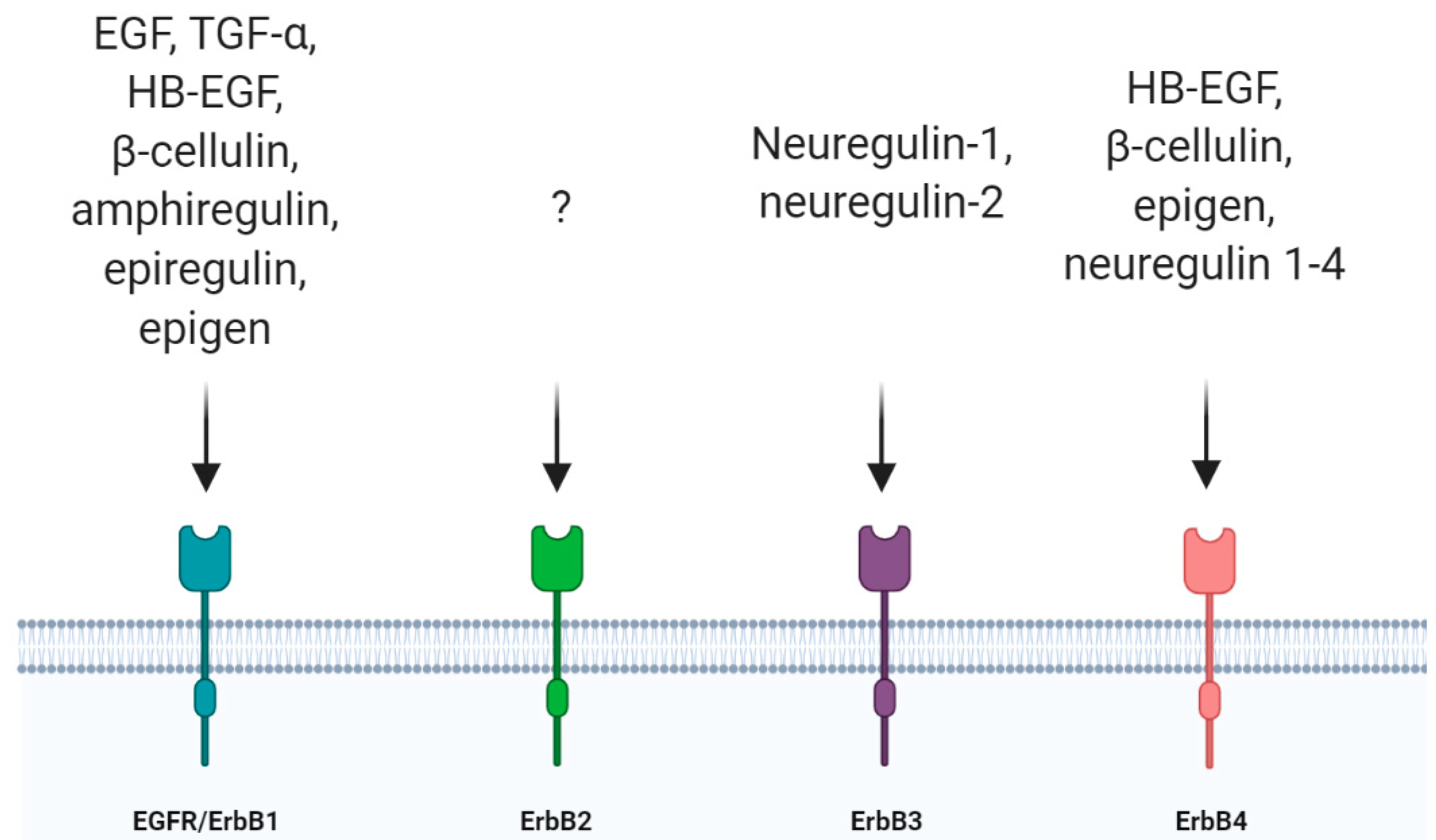
2. EGFR and the Nervous System
3. EGFR Functions in the CNS
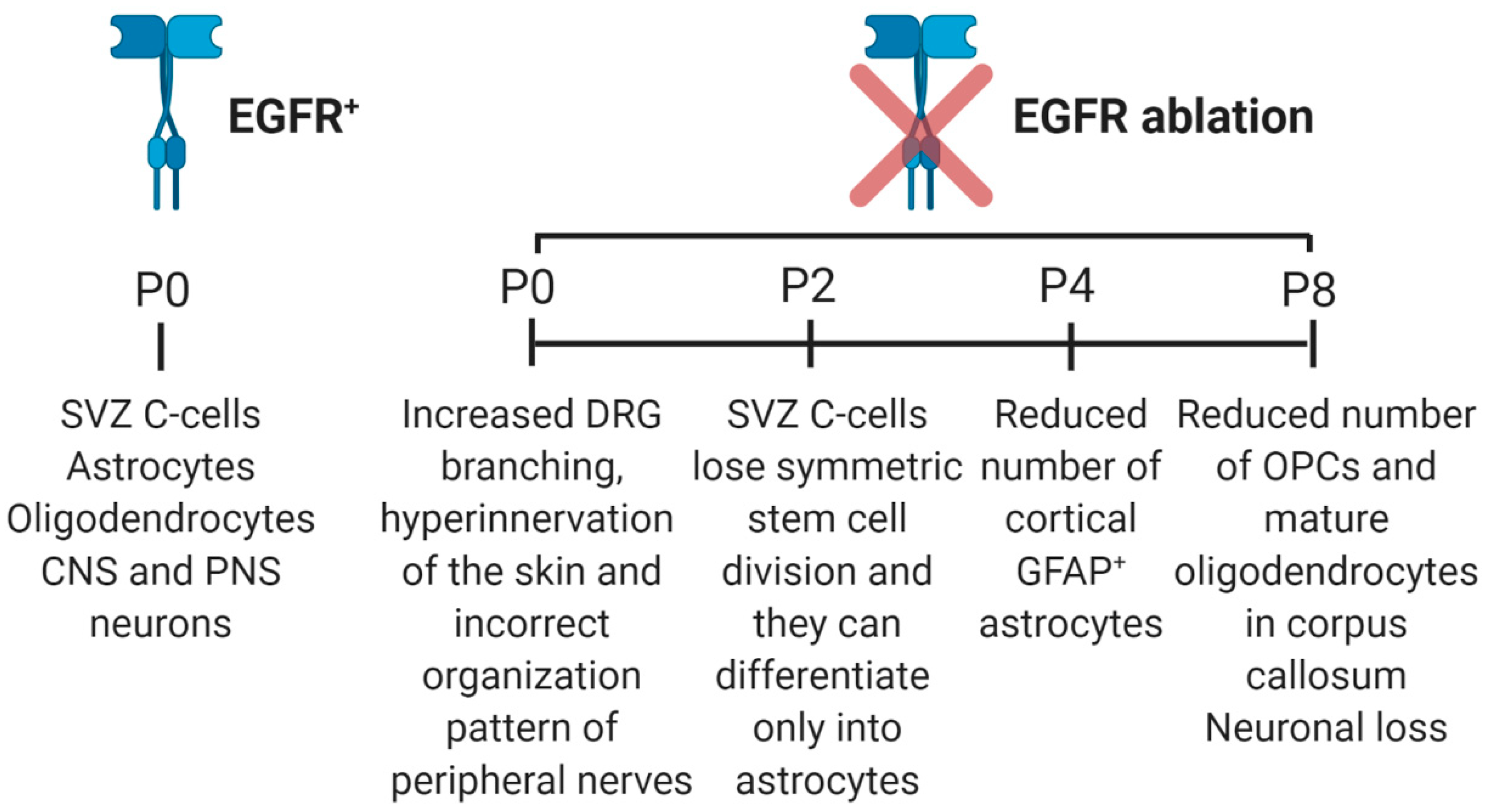
3.1. Role of EGFR in Neural Stem Cell Pool Maintenance
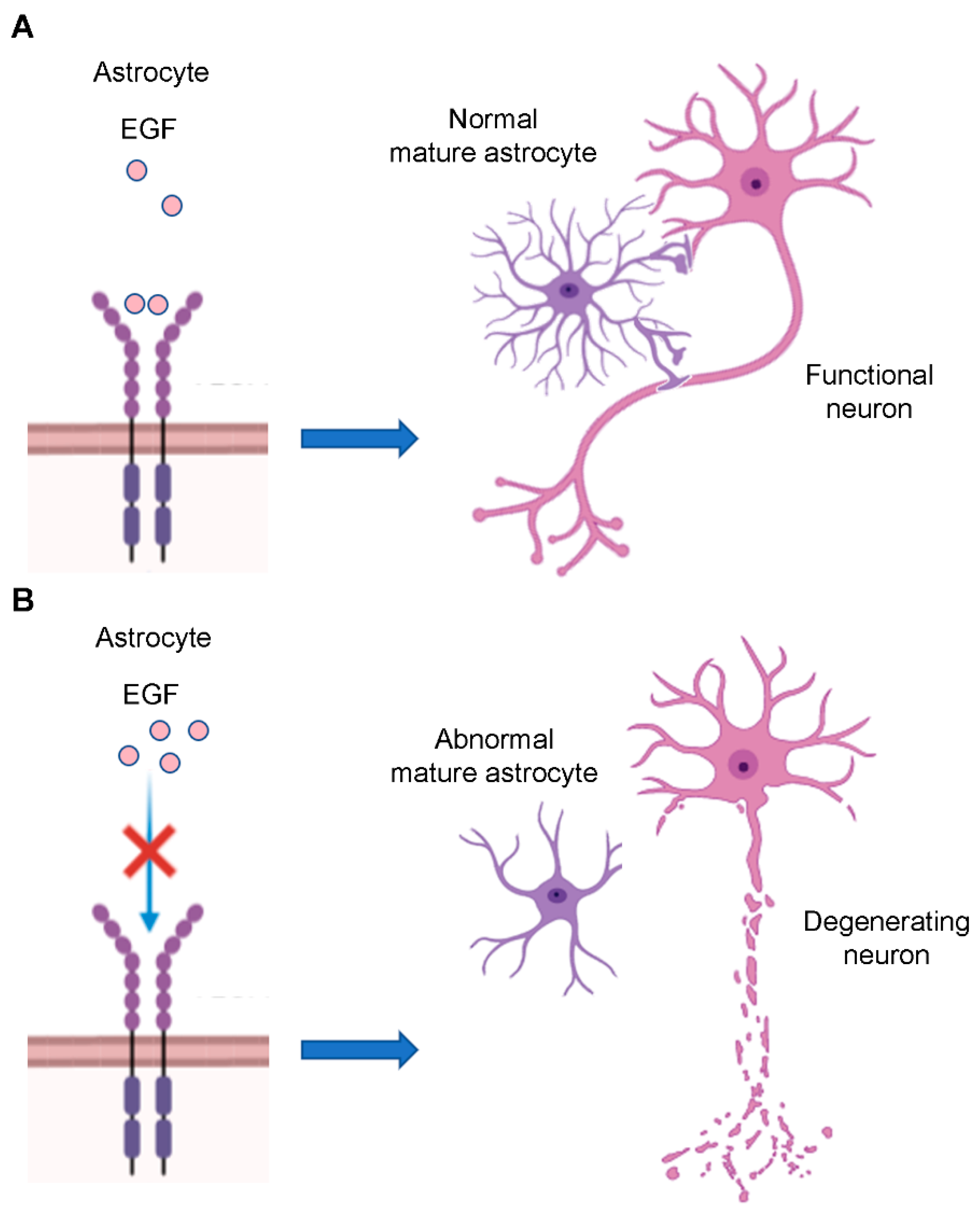
3.2. Role of EGFR in Astrocyte Differentiation and Maturation
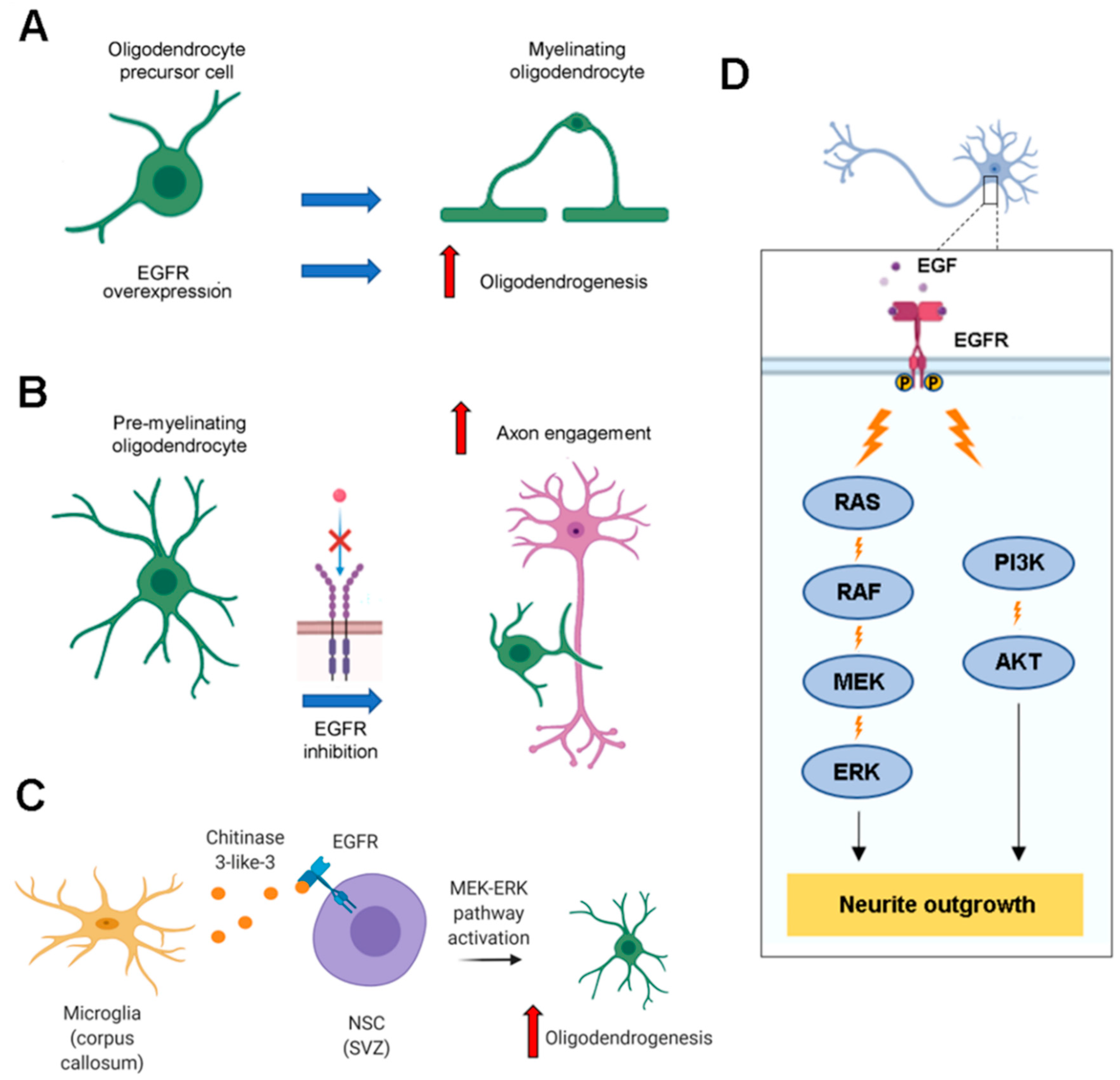
3.3. Role of EGFR in Oligodendrocyte Maturation
3.4. Role of EGFR in Neurite Outgrowth
4. EGFR Functions in the PNS
5. EGFR Functions after Injury and Its Role in Regeneration
6. EGFR in Nervous System Diseases
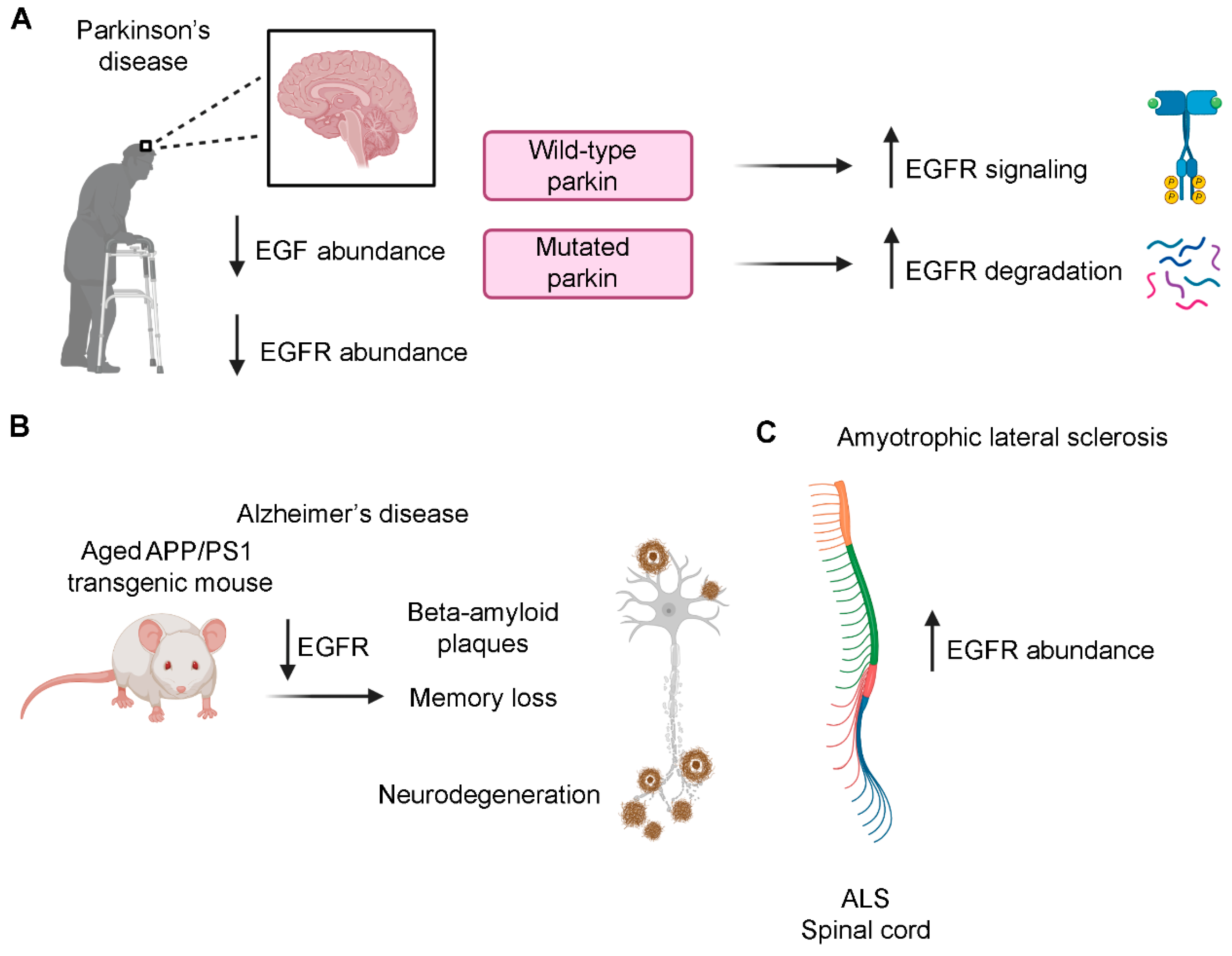
6.1. Parkinson’s Disease
6.2. Alzheimer’s Disease
6.3. Amyotrophic Lateral Sclerosis (ALS)
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of egf receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (egfr) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; King, L., Jr.; Cohen, S. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature 1978, 276, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Wolf, E. The epidermal growth factor receptor ligands at a glance. J. Cell Physiol. 2009, 218, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, S.L.; Mac, A.S.; Henriksen, L.; van Deurs, B.; Grovdal, L.M. Egfr signaling patterns are regulated by its different ligands. Growth Factors 2014, 32, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Black, L.E.; Longo, J.F.; Carroll, S.L. Mechanisms of receptor tyrosine-protein kinase erbb-3 (erbb3) action in human neoplasia. Am. J. Pathol. 2019, 189, 1898–1912. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.C.; Fenton, S.E.; Berkowitz, E.A.; Hissong, M.A. Transforming growth factor alpha: Expression, regulation, and biological activities. Pharmacol. Rev. 1995, 47, 51–85. [Google Scholar]
- Barnard, J.A.; Beauchamp, R.D.; Russell, W.E.; Dubois, R.N.; Coffey, R.J. Epidermal growth factor-related peptides and their relevance to gastrointestinal pathophysiology. Gastroenterology 1995, 108, 564–580. [Google Scholar] [CrossRef]
- Estrada, C.; Villalobo, A. Epidermal Growth Factor Receptor in the Adult Brain. In The Cell Cycle in the Central Nervous System; Janigro, D., Ed.; Humana Press: Totowa, NJ, USA, 2006; pp. 265–277. [Google Scholar]
- Klapper, L.N.; Kirschbaum, M.H.; Sela, M.; Yarden, Y. Biochemical and clinical implications of the erbb/her signaling network of growth factor receptors. Adv. Cancer Res. 2000, 77, 25–79. [Google Scholar]
- Novak, U.; Walker, F.; Kaye, A. Expression of egfr-family proteins in the brain: Role in development, health and disease. J. Clin. Neurosci. 2001, 8, 106–111. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the erbb signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. Erbb receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Woelk, T.; Puri, C.; Maspero, E.; Tacchetti, C.; Transidico, P.; Di Fiore, P.P.; Polo, S. Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. USA 2005, 102, 2760–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, J.; Spits, M.; Neefjes, J.; Berlin, I. The egfr odyssey—From activation to destruction in space and time. J. Cell Sci. 2017, 130, 4087–4096. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Algisi, V.; Nappo, G.; Conte, A.; Pascolutti, R.; Cuomo, A.; Bonaldi, T.; Argenzio, E.; Verhoef, L.G.; Maspero, E.; et al. Threshold-controlled ubiquitination of the egfr directs receptor fate. EMBO J. 2013, 32, 2140–2157. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Kirkpatrick, D.; Jiang, X.; Gygi, S.; Sorkin, A. Differential regulation of egf receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 2006, 21, 737–748. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The escrt pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Elkin, S.R.; Lakoduk, A.M.; Schmid, S.L. Endocytic pathways and endosomal trafficking: A primer. Wien Med. Wochenschr. 2016, 166, 196–204. [Google Scholar] [CrossRef] [Green Version]
- Bilic, J.; Huang, Y.L.; Davidson, G.; Zimmermann, T.; Cruciat, C.M.; Bienz, M.; Niehrs, C. Wnt induces lrp6 signalosomes and promotes dishevelled-dependent lrp6 phosphorylation. Science 2007, 316, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, R.; De Robertis, E.M. Endocytic control of growth factor signalling: Multivesicular bodies as signalling organelles. Nat. Rev. Mol. Cell Biol. 2011, 13, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilo, B.Z. Regulating the dynamics of egf receptor signaling in space and time. Development 2005, 132, 4017–4027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wassarman, D.A.; Therrien, M.; Rubin, G.M. The ras signaling pathway in drosophila. Curr. Opin. Genet. Dev. 1995, 5, 44–50. [Google Scholar] [CrossRef]
- Perrimon, N.; Perkins, L.A. There must be 50 ways to rule the signal: The case of the drosophila egf receptor. Cell 1997, 89, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Voas, M.G.; Rebay, I. Signal integration during development: Insights from the drosophila eye. Dev. Dyn. 2004, 229, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Chia, C.M.; Winston, R.M.; Handyside, A.H. Egf, tgf-alpha and egfr expression in human preimplantation embryos. Development 1995, 121, 299–307. [Google Scholar]
- Chen, J.; Zeng, F.; Forrester, S.J.; Eguchi, S.; Zhang, M.-Z.; Harris, R.C. Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol. Rev. 2016, 96, 1025–1069. [Google Scholar] [CrossRef]
- Peus, D.; Vasa, R.A.; Meves, A.; Beyerle, A.; Pittelkow, M.R. Uvb-induced epidermal growth factor receptor phosphorylation is critical for downstream signaling and keratinocyte survival. Photochem. Photobiol. 2000, 72, 135–140. [Google Scholar] [CrossRef]
- Repertinger, S.K.; Campagnaro, E.; Fuhrman, J.; El-Abaseri, T.; Yuspa, S.H.; Hansen, L.A. Egfr enhances early healing after cutaneous incisional wounding. J. Investig. Dermatol. 2004, 123, 982–989. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, T.S.; Levchenko, V.; O’Connor, P.M.; Ilatovskaya, D.V.; Palygin, O.; Mori, T.; Mattson, D.L.; Sorokin, A.; Lombard, J.H.; Cowley, A.W., Jr.; et al. Deficiency of renal cortical egf increases enac activity and contributes to salt-sensitive hypertension. J. Am. Soc. Nephrol. 2013, 24, 1053–1062. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, I.E.; Kenigsberg, R.L. Localization and characterization of epidermal growth-factor receptors in the developing rat medial septal area in culture. Brain Res. 1994, 656, 115–126. [Google Scholar] [CrossRef]
- Seroogy, K.B.; Numan, S.; Gall, C.M.; Lee, D.C.; Kornblum, H.I. Expression of egf receptor mrna in rat nigrostriatal system. Neuroreport 1994, 6, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Seroogy, K.B.; Gall, C.M.; Lee, D.C.; Kornblum, H.I. Proliferative zones of postnatal rat brain express epidermal growth factor receptor mrna. Brain Res. 1995, 670, 157–164. [Google Scholar] [CrossRef]
- Kornblum, H.I.; Gall, C.M.; Seroogy, K.B.; Lauterborn, J.C. A subpopulation of striatal gabaergic neurons expresses the epidermal growth factor receptor. Neuroscience 1995, 69, 1025–1029. [Google Scholar] [CrossRef]
- Kornblum, H.I.; Hussain, R.J.; Bronstein, J.M.; Gall, C.M.; Lee, D.C.; Seroogy, K.B. Prenatal ontogeny of the epidermal growth factor receptor and its ligand, transforming growth factor alpha, in the rat brain. J. Comp. Neurol. 1997, 380, 243–261. [Google Scholar] [CrossRef]
- Huerta, J.J.; Diaz-Trelles, R.; Naves, F.J.; Llamosas, M.M.; Del Valle, M.E.; Vega, J.A. Epidermal growth factor receptor in adult human dorsal root ganglia. Anat. Embryol. 1996, 194, 253–257. [Google Scholar] [CrossRef]
- Vega, J.A.; Vazquez, E.; Naves, F.J.; Calzada, B.; del Valle, M.E.; Represa, J.J. Expression of epidermal growth factor receptor (egfr) immunoreactivity in human cutaneous nerves and sensory corpuscles. Anat. Rec. 1994, 240, 125–130. [Google Scholar] [CrossRef]
- Xian, C.J.; Zhou, X.F. Neuronal-glial differential expression of tgf-alpha and its receptor in the dorsal root ganglia in response to sciatic nerve lesion. Exp. Neurol. 1999, 157, 317–326. [Google Scholar] [CrossRef]
- Xian, C.J.; Zhou, X.F. Egf family of growth factors: Essential roles and functional redundancy in the nerve system. Front. Biosci. 2004, 9, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Sibilia, M.; Kroismayr, R.; Lichtenberger, B.M.; Natarajan, A.; Hecking, M.; Holcmann, M. The epidermal growth factor receptor: From development to tumorigenesis. Differentiation 2007, 75, 770–787. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Ann. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hennigan, A.; O’Callaghan, R.M.; Kelly, A.M. Neurotrophins and their receptors: Roles in plasticity, neurodegeneration and neuroprotection. Biochem. Soc. Trans. 2007, 35, 424–427. [Google Scholar] [CrossRef] [PubMed]
- Ginty, D.D.; Segal, R.A. Retrograde neurotrophin signaling: Trk-ing along the axon. Curr. Opin. Neurobiol. 2002, 12, 268–274. [Google Scholar] [CrossRef]
- Bucci, C.; Alifano, P.; Cogli, L. The role of rab proteins in neuronal cells and in the trafficking of neurotrophin receptors. Membranes 2014, 4, 642–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Pinilla, F.; Knauer, D.J.; Nieto-Sampedro, M. Epidermal growth factor receptor immunoreactivity in rat brain. Development and cellular localization. Brain Res. 1988, 438, 385–390. [Google Scholar] [CrossRef]
- Grochowski, C.; Radzikowska, E.; Maciejewski, R. Neural stem cell therapy-brief review. Clin. Neurol. Neurosurg. 2018, 173, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Gritti, A.; Frolichsthal-Schoeller, P.; Galli, R.; Parati, E.A.; Cova, L.; Pagano, S.F.; Bjornson, C.R.; Vescovi, A.L. Epidermal and fibroblast growth factors behave as mitogenic regulators for a single multipotent stem cell-like population from the subventricular region of the adult mouse forebrain. J. Neurosci. 1999, 19, 3287–3297. [Google Scholar] [CrossRef] [Green Version]
- Tropepe, V.; Sibilia, M.; Ciruna, B.G.; Rossant, J.; Wagner, E.F.; van der Kooy, D. Distinct neural stem cells proliferate in response to egf and fgf in the developing mouse telencephalon. Dev. Biol. 1999, 208, 166–188. [Google Scholar] [CrossRef] [Green Version]
- Seri, B.; Herrera, D.G.; Gritti, A.; Ferron, S.; Collado, L.; Vescovi, A.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Composition and organization of the scz: A large germinal layer containing neural stem cells in the adult mammalian brain. Cereb. Cortex. 2006, 16, i103–i111. [Google Scholar] [CrossRef] [Green Version]
- Doetsch, F.; Petreanu, L.; Caille, I.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Egf converts transit-amplifying neurogenic precursors in the adult brain into multipotent stem cells. Neuron 2002, 36, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Dunne, C.; Hewson, J.; Wohl, C.; Wheatley, M.; Peterson, A.C.; Reynolds, B.A. Multipotent cns stem cells are present in the adult mammalian spinal cord and ventricular neuroaxis. J. Neurosci. 1996, 16, 7599–7609. [Google Scholar] [CrossRef]
- Morshead, C.M.; Reynolds, B.A.; Craig, C.G.; McBurney, M.W.; Staines, W.A.; Morassutti, D.; Weiss, S.; van der Kooy, D. Neural stem cells in the adult mammalian forebrain: A relatively quiescent subpopulation of subependymal cells. Neuron 1994, 13, 1071–1082. [Google Scholar] [CrossRef]
- Martens, D.J.; Seaberg, R.M.; van der Kooy, D. In vivo infusions of exogenous growth factors into the fourth ventricle of the adult mouse brain increase the proliferation of neural progenitors around the fourth ventricle and the central canal of the spinal cord. Eur J. Neurosci. 2002, 16, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Robson, J.P.; Wagner, B.; Glitzner, E.; Heppner, F.L.; Steinkellner, T.; Khan, D.; Petritsch, C.; Pollak, D.D.; Sitte, H.H.; Sibilia, M. Impaired neural stem cell expansion and hypersensitivity to epileptic seizures in mice lacking the egfr in the brain. FEBS J. 2018, 285, 3175–3196. [Google Scholar] [CrossRef] [Green Version]
- Doetsch, F.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J. Neurosci. 1997, 17, 5046–5061. [Google Scholar] [CrossRef]
- Kuhn, H.G.; Winkler, J.; Kempermann, G.; Thal, L.J.; Gage, F.H. Epidermal growth factor and fibroblast growth factor-2 have different effects on neural progenitors in the adult rat brain. J. Neurosci. 1997, 17, 5820–5829. [Google Scholar] [CrossRef]
- Doetsch, F.; Caille, I.; Lim, D.A.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Palmer, T.D.; Willhoite, A.R.; Gage, F.H. Vascular niche for adult hippocampal neurogenesis. J. Comp. Neurol. 2000, 425, 479–494. [Google Scholar] [CrossRef]
- Temple, S. The development of neural stem cells. Nature 2001, 414, 112–117. [Google Scholar] [CrossRef]
- Hitoshi, S.; Alexson, T.; Tropepe, V.; Donoviel, D.; Elia, A.J.; Nye, J.S.; Conlon, R.A.; Mak, T.W.; Bernstein, A.; van der Kooy, D. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002, 16, 846–858. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, A.; Rubio, M.E.; Gallo, V. Notch and egfr pathway interaction regulates neural stem cell number and self-renewal. Nature 2010, 467, 323–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazel, C.Y.; Limke, T.L.; Osborne, J.K.; Miura, T.; Cai, J.; Pevny, L.; Rao, M.S. Sox2 expression defines a heterogeneous population of neurosphere-forming cells in the adult murine brain. Aging Cell 2005, 4, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Raff, M. Chromatin remodeling and histone modification in the conversion of oligodendrocyte precursors to neural stem cells. Genes Dev. 2004, 18, 2963–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Zhang, L.; Wen, J.; Wang, S.; Li, M.; Feng, R.; Yang, X.; Li, L. The egf receptor-sox2-egf receptor feedback loop positively regulates the self-renewal of neural precursor cells. Stem Cells 2010, 28, 279–286. [Google Scholar]
- Miyata, T.; Kawaguchi, A.; Okano, H.; Ogawa, M. Asymmetric inheritance of radial glial fibers by cortical neurons. Neuron 2001, 31, 727–741. [Google Scholar] [CrossRef] [Green Version]
- Noctor, S.C.; Flint, A.C.; Weissman, T.A.; Dammerman, R.S.; Kriegstein, A.R. Neurons derived from radial glial cells establish radial units in neocortex. Nature 2001, 409, 714–720. [Google Scholar] [CrossRef]
- Sun, Y.; Goderie, S.K.; Temple, S. Asymmetric distribution of egfr receptor during mitosis generates diverse cns progenitor cells. Neuron 2005, 45, 873–886. [Google Scholar] [CrossRef] [Green Version]
- Lillien, L.; Raphael, H. Bmp and fgf regulate the development of egf-responsive neural progenitor cells. Development 2000, 127, 4993–5005. [Google Scholar]
- Burrows, R.C.; Wancio, D.; Levitt, P.; Lillien, L. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in egfr expression in the cortex. Neuron 1997, 19, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Verkhratsky, A.; Toescu, E.C. Neuronal-glial networks as substrate for cns integration. J. Cell Mol. Med. 2006, 10, 826–836. [Google Scholar] [CrossRef]
- Ge, W.P.; Miyawaki, A.; Gage, F.H.; Jan, Y.N.; Jan, L.Y. Local generation of glia is a major astrocyte source in postnatal cortex. Nature 2012, 484, 376–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, A.M.; Gao, Y.; Amemiya, Y.; Kahn, H.J.; Kitching, R.; Yang, Y.; Sun, P.; Narod, S.A.; Hanna, W.M.; Seth, A.K. A novel ring-type ubiquitin ligase breast cancer-associated gene 2 correlates with outcome in invasive breast cancer. Cancer Res. 2005, 65, 10401–10412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deribe, Y.L.; Wild, P.; Chandrashaker, A.; Curak, J.; Schmidt, M.H.H.; Kalaidzidis, Y.; Milutinovic, N.; Kratchmarova, I.; Buerkle, L.; Fetchko, M.J.; et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase hdac6. Sci. Signal. 2009, 2, ra84. [Google Scholar] [PubMed]
- Fujimoto, I.; Hasegawa, K.; Fujiwara, K.; Yamada, M.; Yoshikawa, K. Necdin controls egfr signaling linked to astrocyte differentiation in primary cortical progenitor cells. Cell Signal. 2016, 28, 94–107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An rna-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.L.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Khankan, R.R.; Caneda, C.; Godoy, M.I.; Haney, M.S.; Krawczyk, M.C.; Bassik, M.C.; Sloan, S.A.; Zhang, Y. Astrocyte-to-astrocyte contact and a positive feedback loop of growth factor signaling regulate astrocyte maturation. Glia 2019, 67, 1571–1597. [Google Scholar] [CrossRef]
- Levison, S.W.; Jiang, F.J.; Stoltzfus, O.K.; Ducceschi, M.H. Il-6-type cytokines enhance epidermal growth factor-stimulated astrocyte proliferation. Glia 2000, 32, 328–337. [Google Scholar] [CrossRef]
- Liu, B.; Neufeld, A.H. Activation of epidermal growth factor receptors directs astrocytes to organize in a network surrounding axons in the developing rat optic nerve. Dev. Biol. 2004, 273, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Neufeld, A.H. Activation of epidermal growth factor receptors in astrocytes: From development to neural injury. J. Neurosci. Res. 2007, 85, 3523–3529. [Google Scholar] [CrossRef]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the egf receptor. Science 1995, 269, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Kornblum, H.I.; Hussain, R.; Wiesen, J.; Miettinen, P.; Zurcher, S.D.; Chow, K.; Derynck, R.; Werb, Z. Abnormal astrocyte development and neuronal death in mice lacking the epidermal growth factor receptor. J. Neurosci. Res. 1998, 53, 697–717. [Google Scholar] [CrossRef]
- Liu, B.; Chen, H.; Johns, T.G.; Neufeld, A.H. Epidermal growth factor receptor activation: An upstream signal for transition of quiescent astrocytes into reactive astrocytes after neural injury. J. Neurosci. 2006, 26, 7532–7540. [Google Scholar] [CrossRef] [PubMed]
- Wagner, B.; Natarajan, A.; Grunaug, S.; Kroismayr, R.; Wagner, E.F.; Sibilia, M. Neuronal survival depends on egfr signaling in cortical but not midbrain astrocytes. EMBO J. 2006, 25, 752–762. [Google Scholar] [CrossRef] [Green Version]
- Sibilia, M.; Steinbach, J.P.; Stingl, L.; Aguzzi, A.; Wagner, E.F. A strain-independent postnatal neurodegeneration in mice lacking the egf receptor. EMBO J. 1998, 17, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johe, K.K.; Hazel, T.G.; Muller, T.; Dugich-Djordjevic, M.M.; McKay, R.D. Single factors direct the differentiation of stem cells from the fetal and adult central nervous system. Genes Dev. 1996, 10, 3129–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the central nervous system: Structure, function, and pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef]
- Matthieu, J.M.; Comte, V.; Tosic, M.; Honegger, P. Myelin gene expression during demyelination and remyelination in aggregating brain cell cultures. J. Neuroimmunol. 1992, 40, 231–234. [Google Scholar] [CrossRef]
- Aguirre, A.; Dupree, J.L.; Mangin, J.M.; Gallo, V. A functional role for egfr signaling in myelination and remyelination. Nat. Neurosci. 2007, 10, 990–1002. [Google Scholar] [CrossRef]
- Yang, J.; Cheng, X.; Qi, J.; Xie, B.; Zhao, X.; Zheng, K.; Zhang, Z.; Qiu, M. Egf enhances oligodendrogenesis from glial progenitor cells. Front. Mol. Neurosci. 2017, 10, 106. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.I.; Mallon, B.S.; Matsui, T.; Ogawa, W.; Rosenzweig, A.; Okamoto, T.; Macklin, W.B. Akt-mediated survival of oligodendrocytes induced by neuregulins. J. Neurosci. 2000, 20, 7622–7630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, E.; Park, D.K.; Tzvetanova, I.D.; Leiton, C.V.; Cho, B.S.; Colognato, H. Tyrosine phosphatases shp1 and shp2 have unique and opposing roles in oligodendrocyte development. J. Neurochem. 2010, 113, 200–212. [Google Scholar] [CrossRef]
- Zhu, Y.; Park, J.; Hu, X.; Zheng, K.; Li, H.; Cao, Q.; Feng, G.S.; Qiu, M. Control of oligodendrocyte generation and proliferation by shp2 protein tyrosine phosphatase. Glia 2010, 58, 1407–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Y.; Zhang, Y.; Lu, Y.; Guo, W.; Liu, P.; Zhou, J.; Xiang, Z.; He, C. Shp-2 promotes the maturation of oligodendrocyte precursor cells through akt and erk1/2 signaling in vitro. PLoS ONE 2011, 6, e21058. [Google Scholar] [CrossRef]
- Holgado-Madruga, M.; Emlet, D.R.; Moscatello, D.K.; Godwin, A.K.; Wong, A.J. A grb2-associated docking protein in egf- and insulin-receptor signalling. Nature 1996, 379, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Neel, B.G. The "gab" in signal transduction. Trends Cell Biol. 2003, 13, 122–130. [Google Scholar] [CrossRef]
- Hayakawa-Yano, Y.; Nishida, K.; Fukami, S.; Gotoh, Y.; Hirano, T.; Nakagawa, T.; Shimazaki, T.; Okano, H. Epidermal growth factor signaling mediated by grb2 associated binder1 is required for the spatiotemporally regulated proliferation of olig2-expressing progenitors in the embryonic spinal cord. Stem Cells 2007, 25, 1410–1422. [Google Scholar] [CrossRef]
- Nocita, E.; Del Giovane, A.; Tiberi, M.; Boccuni, L.; Fiorelli, D.; Sposato, C.; Romano, E.; Basoli, F.; Trombetta, M.; Rainer, A.; et al. Egfr/erbb inhibition promotes opc maturation up to axon engagement by co-regulating pip2 and mbp. Cells 2019, 8, 844. [Google Scholar] [CrossRef] [Green Version]
- Menn, B.; Garcia-Verdugo, J.M.; Yaschine, C.; Gonzalez-Perez, O.; Rowitch, D.; Alvarez-Buylla, A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 2006, 26, 7907–7918. [Google Scholar] [CrossRef]
- Keirstead, H.S.; Blakemore, W.F. The role of oligodendrocytes and oligodendrocyte progenitors in cns remyelination. Adv. Exp. Med. Biol. 1999, 468, 183–197. [Google Scholar]
- Scafidi, J.; Hammond, T.R.; Scafidi, S.; Ritter, J.; Jablonska, B.; Roncal, M.; Szigeti-Buck, K.; Coman, D.; Huang, Y.; McCarter, R.J., Jr.; et al. Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature 2014, 506, 230–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Pluchino, S.; Muzio, L.; Imitola, J.; Deleidi, M.; Alfaro-Cervello, C.; Salani, G.; Porcheri, C.; Brambilla, E.; Cavasinni, F.; Bergamaschi, A.; et al. Persistent inflammation alters the function of the endogenous brain stem cell compartment. Brain 2008, 131, 2564–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starossom, S.C.; Imitola, J.; Wang, Y.; Cao, L.; Khoury, S.J. Subventricular zone microglia transcriptional networks. Brain Behav. Immun. 2011, 25, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starossom, S.C.; Campo Garcia, J.; Woelfle, T.; Romero-Suarez, S.; Olah, M.; Watanabe, F.; Cao, L.; Yeste, A.; Tukker, J.J.; Quintana, F.J.; et al. Chi3l3 induces oligodendrogenesis in an experimental model of autoimmune neuroinflammation. Nat. Commun. 2019, 10, 217. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Perez, O.; Romero-Rodriguez, R.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Epidermal growth factor induces the progeny of subventricular zone type b cells to migrate and differentiate into oligodendrocytes. Stem Cells 2009, 27, 2032–2043. [Google Scholar] [CrossRef] [Green Version]
- Fox, I.J.; Kornblum, H.I. Developmental profile of erbb receptors in murine central nervous system: Implications for functional interactions. J. Neurosci. Res. 2005, 79, 584–597. [Google Scholar] [CrossRef]
- Ivkovic, S.; Canoll, P.; Goldman, J.E. Constitutive egfr signaling in oligodendrocyte progenitors leads to diffuse hyperplasia in postnatal white matter. J. Neurosci. 2008, 28, 914–922. [Google Scholar] [CrossRef]
- Goldshmit, Y.; Greenhalgh, C.J.; Turnley, A.M. Suppressor of cytokine signalling-2 and epidermal growth factor regulate neurite outgrowth of cortical neurons. Eur J. Neurosci. 2004, 20, 2260–2266. [Google Scholar] [CrossRef]
- Goldshmit, Y.; Walters, C.E.; Scott, H.J.; Greenhalgh, C.J.; Turnley, A.M. Socs2 induces neurite outgrowth by regulation of epidermal growth factor receptor activation. J. Biol. Chem. 2004, 279, 16349–16355. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Cui, M.; Zhao, H.; Wang, T.; Shen, Y.; Dong, Q. Tissue kallikrein mediates neurite outgrowth through epidermal growth factor receptor and flotillin-2 pathway in vitro. Cell Signal. 2014, 26, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Peng, L.; Wu, Y.; Li, Y.; Wang, L.; Luo, J.H.; Xu, J. Endocytic adaptor protein hip1r controls intracellular trafficking of epidermal growth factor receptor in neuronal dendritic development. Front. Mol. Neurosci. 2018, 11, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niyomchan, A.; Watcharasit, P.; Visitnonthachai, D.; Homkajorn, B.; Thiantanawat, A.; Satayavivad, J. Insulin attenuates arsenic-induced neurite outgrowth impairments by activating the pi3k/akt/sirt1 signaling pathway. Toxicol. Lett. 2015, 236, 138–144. [Google Scholar] [CrossRef]
- Nishimoto, T.; Kimura, R.; Matsumoto, A.; Sugimoto, H. Streptozotocin induces neurite outgrowth via pi3k-akt and glycogen synthase kinase 3beta in neuro2a cells. Cell Mol. Biol. (Noisy-le-grand) 2016, 62, 74–78. [Google Scholar]
- Ng, T.L.; Rohac, R.; Mitchell, A.J.; Boal, A.K.; Balskus, E.P. An n-nitrosating metalloenzyme constructs the pharmacophore of streptozotocin. Nature 2019, 566, 94–99. [Google Scholar] [CrossRef]
- Xiang, Y.Y.; Dong, H.; Wan, Y.; Li, J.; Yee, A.; Yang, B.B.; Lu, W.Y. Versican g3 domain regulates neurite growth and synaptic transmission of hippocampal neurons by activation of epidermal growth factor receptor. J. Biol. Chem. 2006, 281, 19358–19368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Evangelopoulos, M.E.; Weis, J.; Kruttgen, A. Mevastatin-induced neurite outgrowth of neuroblastoma cells via activation of egfr. J. Neurosci. Res. 2009, 87, 2138–2144. [Google Scholar] [CrossRef]
- Simons, M.; Trotter, J. Wrapping it up: The cell biology of myelination. Curr. Opin. Neurobiol. 2007, 17, 533–540. [Google Scholar] [CrossRef]
- Garratt, A.N.; Voiculescu, O.; Topilko, P.; Charnay, P.; Birchmeier, C. A dual role of erbb2 in myelination and in expansion of the schwann cell precursor pool. J. Cell Biol. 2000, 148, 1035–1046. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.K.; Lin, W.; Hauser, C.; Marchuk, Y.; Getman, D.; Lee, K.F. Rescue of the cardiac defect in erbb2 mutant mice reveals essential roles of erbb2 in peripheral nervous system development. Neuron 1999, 23, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Riethmacher, D.; Sonnenberg-Riethmacher, E.; Brinkmann, V.; Yamaai, T.; Lewin, G.R.; Birchmeier, C. Severe neuropathies in mice with targeted mutations in the erbb3 receptor. Nature 1997, 389, 725–730. [Google Scholar] [CrossRef]
- Werner, M.H.; Nanney, L.B.; Stoscheck, C.M.; King, L.E. Localization of immunoreactive epidermal growth factor receptors in human nervous system. J. Histochem. Cytochem. 1988, 36, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.P.; Wu, J.; Johansson, G.; Rizvi, T.A.; Miller, S.C.; Geiger, H.; Malik, P.; Li, W.; Mukouyama, Y.S.; Cancelas, J.A.; et al. Nf1 mutation expands an egfr-dependent peripheral nerve progenitor that confers neurofibroma tumorigenic potential. Cell Stem Cell 2008, 3, 658–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcez, R.C.; Teixeira, B.L.; Schmitt Sdos, S.; Alvarez-Silva, M.; Trentin, A.G. Epidermal growth factor (egf) promotes the in vitro differentiation of neural crest cells to neurons and melanocytes. Cell Mol. Neurobiol. 2009, 29, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Maklad, A.; Nicolai, J.R.; Bichsel, K.J.; Evenson, J.E.; Lee, T.C.; Threadgill, D.W.; Hansen, L.A. The egfr is required for proper innervation to the skin. J. Investig. Dermatol. 2009, 129, 690–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenspan, J.D. Nociceptors and the peripheral nervous system’s role in pain. J. Hand Ther. 1997, 10, 78–85. [Google Scholar] [CrossRef]
- Martin, L.J.; Smith, S.B.; Khoutorsky, A.; Magnussen, C.A.; Samoshkin, A.; Sorge, R.E.; Cho, C.; Yosefpour, N.; Sivaselvachandran, S.; Tohyama, S.; et al. Epiregulin and egfr interactions are involved in pain processing. J. Clin. Investig. 2017, 127, 3353–3366. [Google Scholar] [CrossRef] [Green Version]
- Saveri, P.; De Luca, M.; Nisi, V.; Pisciotta, C.; Romano, R.; Piscosquito, G.; Reilly, M.M.; Polke, J.M.; Cavallaro, T.; Fabrizi, G.M.; et al. Charcot-marie-tooth type 2b: A new phenotype associated with a novel rab7a mutation and inhibited egfr degradation. Cells 2020, 9, 1028. [Google Scholar] [CrossRef]
- Spinosa, M.R.; Progida, C.; De Luca, A.; Colucci, A.M.R.; Alifano, P.; Bucci, C. Functional characterization of rab7 mutant proteins associated with charcot-marie-tooth type 2b disease. J. Neurosci. 2008, 28, 1640–1648. [Google Scholar] [CrossRef]
- De Luca, A.; Progida, C.; Spinosa, M.R.; Alifano, P.; Bucci, C. Characterization of the rab7k157n mutant protein associated with charcot-marie-tooth type 2b. Biochem. Biophys. Res. Commun. 2008, 372, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Romano, R.; Rivellini, C.; De Luca, M.; Tonlorenzi, R.; Beli, R.; Manganelli, F.; Nolano, M.; Santoro, L.; Eskelinen, E.L.; Previtali, S.C.; et al. Alteration of the late endocytic pathway in charcot-marie-tooth type 2b disease. Cell Mol. Life Sci. 2020, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Ikeuchi, T.; Hatanaka, H. The neurotrophic action and signaling of epidermal growth factor. Prog. Neurobiol. 1997, 51, 19–37. [Google Scholar] [CrossRef]
- Ahmed, Z.; Read, M.L.; Berry, M.; Logan, A. Satellite glia not drg neurons constitutively activate egfr but egfr inactivation is not correlated with axon regeneration. Neurobiol. Dis 2010, 39, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W. The struggle to make cns axons regenerate: Why has it been so difficult? Neurochem. Res. 2020, 45, 144–158. [Google Scholar] [CrossRef] [Green Version]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef]
- Koprivica, V.; Cho, K.S.; Park, J.B.; Yiu, G.; Atwal, J.; Gore, B.; Kim, J.A.; Lin, E.; Tessier-Lavigne, M.; Chen, D.F.; et al. Egfr activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science 2005, 310, 106–110. [Google Scholar] [CrossRef]
- Erschbamer, M.; Pernold, K.; Olson, L. Inhibiting epidermal growth factor receptor improves structural, locomotor, sensory, and bladder recovery from experimental spinal cord injury. J. Neurosci. 2007, 27, 6428–6435. [Google Scholar] [CrossRef] [Green Version]
- Leinster, V.H.; Joy, M.T.; Vuononvirta, R.E.; Bolsover, S.R.; Anderson, P.N. Erbb1 epidermal growth factor receptor is a valid target for reducing the effects of multiple inhibitors of axonal regeneration. Exp. Neurol. 2013, 239, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, Z.; Jacques, S.J.; Berry, M.; Logan, A. Epidermal growth factor receptor inhibitors promote cns axon growth through off-target effects on glia. Neurobiol. Dis. 2009, 36, 142–150. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.W.; Li, J.J.; Wang, L.; Zhang, J.P.; Wu, J.J.; Mao, X.Q.; Shi, G.F.; Wang, Q.; Wang, F.; Zou, J. Epidermal growth factor receptor inhibitor ameliorates excessive astrogliosis and improves the regeneration microenvironment and functional recovery in adult rats following spinal cord injury. J. Neuroinflamm. 2014, 11, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Kitada, M.; Yamaguchi, M.; Dezawa, M.; Ide, C. Increase in bfgf-responsive neural progenitor population following contusion injury of the adult rodent spinal cord. Neurosci. Lett. 2006, 397, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Barnabe-Heider, F.; Frisen, J. Stem cells for spinal cord repair. Cell Stem Cell 2008, 3, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.M.; Xiao, Z.F.; Li, X.; Zhao, Y.N.; Wu, X.M.; Han, J.; Chen, B.; Li, J.Y.; Fan, C.X.; Xu, B.; et al. Vascular endothelial growth factor activates neural stem cells through epidermal growth factor receptor signal after spinal cord injury. CNS Neurosci. Ther. 2019, 25, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Ju, P.; Zhang, S.; Yeap, Y.; Feng, Z. Induction of neuronal phenotypes from ng2+ glial progenitors by inhibiting epidermal growth factor receptor in mouse spinal cord injury. Glia 2012, 60, 1801–1814. [Google Scholar] [CrossRef]
- Morrens, J.; Van Den Broeck, W.; Kempermann, G. Glial cells in adult neurogenesis. Glia 2012, 60, 159–174. [Google Scholar] [CrossRef]
- Zhang, S.; Ju, P.; Tjandra, E.; Yeap, Y.; Owlanj, H.; Feng, Z. Inhibition of epidermal growth factor receptor improves myelination and attenuates tissue damage of spinal cord injury. Cell Mol. Neurobiol. 2016, 36, 1169–1178. [Google Scholar] [CrossRef]
- Douglas, M.R.; Morrison, K.C.; Jacques, S.J.; Leadbeater, W.E.; Gonzalez, A.M.; Berry, M.; Logan, A.; Ahmed, Z. Off-target effects of epidermal growth factor receptor antagonists mediate retinal ganglion cell disinhibited axon growth. Brain 2009, 132, 3102–3121. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Neufeld, A.H. Activation of epidermal growth factor receptor causes astrocytes to form cribriform structures. Glia 2004, 46, 153–168. [Google Scholar] [CrossRef]
- Berry, M.; Ahmed, Z.; Douglas, M.R.; Logan, A. Epidermal growth factor receptor antagonists and cns axon regeneration: Mechanisms and controversies. Brain Res. Bull. 2011, 84, 289–299. [Google Scholar] [CrossRef]
- Smith, G.M.; Strunz, C. Growth factor and cytokine regulation of chondroitin sulfate proteoglycans by astrocytes. Glia 2005, 52, 209–218. [Google Scholar] [CrossRef]
- Ahmed, Z.; Suggate, E.L.; Brown, E.R.; Dent, R.G.; Armstrong, S.J.; Barrett, L.B.; Berry, M.; Logan, A. Schwann cell-derived factor-induced modulation of the ngr/p75ntr/egfr axis disinhibits axon growth through cns myelin in vivo and in vitro. Brain 2006, 129, 1517–1533. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.F.; Zhou, H.; Hu, C.Y.; Liang, Y.Q.; Hu, L.; Chen, D. The mechanisms of egfr in the regulation of axon regeneration. Cell Biochem. Funct. 2014, 32, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult cns by modulation of the pten/mtor pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the egfr in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Nawa, H. Erbb1-4-dependent egf/neuregulin signals and their cross talk in the central nervous system: Pathological implications in schizophrenia and parkinson’s disease. Front. Cell Neurosci. 2013, 7, 4. [Google Scholar] [CrossRef] [Green Version]
- Iwakura, Y.; Piao, Y.S.; Mizuno, M.; Takei, N.; Kakita, A.; Takahashi, H.; Nawa, H. Influences of dopaminergic lesion on epidermal growth factor-erbb signals in parkinson’s disease and its model: Neurotrophic implication in nigrostriatal neurons. J. Neurochem. 2005, 93, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Husnjak, K.; Dikic, I. Egfr trafficking: Parkin’ in a jam. Nat. Cell Biol. 2006, 8, 787–788. [Google Scholar] [CrossRef] [PubMed]
- Fallon, L.; Belanger, C.M.; Corera, A.T.; Kontogiannea, M.; Regan-Klapisz, E.; Moreau, F.; Voortman, J.; Haber, M.; Rouleau, G.; Thorarinsdottir, T.; et al. A regulated interaction with the uim protein eps15 implicates parkin in egf receptor trafficking and pi(3)k-akt signalling. Nat. Cell Biol. 2006, 8, 834–842. [Google Scholar] [CrossRef]
- Yang, Y.; Gehrke, S.; Haque, M.E.; Imai, Y.; Kosek, J.; Yang, L.; Beal, M.F.; Nishimura, I.; Wakamatsu, K.; Ito, S.; et al. Inactivation of drosophila dj-1 leads to impairments of oxidative stress response and phosphatidylinositol 3-kinase/akt signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 13670–13675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in lrrk2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Healy, D.G.; Falchi, M.; O’Sullivan, S.S.; Bonifati, V.; Durr, A.; Bressman, S.; Brice, A.; Aasly, J.; Zabetian, C.P.; Goldwurm, S.; et al. Phenotype, genotype, and worldwide genetic penetrance of lrrk2-associated parkinson’s disease: A case-control study. Lancet Neurol. 2008, 7, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Rivero-Rios, P.; Romo-Lozano, M.; Madero-Perez, J.; Thomas, A.P.; Biosa, A.; Greggio, E.; Hilfiker, S. The g2019s variant of leucine-rich repeat kinase 2 (lrrk2) alters endolysosomal trafficking by impairing the function of the gtpase rab8a. J. Biol. Chem. 2019, 294, 4738–4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Suaga, P.; Rivero-Rios, P.; Fdez, E.; Blanca Ramirez, M.; Ferrer, I.; Aiastui, A.; Lopez De Munain, A.; Hilfiker, S. Lrrk2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing rab7 activity. Hum Mol. Genet. 2014, 23, 6779–6796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panpalli Ates, M.; Karaman, Y.; Guntekin, S.; Ergun, M.A. Analysis of genetics and risk factors of alzheimer’s disease. Neuroscience 2016, 325, 124–131. [Google Scholar] [CrossRef]
- Barthet, G.; Georgakopoulos, A.; Robakis, N.K. Cellular mechanisms of gamma-secretase substrate selection, processing and toxicity. Prog. Neurobiol. 2012, 98, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Berthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing v717f beta-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Hsiao, K.; Chapman, P.; Nilsen, S.; Eckman, C.; Harigaya, Y.; Younkin, S.; Yang, F.; Cole, G. Correlative memory deficits, abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saura, C.A.; Chen, G.; Malkani, S.; Choi, S.Y.; Takahashi, R.H.; Zhang, D.; Gouras, G.K.; Kirkwood, A.; Morris, R.G.; Shen, J. Conditional inactivation of presenilin 1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J. Neurosci. 2005, 25, 6755–6764. [Google Scholar] [CrossRef] [Green Version]
- Wines-Samuelson, M.; Schulte, E.C.; Smith, M.J.; Aoki, C.; Liu, X.; Kelleher, R.J., 3rd; Shen, J. Characterization of age-dependent and progressive cortical neuronal degeneration in presenilin conditional mutant mice. PLoS ONE 2010, 5, e10195. [Google Scholar] [CrossRef] [PubMed]
- Beglopoulos, V.; Sun, X.; Saura, C.A.; Lemere, C.A.; Kim, R.D.; Shen, J. Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J. Biol. Chem. 2004, 279, 46907–46914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilig, E.A.; Xia, W.; Shen, J.; Kelleher, R.J., 3rd. A presenilin-1 mutation identified in familial alzheimer disease with cotton wool plaques causes a nearly complete loss of gamma-secretase activity. J. Biol. Chem. 2010, 285, 22350–22359. [Google Scholar] [CrossRef] [Green Version]
- Heilig, E.A.; Gutti, U.; Tai, T.; Shen, J.; Kelleher, R.J., 3rd. Trans-dominant negative effects of pathogenic psen1 mutations on gamma-secretase activity and abeta production. J. Neurosci. 2013, 33, 11606–11617. [Google Scholar] [CrossRef] [Green Version]
- Bruban, J.; Voloudakis, G.; Huang, Q.; Kajiwara, Y.; Al Rahim, M.; Yoon, Y.; Shioi, J.; Gama Sosa, M.A.; Shao, Z.; Georgakopoulos, A.; et al. Presenilin 1 is necessary for neuronal, but not glial, egfr expression and neuroprotection via gamma-secretase-independent transcriptional mechanisms. FASEB J. 2015, 29, 3702–3712. [Google Scholar] [CrossRef] [Green Version]
- Farkas, L.M.; Krieglstein, K. Heparin-binding epidermal growth factor-like growth factor (hb-egf) regulates survival of midbrain dopaminergic neurons. J. Neural Transm. 2002, 109, 267–277. [Google Scholar] [CrossRef]
- Jin, K.; Mao, X.O.; Del Rio Guerra, G.; Jin, L.; Greenberg, D.A. Heparin-binding epidermal growth factor-like growth factor stimulates cell proliferation in cerebral cortical cultures through phosphatidylinositol 3’-kinase and mitogen-activated protein kinase. J. Neurosci. Res. 2005, 81, 497–505. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Young, A.B. Excitatory amino acids and alzheimer’s disease. Neurobiol. Aging 1989, 10, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Pimplikar, S.W.; Nixon, R.A.; Robakis, N.K.; Shen, J.; Tsai, L.H. Amyloid-independent mechanisms in alzheimer’s disease pathogenesis. J. Neurosci. 2010, 30, 14946–14954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthet, G.; Dunys, J.; Shao, Z.; Xuan, Z.; Ren, Y.; Xu, J.; Arbez, N.; Mauger, G.; Bruban, J.; Georgakopoulos, A.; et al. Presenilin mediates neuroprotective functions of ephrinb and brain-derived neurotrophic factor and regulates ligand-induced internalization and metabolism of ephb2 and trkb receptors. Neurobiol. Aging 2013, 34, 499–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Nunan, J.; Small, D.H. Regulation of app cleavage by alpha-, beta- and gamma-secretases. FEBS Lett. 2000, 483, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Birecree, E.; Whetsell, W.O., Jr.; Stoscheck, C.; King, L.E., Jr.; Nanney, L.B. Immunoreactive epidermal growth factor receptors in neuritic plaques from patients with alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1988, 47, 549–560. [Google Scholar] [CrossRef]
- Chiang, H.C.; Wang, L.; Xie, Z.; Yau, A.; Zhong, Y. Pi3 kinase signaling is involved in abeta-induced memory loss in drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 7060–7065. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Chiang, H.C.; Wu, W.; Liang, B.; Xie, Z.; Yao, X.; Ma, W.; Du, S.; Zhong, Y. Epidermal growth factor receptor is a preferred target for treating amyloid-beta-induced memory loss. Proc. Natl. Acad. Sci. USA 2012, 109, 16743–16748. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liang, B.; Zhong, Y. Reduced egfr level potentially mediates the abeta42-induced neuronal loss in transgenic fruit fly and mouse. Protein Cell 2013, 4, 647–649. [Google Scholar] [CrossRef] [Green Version]
- Solca, F.; Dahl, G.; Zoephel, A.; Bader, G.; Sanderson, M.; Klein, C.; Kraemer, O.; Himmelsbach, F.; Haaksma, E.; Adolf, G.R. Target binding properties and cellular activity of afatinib (bibw 2992), an irreversible erbb family blocker. J. Pharmacol. Exp. Ther. 2012, 343, 342–350. [Google Scholar] [CrossRef]
- Chen, Y.J.; Hsu, C.C.; Shiao, Y.J.; Wang, H.T.; Lo, Y.L.; Lin, A.M.Y. Anti-inflammatory effect of afatinib (an egfr-tki) on ogd-induced neuroinflammation. Sci. Rep. 2019, 9, 2516. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Alcantara, S.; Ballabriga, J.; Olive, M.; Blanco, R.; Rivera, R.; Carmona, M.; Berruezo, M.; Pitarch, S.; Planas, A.M. Transforming growth factor-alpha (tgf-alpha) and epidermal growth factor-receptor (egf-r) immunoreactivity in normal and pathologic brain. Prog. Neurobiol. 1996, 49, 99–123. [Google Scholar] [CrossRef]
- Chio, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; Eurals, C. Prognostic factors in als: A critical review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offen, D.; Barhum, Y.; Melamed, E.; Embacher, N.; Schindler, C.; Ransmayr, G. Spinal cord mrna profile in patients with als: Comparison with transgenic mice expressing the human sod-1 mutant. J. Mol. Neurosci. 2009, 38, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Le Pichon, C.E.; Dominguez, S.L.; Solanoy, H.; Ngu, H.; Lewin-Koh, N.; Chen, M.; Eastham-Anderson, J.; Watts, R.; Scearce-Levie, K. Egfr inhibitor erlotinib delays disease progression but does not extend survival in the sod1 mouse model of als. PLoS ONE 2013, 8, e62342. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romano, R.; Bucci, C. Role of EGFR in the Nervous System. Cells 2020, 9, 1887. https://doi.org/10.3390/cells9081887
Romano R, Bucci C. Role of EGFR in the Nervous System. Cells. 2020; 9(8):1887. https://doi.org/10.3390/cells9081887
Chicago/Turabian StyleRomano, Roberta, and Cecilia Bucci. 2020. "Role of EGFR in the Nervous System" Cells 9, no. 8: 1887. https://doi.org/10.3390/cells9081887
APA StyleRomano, R., & Bucci, C. (2020). Role of EGFR in the Nervous System. Cells, 9(8), 1887. https://doi.org/10.3390/cells9081887