Aberrant Methylation of LINE-1 Transposable Elements: A Search for Cancer Biomarkers


Abstract
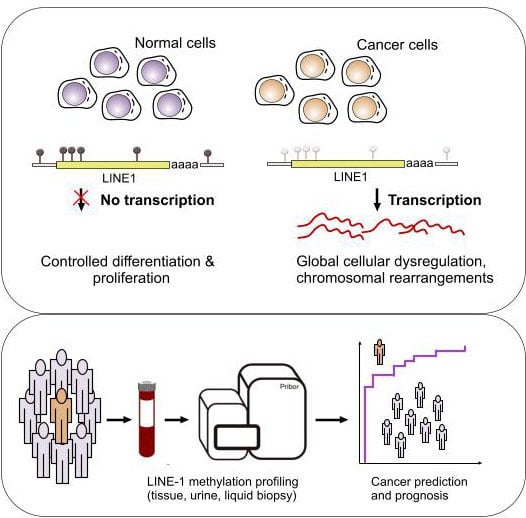
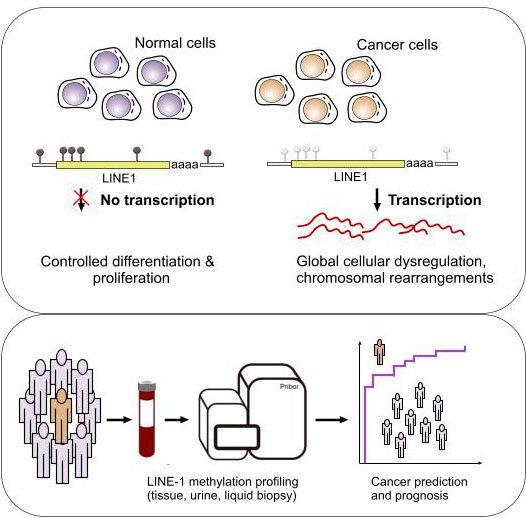
:
1. Introduction
2. LINE-1 Retrotransposons in the Human Genome
2.1. L1 Methylation in Cancer Diagnostics
2.2. L1 Methylation in Cancer Prognosis
3. Circulating DNA as a Source of Cancer Biomarkers
3.1. Methylation of Circulating L1 in the Healthy State
3.2. Perspectives of Using Aberrantly Methylated Circulating L1 for Cancer Diagnostics and Prognosis
4. Final Remarks
Funding
Conflicts of Interest
References
- Hotchkiss, R.D. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J. Biol. Chem. 1948, 175, 315–332. [Google Scholar] [PubMed]
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Deaton, A.M.; Bird, A. CpG islands and the regulation of transcription. Genes Dev. 2011, 25, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; He, F.; Hu, S.; Yu, J. On the nature of human housekeeping genes. Trends Genet. 2008, 24, 481–484. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Luo, H.; Krawczyk, M.; Wei, W.; Wang, W.; Wang, J.; Flagg, K.; Hou, J.; Zhang, H.; Yi, S.; et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 7414–7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef] [PubMed]
- Bouras, E.; Karakioulaki, M.; Bougioukas, K.I.; Aivaliotis, M.; Tzimagiorgis, G.; Chourdakis, M. Gene promoter methylation and cancer: An umbrella review. Gene 2019, 710, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.; Sidransky, D.; Brait, M. Tissue and Cell-Free DNA-Based Epigenomic Approaches for Cancer Detection. Clin. Chem. 2020, 66, 105–116. [Google Scholar] [CrossRef]
- Dunn, B.K. Hypomethylation: One side of a larger picture. Ann. N. Y. Acad. Sci. 2003, 983, 28–42. [Google Scholar] [CrossRef]
- Babaian, A.; Mager, D.L. Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 2016, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Burns, K.H. Transposable elements in cancer. Nat. Rev. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef]
- Deininger, P.L.; Batzer, M.A. Mammalian retroelements. Genome Res. 2002, 12, 1455–1465. [Google Scholar] [CrossRef] [Green Version]
- Kazazian, H.H., Jr. Genetics. L1 retrotransposons shape the mammalian genome. Science 2000, 289, 1152–1153. [Google Scholar] [CrossRef]
- Lavasanifar, A.; Sharp, C.N.; Korte, E.A.; Yin, T.; Hosseinnejad, K.; Jortani, S.A. Long interspersed nuclear element-1 mobilization as a target in cancer diagnostics, prognostics and therapeutics. Clin. Chim. Acta 2019, 493, 52–62. [Google Scholar] [CrossRef]
- Burns, K.H. Our Conflict with Transposable Elements and Its Implications for Human Disease. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 51–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luan, D.D.; Korman, M.H.; Jakubczak, J.L.; Eickbush, T.H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 1993, 72, 595–605. [Google Scholar] [CrossRef]
- Denli, A.M.; Narvaiza, I.; Kerman, B.E.; Pena, M.; Benner, C.; Marchetto, M.C.; Diedrich, J.K.; Aslanian, A.; Ma, J.; Moresco, J.J.; et al. Primate-specific ORF0 contributes to retrotransposon-mediated diversity. Cell 2015, 163, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodic, N. LINE-1 activity and regulation in cancer. Front. Biosci. (Landmark Ed.) 2018, 23, 1680–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, C.R.; Collier, P.; Macfarlane, C.; Malig, M.; Kidd, J.M.; Eichler, E.E.; Badge, R.M.; Moran, J.V. LINE-1 retrotransposition activity in human genomes. Cell 2010, 141, 1159–1170. [Google Scholar] [CrossRef] [Green Version]
- Brouha, B.; Schustak, J.; Badge, R.M.; Lutz-Prigge, S.; Farley, A.H.; Moran, J.V.; Kazazian, H.H. Hot L1s account for the bulk of retrotransposition in the human population. Proc. Natl. Acad. Sci. USA 2003, 100, 5280–5285. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G.P. Defining driver DNA methylation changes in human cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef] [Green Version]
- Cajuso, T.; Sulo, P.; Tanskanen, T.; Katainen, R.; Taira, A.; Hänninen, U.A.; Kondelin, J.; Forsström, L.; Välimäki, N.; Aavikko, M. Retrotransposon insertions can initiate colorectal cancer and are associated with poor survival. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Helman, E.; Lawrence, M.S.; Stewart, C.; Sougnez, C.; Getz, G.; Meyerson, M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014, 24, 1053–1063. [Google Scholar] [CrossRef] [Green Version]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K.W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52, 643–645. [Google Scholar]
- Shukla, R.; Upton, K.R.; Muñoz-Lopez, M.; Gerhardt, D.J.; Fisher, M.E.; Nguyen, T.; Brennan, P.M.; Baillie, J.K.; Collino, A.; Ghisletti, S. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell 2013, 153, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robberecht, C.; Voet, T.; Zamani Esteki, M.; Nowakowska, B.A.; Vermeesch, J.R. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res. 2013, 23, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Sharif, F.A. Frequency of balanced reciprocal translocations from couples with recurrent miscarriages correlates with the density of Alu and L1 repeat elements: Literature finding-based study. Middle East. J. Med. Genet. 2019, 8, 61. [Google Scholar] [CrossRef]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, Y.; Murata, A.; Watanabe, M.; Baba, H. Clinical implications of the LINE-1 methylation levels in patients with gastrointestinal cancer. Surg. Today 2014, 44, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Iskow, R.C.; McCabe, M.T.; Mills, R.E.; Torene, S.; Pittard, W.S.; Neuwald, A.F.; Van Meir, E.G.; Vertino, P.M.; Devine, S.E. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell 2010, 141, 1253–1261. [Google Scholar] [CrossRef] [Green Version]
- Tufarelli, C.; Badge, R.M. Retrotransposon-Driven Transcription and Cancer. In Human Retrotransposons in Health and Disease; Springer: Berlin/Heidelberg, Germany, 2017; pp. 259–273. [Google Scholar]
- Zheng, Y.; Joyce, B.T.; Liu, L.; Zhang, Z.; Kibbe, W.A.; Zhang, W.; Hou, L. Prediction of genome-wide DNA methylation in repetitive elements. Nucleic Acids Res. 2017, 45, 8697–8711. [Google Scholar] [CrossRef] [Green Version]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Ishak, C.A.; Classon, M.; De Carvalho, D.D. Deregulation of Retroelements as an Emerging Therapeutic Opportunity in Cancer. Trends Cancer 2018, 4, 583–597. [Google Scholar] [CrossRef]
- Tubio, J.M.C.; Li, Y.; Ju, Y.S.; Martincorena, I.; Cooke, S.L.; Tojo, M.; Gundem, G.; Pipinikas, C.P.; Zamora, J.; Raine, K.; et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science 2014, 345, 1251343. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Iskow, R.; Yang, L.; Gokcumen, O.; Haseley, P.; Luquette, L.J., III; Lohr, J.G.; Harris, C.C.; Ding, L.; Wilson, R.K.; et al. Landscape of somatic retrotransposition in human cancers. Science 2012, 337, 967–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, B.; Kimhi, S.; Howard, G.; Eden, A.; Lyko, F. Demethylation of a LINE-1 antisense promoter in the cMet locus impairs Met signalling through induction of illegitimate transcription. Oncogene 2010, 29, 5775–5784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, A.; Parle-McDermott, A. DNA methylation: A timeline of methods and applications. Front. Genet. 2011, 2, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurinomaru, T.; Kurita, R. Bisulfite-free approaches for DNA methylation profiling. Anal. Methods 2017, 9, 1537–1549. [Google Scholar] [CrossRef]
- Niya, M.H.K.; Roshan-zamir, N.; Mortazavi, E. DNA Methylation Tools and Strategies: Methods in a Review. Asian Pac. J. Cancer Biol. 2019, 4, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Laird, P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000, 28, e32-00. [Google Scholar] [CrossRef] [Green Version]
- Radpour, R.; Kohler, C.; Haghighi, M.; Fan, A.; Holzgreve, W.; Zhong, X. Methylation profiles of 22 candidate genes in breast cancer using high-throughput MALDI-TOF mass array. Oncogene 2009, 28, 2969–2978. [Google Scholar] [CrossRef] [Green Version]
- Rauch, T.; Li, H.; Wu, X.; Pfeifer, G.P. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006, 66, 7939–7947. [Google Scholar] [CrossRef] [Green Version]
- de Maat, M.F.; Umetani, N.; Sunami, E.; Turner, R.R.; Hoon, D.S. Assessment of methylation events during colorectal tumor progression by absolute quantitative analysis of methylated alleles. Mol. Cancer Res. 2007, 5, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barchitta, M.; Quattrocchi, A.; Maugeri, A.; Vinciguerra, M.; Agodi, A. LINE-1 hypomethylation in blood and tissue samples as an epigenetic marker for cancer risk: A systematic review and meta-analysis. PLoS ONE 2014, 9, e109478. [Google Scholar] [CrossRef] [PubMed]
- van Hoesel, A.Q.; van de Velde, C.J.; Kuppen, P.J.; Liefers, G.J.; Putter, H.; Sato, Y.; Elashoff, D.A.; Turner, R.R.; Shamonki, J.M.; de Kruijf, E.M.; et al. Hypomethylation of LINE-1 in primary tumor has poor prognosis in young breast cancer patients: A retrospective cohort study. Breast Cancer Res. Treat. 2012, 134, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Seo, A.N.; Jung, H.Y.; Gwak, J.M.; Jung, N.; Cho, N.Y.; Kang, G.H. Alu and LINE-1 hypomethylation is associated with HER2 enriched subtype of breast cancer. PLoS ONE 2014, 9, e100429. [Google Scholar] [CrossRef]
- Gao, X.D.; Qu, J.H.; Chang, X.J.; Lu, Y.Y.; Bai, W.L.; Wang, H.; Xu, Z.X.; An, L.J.; Wang, C.P.; Zeng, Z.; et al. Hypomethylation of long interspersed nuclear element-1 promoter is associated with poor outcomes for curative resected hepatocellular carcinoma. Liver Int. 2014, 34, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Utsunomiya, T.; Ikemoto, T.; Yamada, S.; Morine, Y.; Imura, S.; Arakawa, Y.; Takasu, C.; Ishikawa, D.; Imoto, I.; et al. Hypomethylation of long interspersed nuclear element-1 (LINE-1) is associated with poor prognosis via activation of c-MET in hepatocellular carcinoma. Ann. Surg. Oncol. 2014, 21 (Suppl. 4), S729–S735. [Google Scholar] [CrossRef]
- Anwar, S.L.; Hasemeier, B.; Schipper, E.; Vogel, A.; Kreipe, H.; Lehmann, U. LINE-1 hypomethylation in human hepatocellular carcinomas correlates with shorter overall survival and CIMP phenotype. PLoS ONE 2019, 14, e0216374. [Google Scholar] [CrossRef] [Green Version]
- Shigaki, H.; Baba, Y.; Watanabe, M.; Iwagami, S.; Miyake, K.; Ishimoto, T.; Iwatsuki, M.; Baba, H. LINE-1 hypomethylation in noncancerous esophageal mucosae is associated with smoking history. Ann. Surg. Oncol. 2012, 19, 4238–4243. [Google Scholar] [CrossRef]
- Iwagami, S.; Baba, Y.; Watanabe, M.; Shigaki, H.; Miyake, K.; Ishimoto, T.; Iwatsuki, M.; Sakamaki, K.; Ohashi, Y.; Baba, H. LINE-1 hypomethylation is associated with a poor prognosis among patients with curatively resected esophageal squamous cell carcinoma. Ann. Surg. 2013, 257, 449–455. [Google Scholar] [CrossRef]
- Zhu, J.; Ling, Y.; Xu, Y.; Lu, M.Z.; Liu, Y.P.; Zhang, C.S. Elevated expression of MDR1 associated with Line-1 hypomethylation in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 14392–14400. [Google Scholar]
- Antelo, M.; Balaguer, F.; Shia, J.; Shen, Y.; Hur, K.; Moreira, L.; Cuatrecasas, M.; Bujanda, L.; Giraldez, M.D.; Takahashi, M.; et al. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer. PLoS ONE 2012, 7, e45357. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Yamauchi, M.; Nishihara, R.; Lochhead, P.; Qian, Z.R.; Kuchiba, A.; Kim, S.A.; Mima, K.; Sukawa, Y.; Jung, S.; et al. Tumor LINE-1 methylation level and microsatellite instability in relation to colorectal cancer prognosis. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, M.; Kotake, M.; Bando, H.; Yamada, T.; Takemura, H.; Minamoto, T. Prognostic and predictive significance of long interspersed nucleotide element-1 methylation in advanced-stage colorectal cancer. BMC Cancer 2016, 16, 945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swets, M.; Zaalberg, A.; Boot, A.; van Wezel, T.; Frouws, M.A.; Bastiaannet, E.; Gelderblom, H.; van de Velde, C.J.; Kuppen, P.J. Tumor LINE-1 Methylation Level in Association with Survival of Patients with Stage II Colon Cancer. Int. J. Mol. Sci. 2016, 18, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupcinskas, J.; Steponaitiene, R.; Langner, C.; Smailyte, G.; Skieceviciene, J.; Kupcinskas, L.; Malfertheiner, P.; Link, A. LINE-1 hypomethylation is not a common event in preneoplastic stages of gastric carcinogenesis. Sci. Rep. 2017, 7, 4828. [Google Scholar] [CrossRef]
- Bae, J.M.; Shin, S.H.; Kwon, H.J.; Park, S.Y.; Kook, M.C.; Kim, Y.W.; Cho, N.Y.; Kim, N.; Kim, T.Y.; Kim, D.; et al. ALU and LINE-1 hypomethylations in multistep gastric carcinogenesis and their prognostic implications. Int. J. Cancer 2012, 131, 1323–1331. [Google Scholar] [CrossRef]
- Ikeda, K.; Shiraishi, K.; Eguchi, A.; Shibata, H.; Yoshimoto, K.; Mori, T.; Baba, Y.; Baba, H.; Suzuki, M. Long interspersed nucleotide element 1 hypomethylation is associated with poor prognosis of lung adenocarcinoma. Ann. Thorac. Surg. 2013, 96, 1790–1794. [Google Scholar] [CrossRef]
- Rhee, Y.Y.; Lee, T.H.; Song, Y.S.; Wen, X.; Kim, H.; Jheon, S.; Lee, C.T.; Kim, J.; Cho, N.Y.; Chung, J.H.; et al. Prognostic significance of promoter CpG island hypermethylation and repetitive DNA hypomethylation in stage I lung adenocarcinoma. Virchows Arch. 2015, 466, 675–683. [Google Scholar] [CrossRef]
- Imperatori, A.; Sahnane, N.; Rotolo, N.; Franzi, F.; Nardecchia, E.; Libera, L.; Romualdi, C.; Cattoni, M.; Sessa, F.; Dominioni, L.; et al. LINE-1 hypomethylation is associated to specific clinico-pathological features in Stage I non-small cell lung cancer. Lung Cancer 2017, 108, 83–89. [Google Scholar] [CrossRef]
- Furlan, C.; Polesel, J.; Barzan, L.; Franchin, G.; Sulfaro, S.; Romeo, S.; Colizzi, F.; Rizzo, A.; Baggio, V.; Giacomarra, V.; et al. Prognostic significance of LINE-1 hypomethylation in oropharyngeal squamous cell carcinoma. Clin. Epigenet. 2017, 9, 58. [Google Scholar] [CrossRef]
- Sunami, E.; de Maat, M.; Vu, A.; Turner, R.R.; Hoon, D.S. LINE-1 hypomethylation during primary colon cancer progression. PLoS ONE 2011, 6, e18884. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Jensen, C.D.; Marks, A.R.; Zhao, W.K.; Lee, J.K.; Doubeni, C.A.; Zauber, A.G.; de Boer, J.; Fireman, B.H.; Schottinger, J.E.; et al. Adenoma detection rate and risk of colorectal cancer and death. N. Engl. J. Med. 2014, 370, 1298–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.K.; Saunders, N.A.; Barbour, A.P.; Hill, M.M. Early diagnostic biomarkers for esophageal adenocarcinoma--the current state of play. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1185–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalitchagorn, K.; Shuangshoti, S.; Hourpai, N.; Kongruttanachok, N.; Tangkijvanich, P.; Thong-ngam, D.; Voravud, N.; Sriuranpong, V.; Mutirangura, A. Distinctive pattern of LINE-1 methylation level in normal tissues and the association with carcinogenesis. Oncogene 2004, 23, 8841–8846. [Google Scholar] [CrossRef] [Green Version]
- Schulz, W.A.; Elo, J.P.; Florl, A.R.; Pennanen, S.; Santourlidis, S.; Engers, R.; Buchardt, M.; Seifert, H.H.; Visakorpi, T. Genomewide DNA hypomethylation is associated with alterations on chromosome 8 in prostate carcinoma. Genes Chromosomes Cancer 2002, 35, 58–65. [Google Scholar] [CrossRef]
- Santourlidis, S.; Florl, A.; Ackermann, R.; Wirtz, H.C.; Schulz, W.A. High frequency of alterations in DNA methylation in adenocarcinoma of the prostate. Prostate 1999, 39, 166–174. [Google Scholar] [CrossRef]
- Pattamadilok, J.; Huapai, N.; Rattanatanyong, P.; Vasurattana, A.; Triratanachat, S.; Tresukosol, D.; Mutirangura, A. LINE-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 711–717. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef] [Green Version]
- van Bemmel, D.; Lenz, P.; Liao, L.M.; Baris, D.; Sternberg, L.R.; Warner, A.; Johnson, A.; Jones, M.; Kida, M.; Schwenn, M.; et al. Correlation of LINE-1 methylation levels in patient-matched buffy coat, serum, buccal cell, and bladder tumor tissue DNA samples. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1143–1148. [Google Scholar] [CrossRef] [Green Version]
- Ilie, M.; Hofman, P. Pros: Can tissue biopsy be replaced by liquid biopsy? Transl. Lung Cancer Res. 2016, 5, 420–423. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus tissue biopsy for detecting acquired resistance and tumor heterogeneity in gastrointestinal cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of ctDNA to support patient selection for early phase clinical trials: The TARGET study. Nat. Med. 2019, 25, 738–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rykova, E.Y.; Morozkin, E.S.; Ponomaryova, A.A.; Loseva, E.M.; Zaporozhchenko, I.A.; Cherdyntseva, N.V.; Vlassov, V.V.; Laktionov, P.P. Cell-free and cell-bound circulating nucleic acid complexes: Mechanisms of generation, concentration and content. Expert Opin. Biol. Ther. 2012, 12 (Suppl. 1), S141–S153. [Google Scholar] [CrossRef]
- Warton, K.; Samimi, G. Methylation of cell-free circulating DNA in the diagnosis of cancer. Front. Mol. Biosci. 2015, 2, 13. [Google Scholar] [CrossRef]
- Rykova, E.Y.; Ponomaryova, A.A.; Zaporozhchenko, I.A.; Vlassov, V.V.; Cherdyntseva, N.V.; Laktionov, P.P. Circulating DNA-based lung cancer diagnostics and follow-up: Looking for epigenetic markers. Transl. Cancer Res. 2018, 7, S153–S170. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef]
- Jorda, M.; Diez-Villanueva, A.; Mallona, I.; Martin, B.; Lois, S.; Barrera, V.; Esteller, M.; Vavouri, T.; Peinado, M.A. The epigenetic landscape of Alu repeats delineates the structural and functional genomic architecture of colon cancer cells. Genome Res. 2017, 27, 118–132. [Google Scholar] [CrossRef] [Green Version]
- Xiao-Jie, L.; Hui-Ying, X.; Qi, X.; Jiang, X.; Shi-Jie, M. LINE-1 in cancer: Multifaceted functions and potential clinical implications. Genet. Med. 2016, 18, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Farhat, F.S.; Houhou, W. Targeted therapies in non-small cell lung carcinoma: What have we achieved so far? Ther. Adv. Med. Oncol. 2013, 5, 249–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robles, A.I.; Harris, C.C. Integration of multiple “OMIC” biomarkers: A precision medicine strategy for lung cancer. Lung Cancer 2017, 107, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Maarri, O.; Walier, M.; Behne, F.; van Uum, J.; Singer, H.; Diaz-Lacava, A.; Nusgen, N.; Niemann, B.; Watzka, M.; Reinsberg, J.; et al. Methylation at global LINE-1 repeats in human blood are affected by gender but not by age or natural hormone cycles. PLoS ONE 2011, 6, e16252. [Google Scholar] [CrossRef] [PubMed]
- Bollati, V.; Schwartz, J.; Wright, R.; Litonjua, A.; Tarantini, L.; Suh, H.; Sparrow, D.; Vokonas, P.; Baccarelli, A. Decline in genomic DNA methylation through aging in a cohort of elderly subjects. Mech. Ageing Dev. 2009, 130, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Erichsen, L.; Beermann, A.; Arauzo-Bravo, M.J.; Hassan, M.; Dkhil, M.A.; Al-Quraishy, S.; Hafiz, T.A.; Fischer, J.C.; Santourlidis, S. Genome-wide hypomethylation of LINE-1 and Alu retroelements in cell-free DNA of blood is an epigenetic biomarker of human aging. Saudi J. Biol. Sci. 2018, 25, 1220–1226. [Google Scholar] [CrossRef]
- Terry, D.M.; Devine, S.E. Aberrantly High Levels of Somatic LINE-1 Expression and Retrotransposition in Human Neurological Disorders. Front. Genet. 2019, 10, 1244. [Google Scholar] [CrossRef] [Green Version]
- Ghanjati, F.; Beermann, A.; Hermanns, T.; Poyet, C.; Arauzo-Bravo, M.J.; Seifert, H.H.; Schmidtpeter, M.; Goering, W.; Sorg, R.; Wernet, P.; et al. Unreserved application of epigenetic methods to define differences of DNA methylation between urinary cellular and cell-free DNA. Cancer Biomark. 2014, 14, 295–302. [Google Scholar] [CrossRef]
- Wolff, E.M.; Byun, H.M.; Han, H.F.; Sharma, S.; Nichols, P.W.; Siegmund, K.D.; Yang, A.S.; Jones, P.A.; Liang, G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010, 6, e1000917. [Google Scholar] [CrossRef]
- Zhong, H.H.; Hu, S.J.; Yu, B.; Jiang, S.S.; Zhang, J.; Luo, D.; Yang, M.W.; Su, W.Y.; Shao, Y.L.; Deng, H.L.; et al. Apoptosis in the aging liver. Oncotarget 2017, 8, 102640–102652. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wangsri, S.; Subbalekha, K.; Kitkumthorn, N.; Mutirangura, A. Patterns and possible roles of LINE-1 methylation changes in smoke-exposed epithelia. PLoS ONE 2012, 7, e45292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Killian, J.K.; Yang, M.; Walker, R.L.; Hong, J.A.; Zhang, M.; Davis, S.; Zhang, Y.; Hussain, M.; Xi, S.; et al. Epigenomic alterations and gene expression profiles in respiratory epithelia exposed to cigarette smoke condensate. Oncogene 2010, 29, 3650–3664. [Google Scholar] [CrossRef]
- Zeidler, R.; Albermann, K.; Lang, S. Nicotine and apoptosis. Apoptosis 2007, 12, 1927–1943. [Google Scholar] [CrossRef] [PubMed]
- Pox, C.P.; Altenhofen, L.; Brenner, H.; Theilmeier, A.; Von Stillfried, D.; Schmiegel, W. Efficacy of a nationwide screening colonoscopy program for colorectal cancer. Gastroenterology 2012, 142, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.M.; Lee, Y.C.; Tu, C.H.; Chen, C.C.; Tseng, P.H.; Liang, J.T.; Shun, C.T.; Lin, J.T.; Wu, M.S. Association between early stage colon neoplasms and false-negative results from the fecal immunochemical test. Clin. Gastroenterol. Hepatol. 2013, 11, 832–838. [Google Scholar] [CrossRef]
- Hirai, H.W.; Tsoi, K.K.; Chan, J.Y.; Wong, S.H.; Ching, J.Y.; Wong, M.C.; Wu, J.C.; Chan, F.K.; Sung, J.J.; Ng, S.C. Systematic review with meta-analysis: Faecal occult blood tests show lower colorectal cancer detection rates in the proximal colon in colonoscopy-verified diagnostic studies. Aliment. Pharmacol. Ther. 2016, 43, 755–764. [Google Scholar] [CrossRef]
- Smith, R.A.; Andrews, K.; Brooks, D.; DeSantis, C.E.; Fedewa, S.A.; Lortet-Tieulent, J.; Manassaram-Baptiste, D.; Brawley, O.W.; Wender, R.C. Cancer screening in the United States, 2016: A review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J. Clin. 2016, 66, 96–114. [Google Scholar] [CrossRef]
- von Wagner, C.; Baio, G.; Raine, R.; Snowball, J.; Morris, S.; Atkin, W.; Obichere, A.; Handley, G.; Logan, R.F.; Rainbow, S.; et al. Inequalities in participation in an organized national colorectal cancer screening programme: Results from the first 2.6 million invitations in England. Int. J. Epidemiol. 2011, 40, 712–718. [Google Scholar] [CrossRef] [Green Version]
- Taber, J.M.; Aspinwall, L.G.; Heichman, K.A.; Kinney, A.Y. Preferences for blood-based colon cancer screening differ by race/ethnicity. Am. J. Health Behav. 2014, 38, 351–361. [Google Scholar] [CrossRef]
- Nagai, Y.; Sunami, E.; Yamamoto, Y.; Hata, K.; Okada, S.; Murono, K.; Yasuda, K.; Otani, K.; Nishikawa, T.; Tanaka, T.; et al. LINE-1 hypomethylation status of circulating cell-free DNA in plasma as a biomarker for colorectal cancer. Oncotarget 2017, 8, 11906–11916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunoki, A.; Kawakami, K.; Kotake, M.; Kaneko, M.; Kitamura, H.; Ooi, A.; Watanabe, G.; Minamoto, T. LINE-1 methylation shows little intra-patient heterogeneity in primary and synchronous metastatic colorectal cancer. BMC Cancer 2012, 12, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, Y.Y.; Kim, M.J.; Bae, J.M.; Koh, J.M.; Cho, N.Y.; Juhnn, Y.S.; Kim, D.; Kang, G.H. Clinical outcomes of patients with microsatellite-unstable colorectal carcinomas depend on L1 methylation level. Ann. Surg. Oncol. 2012, 19, 3441–3448. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, I.V.; Kapitskaya, K.Y.; Rykova, E.Y.; Ponomaryova, A.A.; Cherdyntseva, N.V.; Vlassov, V.V.; Laktionov, P.P.; Azhikina, T.L. Hypomethylation of human-specific family of LINE-1 retrotransposons in circulating DNA of lung cancer patients. Lung Cancer 2016, 99, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Ponomaryova, A.; Rykova, E.; Cherdyntseva, N.; Bondar, A.; Dobrodeev, A.; Zavyalov, A.; Tuzikov, S.; Bryzgalov, L.; Merkulova, T.; Vlassov, V. Epigenetic probes for lung cancer monitoring: Line-1 methylation pattern in blood-circulating DNA. Russ. J. Genet. Appl. Res. 2016, 6, 99–104. [Google Scholar] [CrossRef]
- Ponomaryova, A.A.; Cherdyntseva, N.V.; Bondar, A.A.; Dobrodeev, A.Y.; Zavyalov, A.A.; Tuzikov, S.A.; Vlassov, V.V.; Choinzonov, E.L.; Laktionov, P.P.; Rykova, E.Y. Dynamics of LINE-1 Retrotransposon Methylation Levels in Circulating DNA from Lung Cancer Patients Undergoing Antitumor Therapy. Mol. Biol. (Mosk) 2017, 51, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Ponomaryova, A.A.; Rykova, E.Y.; Azhikina, T.L.; Bondar, A.A.; Cheremisina, O.V.; Rodionov, E.O.; Boyarko, V.V.; Laktionov, P.P.; Cherdyntseva, N.V. Long interspersed nuclear element-1 methylation status in the circulating DNA from blood of patients with malignant and chronic inflammatory lung diseases. Eur. J. Cancer Prev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wedge, E.; Hansen, J.W.; Garde, C.; Asmar, F.; Tholstrup, D.; Kristensen, S.S.; Munch-Petersen, H.D.; Ralfkiaer, E.; Brown, P.; Gronbaek, K.; et al. Global hypomethylation is an independent prognostic factor in diffuse large B cell lymphoma. Am. J. Hematol. 2017, 92, 689–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshimoto, S.; Kuo, C.T.; Chong, K.K.; Takeshima, T.L.; Takei, Y.; Li, M.W.; Huang, S.K.; Sim, M.S.; Morton, D.L.; Hoon, D.S. AIM1 and LINE-1 epigenetic aberrations in tumor and serum relate to melanoma progression and disease outcome. J. Investig. Dermatol. 2012, 132, 1689–1697. [Google Scholar] [CrossRef] [Green Version]
- Gold, B.; Cankovic, M.; Furtado, L.V.; Meier, F.; Gocke, C.D. Do circulating tumor cells, exosomes, and circulating tumor nucleic acids have clinical utility? A report of the association for molecular pathology. J. Mol. Diagn. 2015, 17, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Pantel, K.; Alix-Panabieres, C. Tumour microenvironment: Informing on minimal residual disease in solid tumours. Nat. Rev. Clin. Oncol. 2017, 14, 325–326. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.-F.; de Castro Abreu, A.L.; Chihara, Y.; Tsai, Y.; Andreu-Vieyra, C.; Daneshmand, S.; Skinner, E.C.; Jones, P.A.; Siegmund, K.D.; Liang, G. A panel of three markers hyper-and hypomethylated in urine sediments accurately predicts bladder cancer recurrence. Clin. Cancer Res. 2014, 20, 1978–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivioja, T.; Vähärautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2012, 9, 72–74. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Methods | Diagnostic Value (*) | Prognostic Value | Ref. |
|---|---|---|---|---|
| Breast cancer | Pyrosequencing | Normal tissues—92%, IBC tissues—86% | L1 hypomethylation was significantly associated with decreased OS (HR 2.19, 95% CI 1.17–4.09), decreased DFS (HR 2.05, 95% CI 1.14–3.67), and increased DR (HR 2.83, 95 % CI 1.53–5.21) in younger (≤55 years) but not in older patients (>55 years) | [54] |
| Pyrosequencing | Normal tissues—64%, IBC tissues—61% | - | [55] | |
| Hepatocellular carcinoma | Bisulfite-specific PCR and DNA sequencing analysis | Normal tissues—60%, tumor tissues—34% | Patients with L1 hypomethylation had decreased median postresection TFS (22 months [95% CI: 13.3–30.7]) and OS (35 months [95% CI: 24.0–46.1]) compared to those with L1 hypermethylation (40 and 60 months, respectively) | [56] |
| Pyrosequencing | Normal tissues—68%, tumor tissues—48% | - | [57] | |
| Normal tissues—57%, tumor tissues—46% | - | [58] | ||
| Esophageal cell carcinoma | Pyrosequencing | Normal tissues—82%, tumor tissues—64% | - | [59] |
| Normal tissues—79%, tumor tissues—63% | L1 methylation was significantly associated with DFS (univariate HR 2.32, 95% CI 1.38–3.84, methylation level [quartile] < 56%; multivariate HR 1.81, 95% CI 1.06–3.05) and CSS (univariate HR 2.21, 95% CI 1.33–3.60; multivariate HR 1.87, 95% CI 1.12–3.08) | [60] | ||
| Quantitative real-time MSP | Normal tissues—90%, tumor tissues—78% | Cumulative survival was significantly shorter for ESCC patients with L1 methylation level ≤ 78% than for those with > 78% (34 vs. 43 months) | [61] | |
| Colorectal cancer | Pyrosequencing | Normal tissues—77%, tumor tissues—57% | OS was significantly longer in patients with L1 methylation level ≥ 65% | [62] |
| MSP-PCR, pyrosequencing after assay validation | - | L1 hypomethylation was significantly associated with higher CRC-specific mortality (for 10% decrease in L1 methylation: HR 2.45, 95% CI 1.64–3.66) | [63] | |
| MethyLight assay | - | PFS, OS, and 5-year OPS were significantly shorter in patients with low L1 methylation than in those with high L1 methylation (HR 1.00 vs. HR 2.74 [95% CI 1.19–6.29]) | [64] | |
| Quantitative PCR | - | L1 hypomethylation was significantly associated with lower OS (HR 1.68, 95% CI 1.03–2.75); the association was stronger in patients > 65 years (HR 2.00, 95% CI 1.13–3.52) | [65] | |
| Gastric and colon cancers | Pyrosequencing | Colon: normal tissues—67%, tumor tissues—61% Gastric: normal tissues—66%, tumor tissues—62% | - | [66] |
| Gastric cancer | Pyrosequencing | Chronic gastritis—62%, cancer—52% | L1 hypomethylation level (<51%) was significantly associated with shorter DFS and OS | [67] |
| Lung cancer | Pyrosequencing | Normal tissues—74%, ADC tissues—67% | Patients with low L1 methylation levels (19–69%) had significantly higher recurrence rates and shorter DFS compared to those with high methylation levels (74–81%) | [68] |
| - | L1 hypomethylation (<52%) was significantly associated with lower survival rates in patients with ADC stage I | [69] | ||
| Bisulfite-PCR, pyrosequencing | Normal tissues—70%, ADC tissues—63%, SCC tissues—38% | L1 hypomethylation (≤58%) was independently associated with poor prognosis (p = 0.025) | [70] | |
| Oropharyngeal squamous cell carcinoma | Quantitative MSP-PCR | - | L1 hypomethylation (<50% vs. ≥70%) was significantly associated with higher risk of early disease relapse (OR = 3.51; 95% CI 1.03–12.00) | [71] |
| Tumor Location | Clinical Samples | Method | Results | Ref. |
|---|---|---|---|---|
| Colorectal (patients before treatment) | Plasma | AQAMA qPCR | Significant decrease of L1 MI in cancer patients compared with healthy subjects Association of L1MI with disease progression (advanced stage and distant metastasis) | [112] |
| Lung (patients before treatment) | Cell surface-bound fraction of blood | MIRA | Significant decrease of L1 MI in cancer patients compared with healthy subjects Hypomethylation of L1 promoters in cancer patients is more pronounced for the L1 human-specific (L1Hs) family | [115] |
| Lung (patients before treatment and after antitumor therapy) | Cell surface-bound fraction of blood | qMSP PCR | Association of L1 MI with tumor histological type Dynamic changes in L1 MI of csb-cirDNA during the follow-up period | [117] |
| Lung (patients before treatment) | Cell surface-bound fraction of blood, plasma | qMSP PCR | Significant decrease of L1 MI in csb-cirDNA in cancer patients compared with healthy subjects | [116] |
| Lung (patients before treatment) | Cell surface-bound fraction of blood, plasma | qMSP PCR | Decrease of L1 MI in cancer patients compared with the joint control group (healthy subjects + patients with bronchitis + COPD patients) and with COPD patients | [118] |
| Diffuse large B cell lymphoma (patients before treatment) | Plasma | Pyrosequencing | Association of L1 hypomethylation with poor overall survival | [119] |
| Melanoma (patients before treatment) | Serum | AQAMA qPCR | Decrease of L1 methylation during disease progression (advanced stage) | [120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomaryova, A.A.; Rykova, E.Y.; Gervas, P.A.; Cherdyntseva, N.V.; Mamedov, I.Z.; Azhikina, T.L. Aberrant Methylation of LINE-1 Transposable Elements: A Search for Cancer Biomarkers. Cells 2020, 9, 2017. https://doi.org/10.3390/cells9092017
Ponomaryova AA, Rykova EY, Gervas PA, Cherdyntseva NV, Mamedov IZ, Azhikina TL. Aberrant Methylation of LINE-1 Transposable Elements: A Search for Cancer Biomarkers. Cells. 2020; 9(9):2017. https://doi.org/10.3390/cells9092017
Chicago/Turabian StylePonomaryova, Anastasia A., Elena Y. Rykova, Polina A. Gervas, Nadezhda V. Cherdyntseva, Ilgar Z. Mamedov, and Tatyana L. Azhikina. 2020. "Aberrant Methylation of LINE-1 Transposable Elements: A Search for Cancer Biomarkers" Cells 9, no. 9: 2017. https://doi.org/10.3390/cells9092017