Genome-Wide Signatures of Selection Detection in Three South China Indigenous Pigs

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and methods
2.1. Ethics Approval
2.2. DNA Sample Collection
2.3. Single-Nucleotide Polymorphism (SNP) Genotyping and Data Quality Control
2.4. Principal Component Analysis
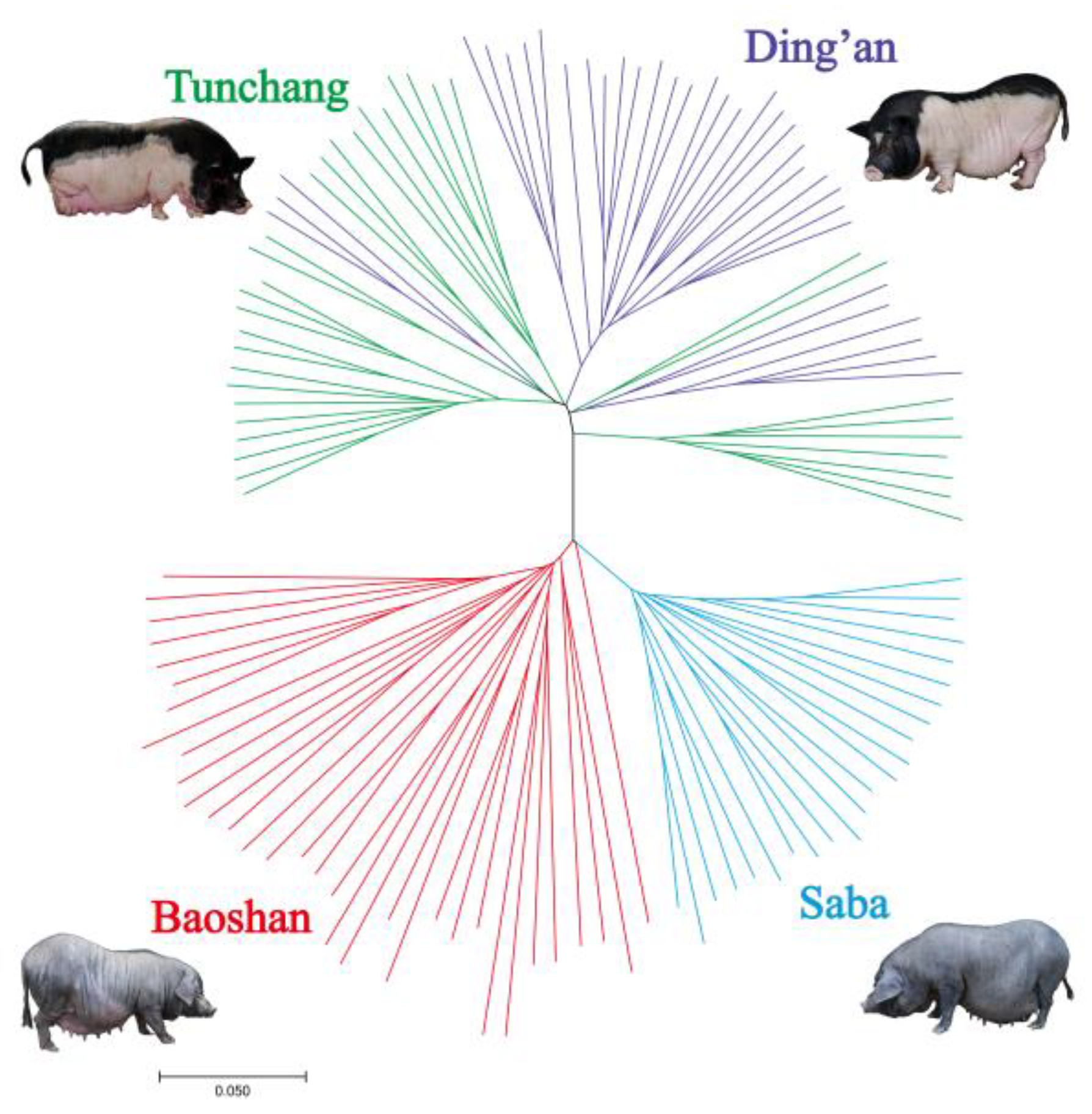
2.5. Phylogenetic Tree
2.6. Identification of Signatures of Selection
2.7. Genome Annotation and Quantitative Trait Loci (QTL) Overlapping with Potential Signatures of Selection
2.8. Gene Ontology (GO) Terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis
3. Results
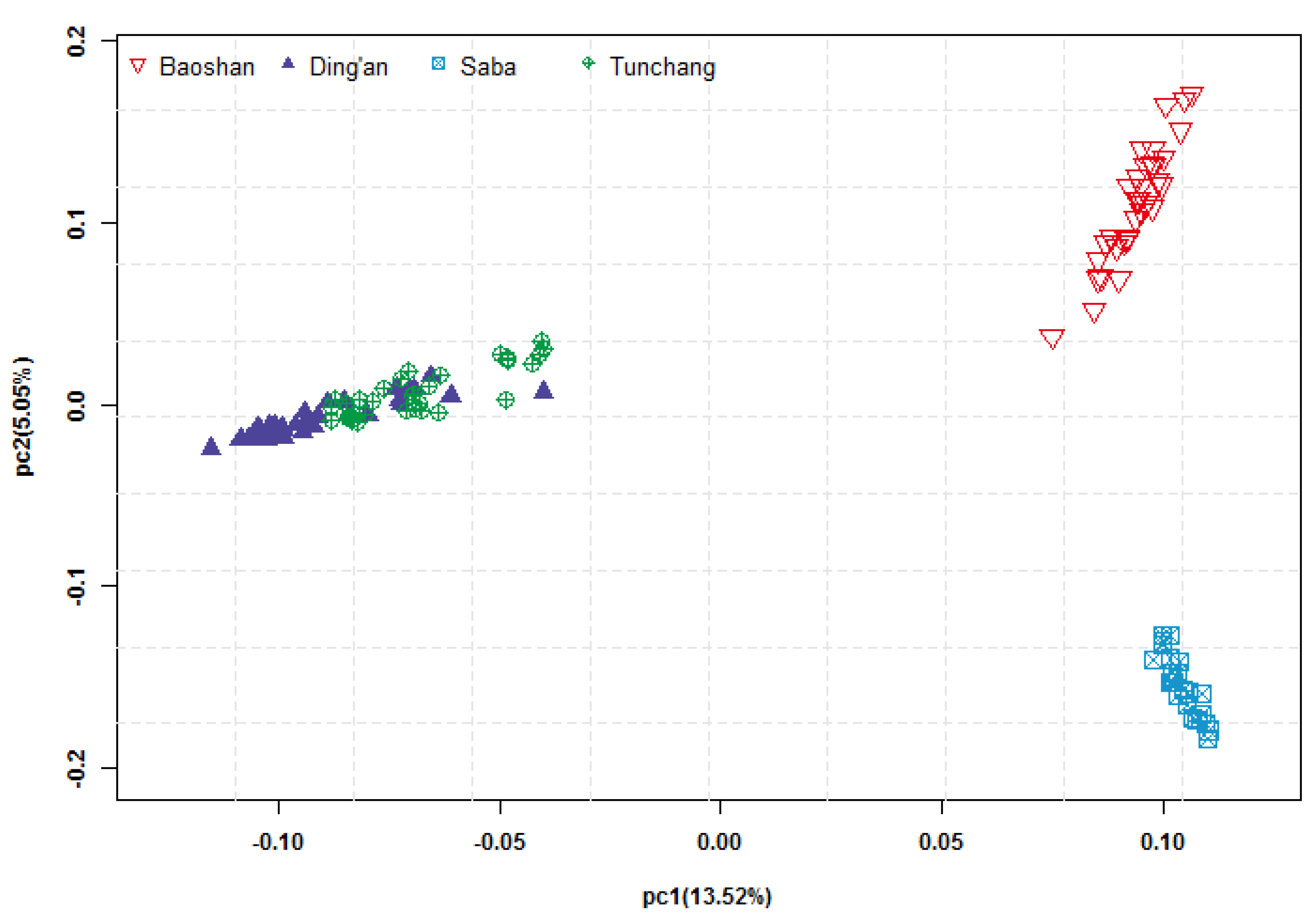
3.1. Genotypes and Population Structure
3.2. Identification of Signatures of Selection
3.3. Genome Annotation and QTL Overlapping with Potential Signatures of Selection
3.4. GO Terms and KEGG Pathway Enrichment Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bökönyi, S. History of domestic mammals in Central and Eastern Europe. Euromoney 1974, 144, 94–98. [Google Scholar]
- Tian, D.; Araki, H.; Stahl, E.; Bergelson, J.; Kreitman, M. Signature of balancing selection in Arabidopsis. Proc. Natl. Acad. Sci. USA 2002, 99, 11525–11530. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, B.; Tang, K.; Lee, E.J.; Chong, S.S.; Lee, C.G. A functional polymorphism within the MRP1 gene locus identified through its genomic signature of positive selection. Hum. Mol. Genet. 2005, 14, 2075–2087. [Google Scholar] [CrossRef]
- Grossman, S.R.; Shylakhter, I.; Karlsson, E.K.; Byrne, E.H.; Morales, S.; Frieden, G.; Hostetter, E.; Angelino, E.; Garber, M.; Zuk, O. A Composite of Multiple Signals Distinguishes Causal Variants in Regions of Positive Selection. Science 2010, 327, 883–886. [Google Scholar] [CrossRef] [PubMed]
- Zeng, K.; Shi, S.; Wu, C. Compound Tests for the Detection of Hitchhiking Under Positive Selection. Mol. Biol. Evol. 2007, 24, 1898–1908. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar] [CrossRef]
- Lewontin, R.C.; Krakauer, J. Distribution of gene frequency as a test of the theory of the selective neutrality of polymorphisms. Genetics 1973, 74, 175–195. [Google Scholar] [PubMed]
- Wright, S. The genetical structure of populations. Ann. Eugen. 1949, 15, 323–354. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Akey, J.M.; Zhang, G.; Zhang, K.; Jin, L.; Shriver, M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Gianola, D.; Simianer, H.; Qanbari, S. A two-step method for detecting selection signatures using genetic markers. Genet. Res. 2010, 92, 141–155. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Wang, Z.; Pan, Y.; Wang, Q.; Xu, N.; Wang, Z. Detection of selection signatures of population-specific genomic regions selected during domestication process in Jinhua pigs. Anim. Genet. 2016, 47, 672–681. [Google Scholar] [CrossRef]
- Moon, S.; Kim, T.H.; Lee, K.T.; Kwak, W.; Lee, T.; Lee, S.W.; Kim, M.J.; Cho, K.; Kim, N.; Chung, W.H.; et al. A genome-wide scan for signatures of directional selection in domesticated pigs. BMC Genom. 2015, 16, 130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, Q.; Yang, Y.; Yang, H.; He, P.; Zhang, Z.; Chen, Z.; Liao, R.; Tu, Y.; Zhang, X.; et al. A genome-wide scan for selection signatures in Yorkshire and Landrace pigs based on sequencing data. Anim. Genet. 2014, 45, 808–816. [Google Scholar] [CrossRef]
- Chen, M.; Wang, J.; Wang, Y.; Wu, Y.; Fu, J.; Liu, J.F. Genome-wide detection of selection signatures in Chinese indigenous Laiwu pigs revealed candidate genes regulating fat deposition in muscle. BMC Genet. 2018, 19, 31. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, W.; Yang, B.; Zhang, Z.; Ai, H.; Ren, J.; Huang, L. Signatures of Selection and Interspecies Introgression in the Genome of Chinese Domestic Pigs. Genome Biol. Evol. 2017, 9, 2592–2603. [Google Scholar] [CrossRef] [PubMed]
- Rubin, C.J.; Megens, H.J.; Martinez Barrio, A.; Maqbool, K.; Sayyab, S.; Schwochow, D.; Wang, C.; Carlborg, O.; Jern, P.; Jorgensen, C.B.; et al. Strong signatures of selection in the domestic pig genome. Proc. Natl. Acad. Sci. USA 2012, 109, 19529–19536. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Huang, L.; Ren, J. Genetic diversity, linkage disequilibrium and selection signatures in Chinese and Western pigs revealed by genome-wide SNP markers. PLoS ONE 2013, 8, e56001. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Z.; Tian, S.L.; Yeung, C.K.L.; Meng, X.H.; Tang, Q.Z.; Niu, L.L.; Wang, X.; Jin, L.; Ma, J.D.; Long, K.; et al. Whole-genome sequencing of Berkshire (European native pig) provides insights into its origin and domestication. Sci. Rep. 2014, 4, 4678. [Google Scholar] [CrossRef]
- Yang, S.; Li, X.; Li, K.; Fan, B.; Tang, Z. A genome-wide scan for signatures of selection in Chinese indigenous and commercial pig breeds. BMC Genet. 2014, 15, 7. [Google Scholar] [CrossRef] [PubMed]
- Ai, H.; Fang, X.; Yang, B.; Huang, Z.; Chen, H.; Mao, L.; Zhang, F.; Zhang, L.; Cui, L.; He, W.; et al. Adaptation and possible ancient interspecies introgression in pigs identified by whole-genome sequencing. Nat. Genet. 2015, 47, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wei, J.; Zhang, Q.; Chen, L.; Wang, J.; Liu, J.; Ding, X. A genome scan for selection signatures in pigs. PLoS ONE 2015, 10, e0116850. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zou, H.; Chen, L.; Long, X.; Lan, J.; Liu, W.; Ma, L.; Wang, C.; Xu, X.; Ren, L. Convergent and divergent genetic changes in the genome of Chinese and European pigs. Sci. Rep. 2017, 7, 8662. [Google Scholar] [CrossRef]
- China National Commission of Animal Genetic Resources. Animal Genetic Resources in China: Pigs; China Agriculture Press: Beijing, China, 2011. [Google Scholar]
- Li, B. Conservation Status and Countermeasure Research of Hainan Pig, Wuzhishan Pig, Duntou Pig. Anhui Agric. Sci. 2013, 41, 11704–11705, 11715. [Google Scholar]
- Ramos, A.M.; Crooijmans, R.P.M.A.; Affara, N.A.; Amaral, A.J.; Archibald, A.L.; Beever, J.E.; Bendixen, C.; Churcher, C.; Clark, R.; Dehais, P.; et al. Design of a High Density SNP Genotyping Assay in the Pig Using SNPs Identified and Characterized by Next Generation Sequencing Technology. PLoS ONE 2009, 4, e6524. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Scheet, P.; Stephens, M. A Fast and Flexible Statistical Model for Large-Scale Population Genotype Data: Applications to Inferring Missing Genotypes and Haplotypic Phase. Am. J. Hum. Genet. 2006, 78, 629–644. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Coop, G.; Novembre, J.; Kudaravalli, S.; Li, J.Z.; Absher, D.; Srinivasan, B.S.; Barsh, G.S.; Myers, R.M.; Feldman, M.W. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009, 19, 826–837. [Google Scholar] [CrossRef]
- Rousset, F. GENEPOP’007: A complete re-implementation of the genepop software for Windows and Linux. Mol. Ecol. Res. 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Ma, Y.; Ding, X.; Qanbari, S.; Weigend, S.; Zhang, Q.; Simianer, H. Properties of different selection signature statistics and a new strategy for combining them. Heredity 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.L.; Fang, C.; Liu, W.J. Identification on novel locus of dairy traits of Kazakh horse in Xinjiang. Gene 2018, 677, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2018, 47, D701–D710. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.D.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Wilkinson, S.; Lu, Z.H.; Megens, H.J.; Archibald, A.L.; Haley, C.; Jackson, I.J.; Groenen, M.A.; Crooijmans, R.P.; Ogden, R.; Wiener, P. Signatures of diversifying selection in European pig breeds. PLoS Genet. 2013, 9, e1003453. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Hamamoto, A.; Takahashi, A.; Saito, Y. Dimerization of melanocortin receptor 1 (MC1R) and MC5R creates a ligand-dependent signal modulation: Potential participation in physiological color change in the flounder. Gen. Comp. Endocrinol. 2016, 230, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kováčik, A.; Bulla, J.; Trakovická, A.; Žitný, J.; Rafayová, A. The effect of the porcine melanocortin-5 receptor (MC5R) gene associated with feed intake, carcass and physico-chemical characteristics. J. Microbiol. Biotechnol. Food Sci. 2012, 1, 498–506. [Google Scholar]
- Beissinger, T.; Kruppa, J.; Cavero, D.; Ha, N.T.; Erbe, M.; Simianer, H. A Simple Test Identifies Selection on Complex Traits. Genetics 2018, 209, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Pavlidis, P.; Jensen, J.D.; Stephan, W.; Stamatakis, A. A critical assessment of storytelling: Gene ontology categories and the importance of validating genomic scans. Mol. Boil. Evol. 2012, 29, 3237–3248. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
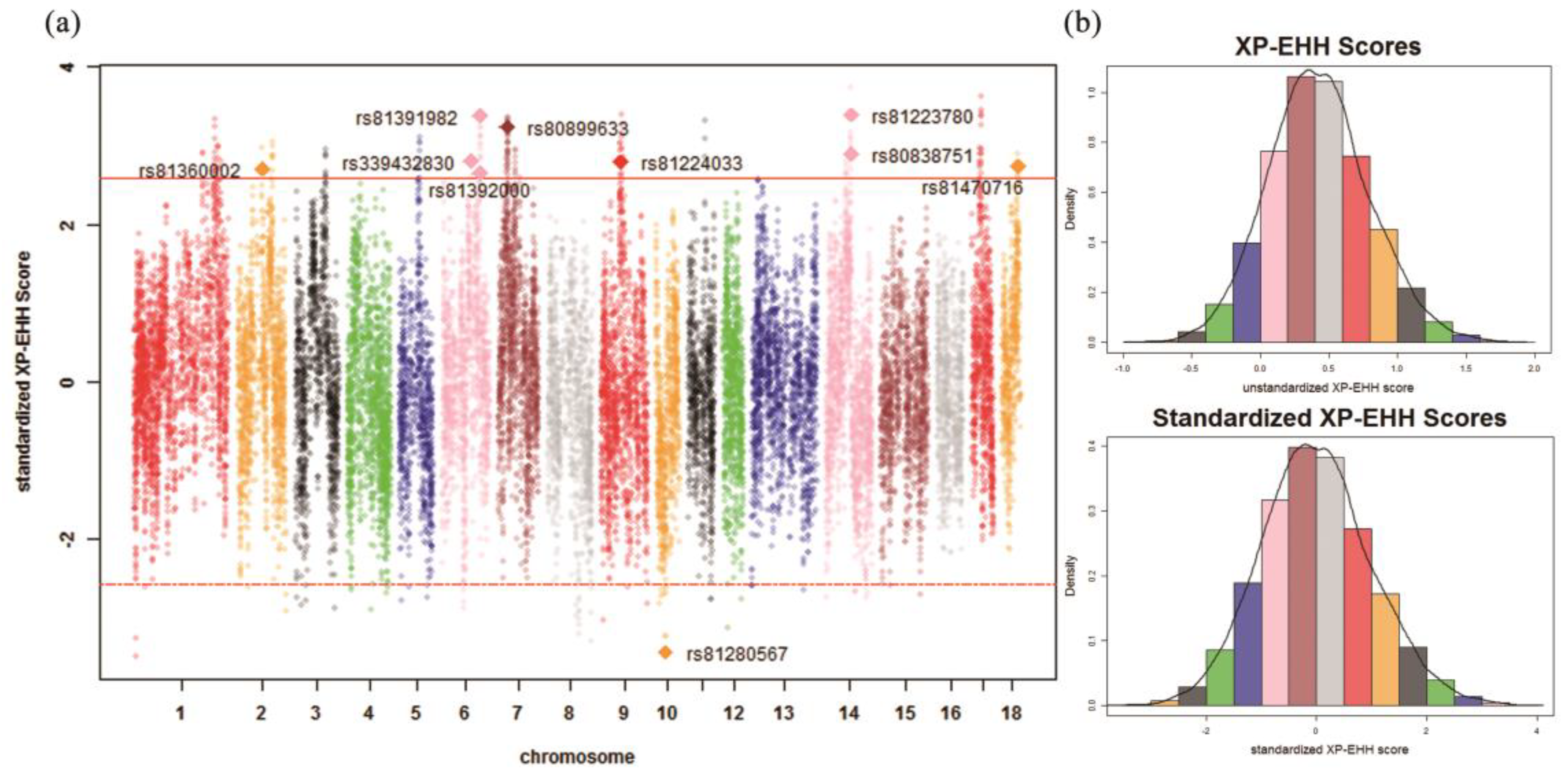
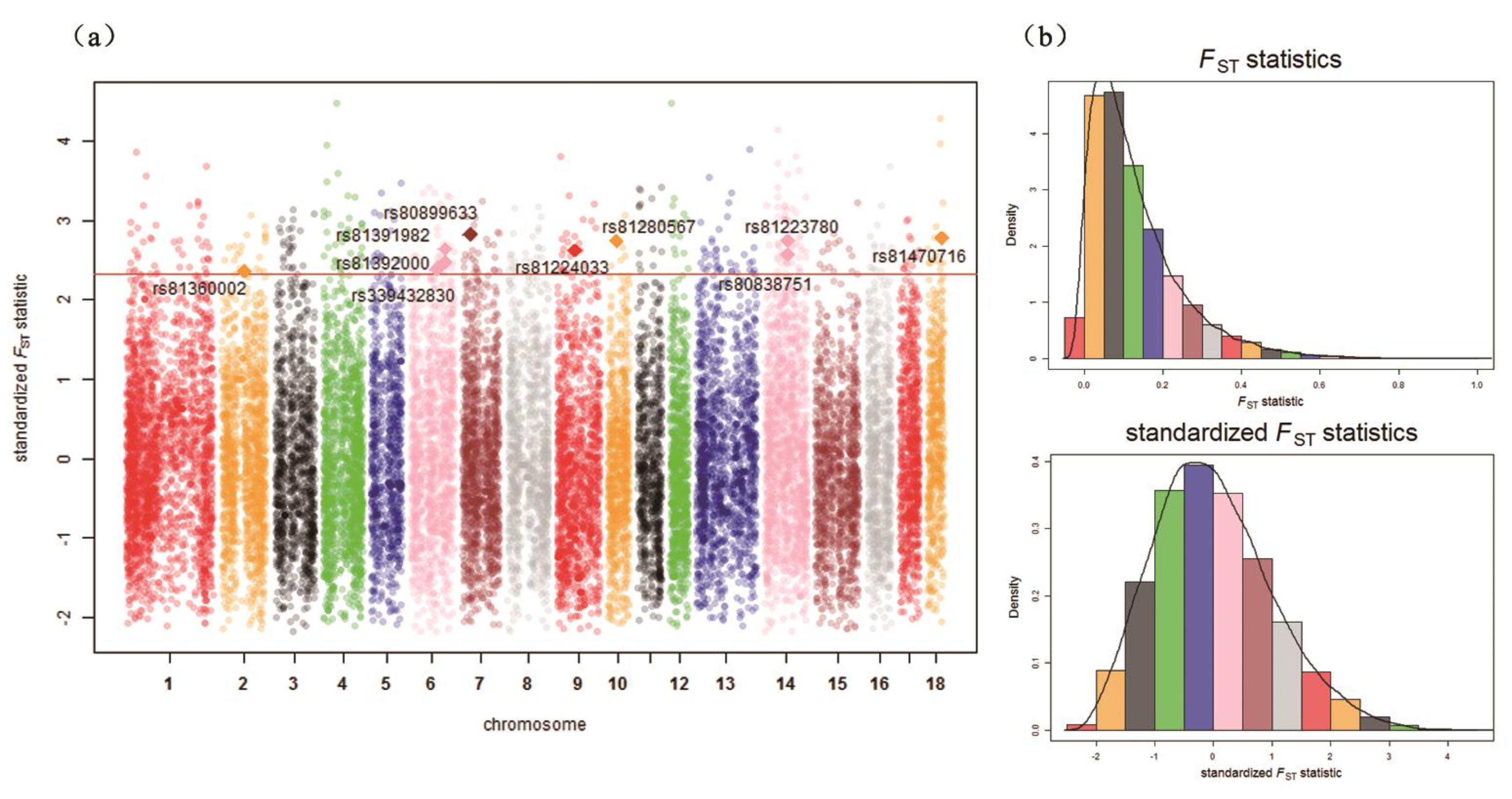
| Chr. 1 | ID | Detected in | iHH 2 in Test 3 | iHH in Ref 4 | Standardized XP-EHH 5 Score | FST | Genes | QTL 6 (Counts) |
|---|---|---|---|---|---|---|---|---|
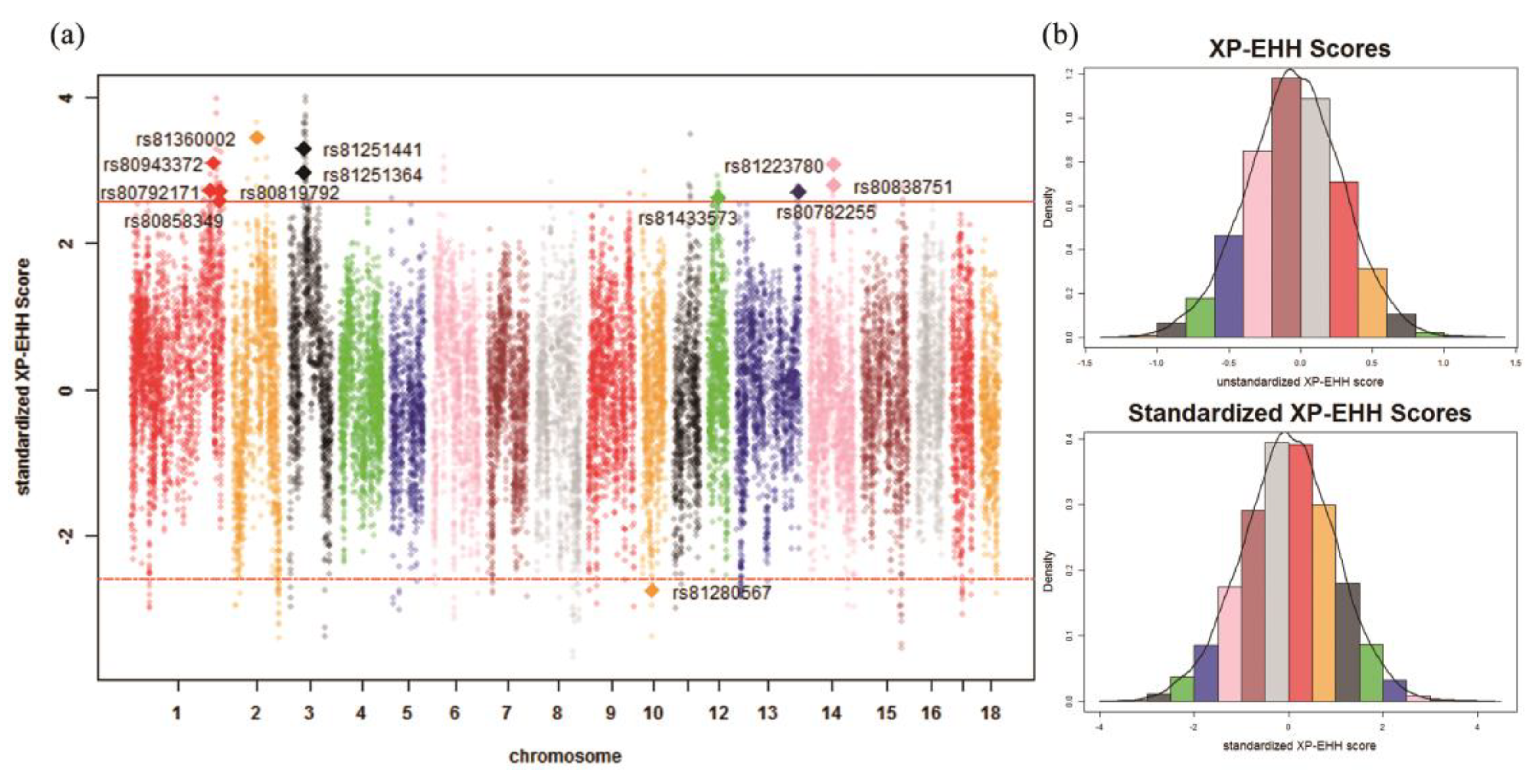
| 2 | rs81360002 | Test | 3.2266 | 0.7763 | 2.7056 | 0.4685 | BTF3, ANKRA2, UTP15, ARHGEF28 | Actinobacillus pleuropneumoniae susceptibility (7) |
| 6 | rs339432830 | Test | 3.9553 | 0.9181 | 2.8027 | 0.4726 | MC5R, RNMT, FAM210A, LDLRAD4, CEP192 | Backfat at last rib (11) |
| 6 | rs81391982 | Test | 1.0048 | 0.1885 | 3.3794 | 0.5273 | PIK3C3 | Backfat at last rib (9) |
| 6 | rs81392000 | Test | 0.9475 | 0.2324 | 2.6532 | 0.4930 | – | Backfat at last rib (9) |
| 7 | rs80899633 | Test | 1.3000 | 0.2568 | 3.2394 | 0.5677 | GRM4, HMGA1, NUDT3, RPS10, PACSIN1, SPDEF | Average backfat thickness (20) |
| 9 | rs81224033 | Test | 1.7863 | 0.4160 | 2.7937 | 0.5233 | PLEKHA6, PPP1R15B, PIK3C2B, MDM4, LRRN2 | Shoulder weight (3) |
| 14 | rs81223780 | Test | 2.4569 | 0.4583 | 3.3948 | 0.5498 | NRG3 | Fat androstenone level (4) |
| 14 | rs80838751 | Test | 2.6767 | 0.5997 | 2.8987 | 0.5129 | NRG3 | Fat androstenone level (4) |
| 18 | rs81470716 | Test | 0.7901 | 0.1880 | 2.7350 | 0.5564 | – | Actinobacillus pleuropneumoniae susceptibility (3) |
| 10 | rs81280567 | Ref | 0.1573 | 0.3664 | –3.4439 | 0.5492 | FRMD3, RASEF | Average daily gain (4) |
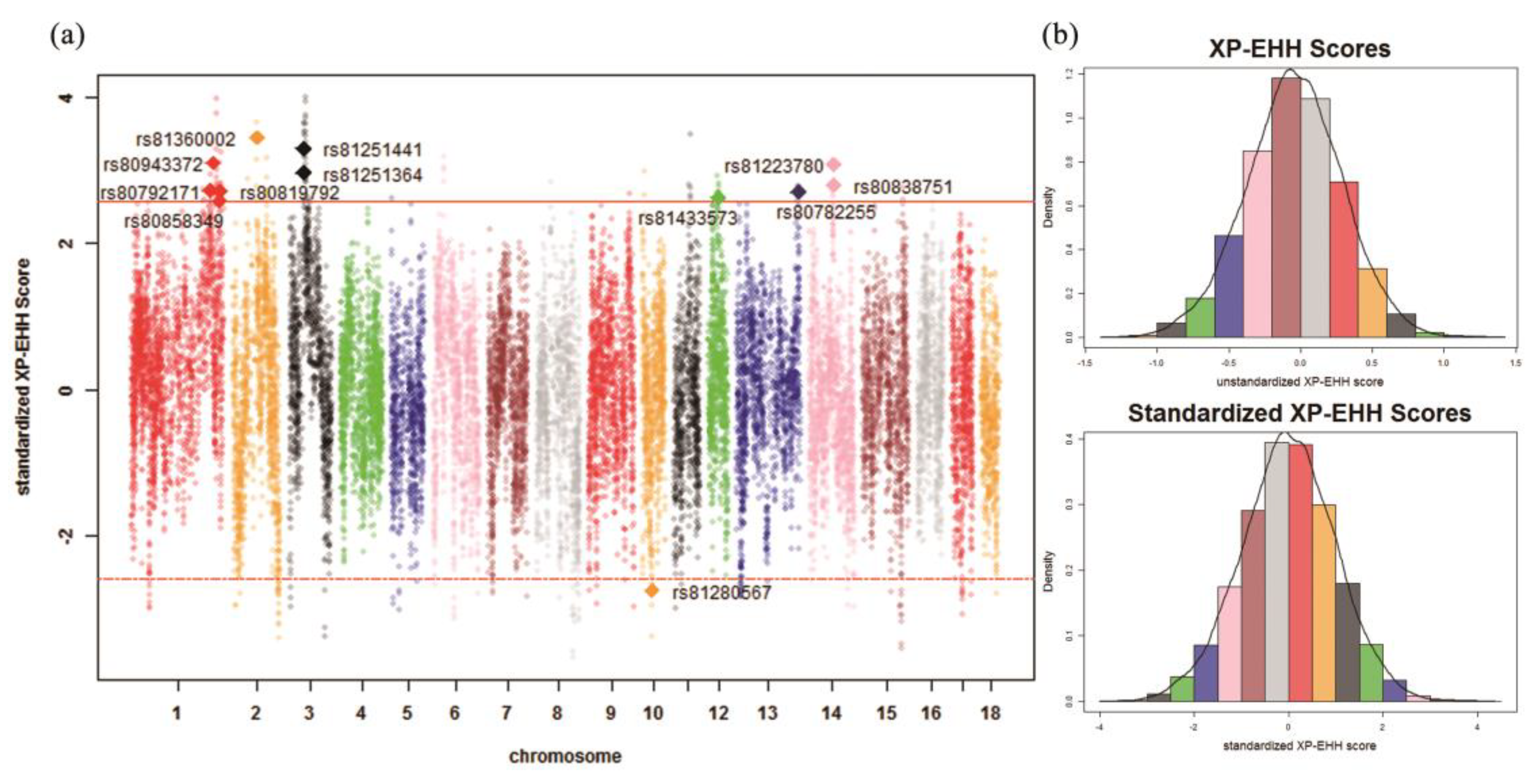
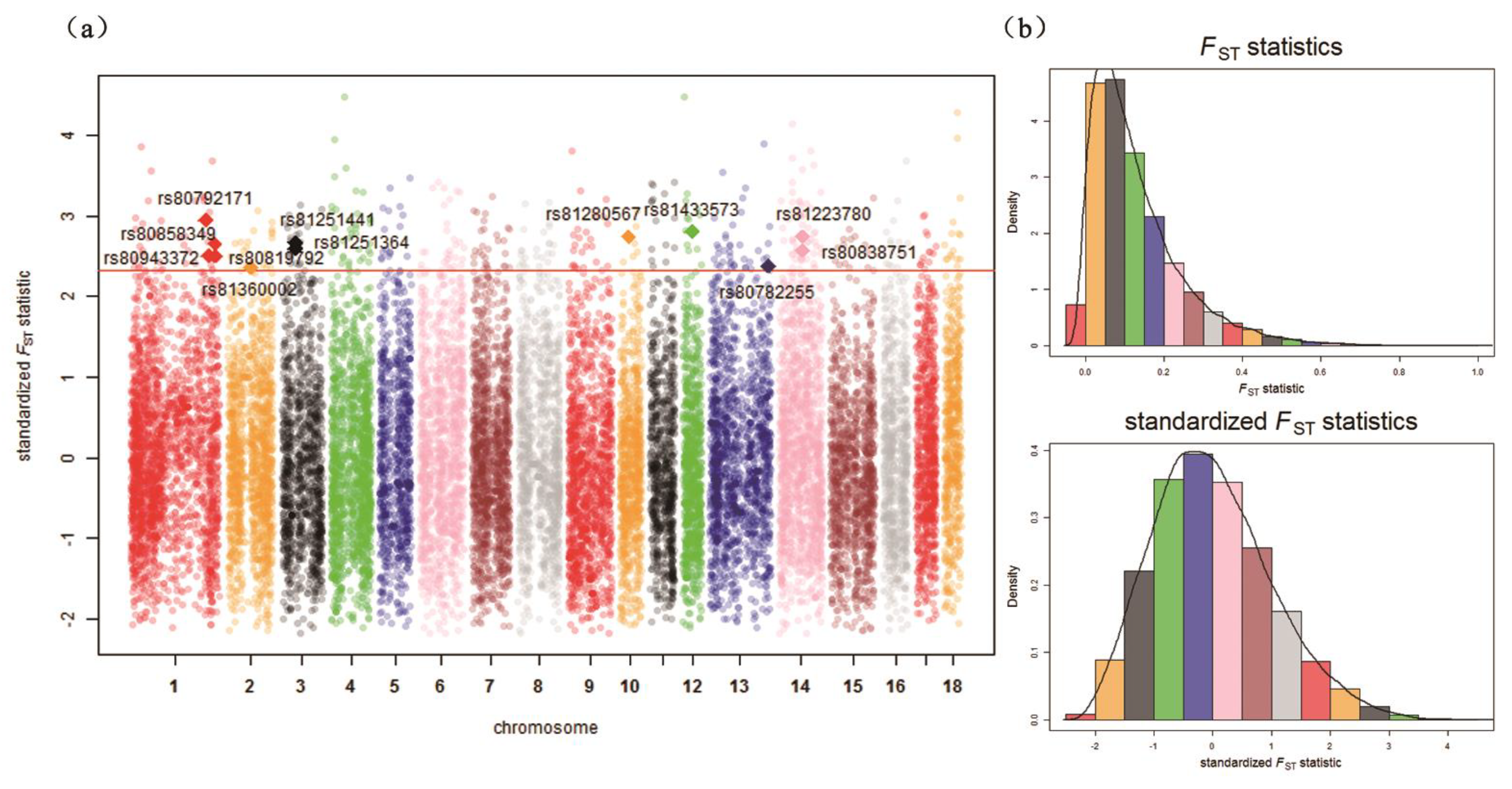
| Chr. 1 | ID | Detected in | iHH 2 in Test 3 | iHH in Ref 4 | Standardized XP-EHH 5 Score | FST | Genes | QTL 6 (Counts) |
|---|---|---|---|---|---|---|---|---|
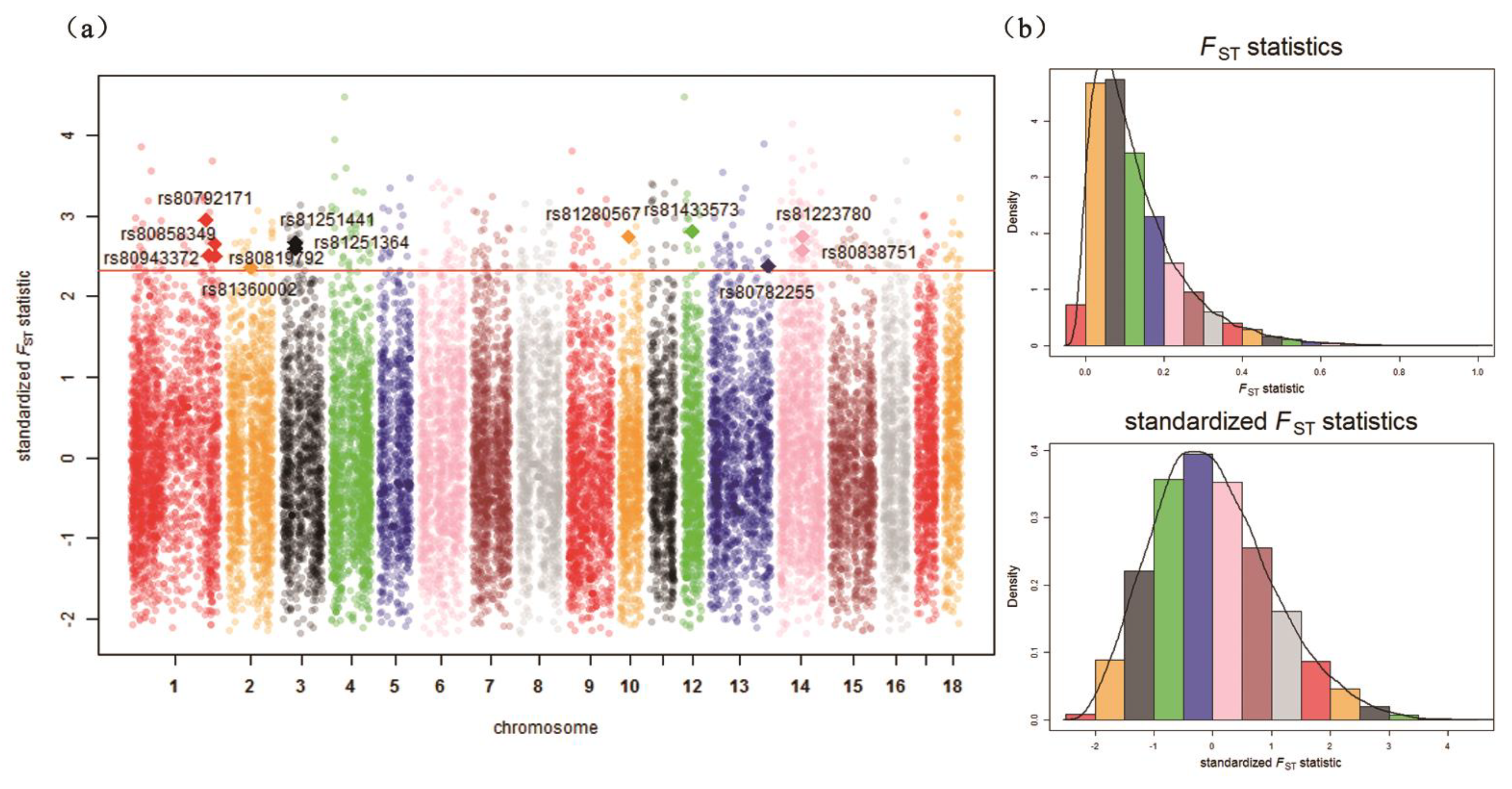
| 1 | rs80792171 | Test | 1.3676 | 0.5713 | 2.7257 | 0.5956 | LRSAM1, FAM129B, STXBP1, CFAP157, PTRH1, TTC16, TOR2A, SH2D3C, CDK9, FPGS, ENG, AK1, ST6GALNAC6, ST6GALNAC4 | Drip loss (15) |
| 1 | rs80943372 | Test | 4.5343 | 1.6659 | 3.1073 | 0.4985 | – | Drip loss (16) |
| 1 | rs80858349 | Test | 2.2043 | 0.9644 | 2.5884 | 0.5304 | – | Drip loss (16) |
| 1 | rs80819792 | Test | 0.9818 | 0.4111 | 2.7187 | 0.4975 | – | Drip loss (16) |
| 2 | rs81360002 | Test | 3.2297 | 1.0560 | 3.4540 | 0.4685 | BTF3, ANKRA2, UTP15, ARHGEF28 | Actinobacillus pleuropneumoniae susceptibility (7) |
| 3 | rs81251364 | Test | 1.8817 | 0.7229 | 2.9747 | 0.5172 | UXS1, C3H2orf40, NCK2 | Average daily gain (6) |
| 3 | rs81251441 | Test | 1.8762 | 0.6455 | 3.3025 | 0.5333 | UXS1, C3H2orf40, NCK2 | Average daily gain (6) |
| 12 | rs81433573 | Test | 0.9817 | 0.4227 | 2.6354 | 0.5630 | ANKFN1, NOG | Muscle moisture percentage (4) |
| 13 | rs80782255 | Test | 1.0648 | 0.4471 | 2.7108 | 0.4731 | – | Body weight (5 weeks) (1) |
| 14 | rs81223780 | Test | 2.5220 | 0.9360 | 3.0773 | 0.5498 | NRG3 | Fat androstenone level (4) |
| 14 | rs80838751 | Test | 2.7541 | 1.1181 | 2.8106 | 0.5129 | NRG3 | Fat androstenone level (4) |
| 10 | rs81280567 | Ref | 0.1720 | 0.4522 | –2.7414 | 0.5492 | FRMD3, RASEF | Average daily gain (4) |
| GO Terms and KEGG Pathways | Count | Genes | p-Value | Test/Ref 1 |
|---|---|---|---|---|
| GO: 0048015~phosphatidylinositol-mediated signaling | 2 | PIK3C2B, PIK3C3 | 0.0215 | Ding’an and Tunchang/Baoshan |
| ssc00604: Glycosphingolipid biosynthesis—ganglio series | 2 | ST6GALNAC6, ST6GALNAC4 | 0.0198 | Ding’an and Tunchang/Saba |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diao, S.; Huang, S.; Chen, Z.; Teng, J.; Ma, Y.; Yuan, X.; Chen, Z.; Zhang, H.; Li, J.; Zhang, Z. Genome-Wide Signatures of Selection Detection in Three South China Indigenous Pigs. Genes 2019, 10, 346. https://doi.org/10.3390/genes10050346
Diao S, Huang S, Chen Z, Teng J, Ma Y, Yuan X, Chen Z, Zhang H, Li J, Zhang Z. Genome-Wide Signatures of Selection Detection in Three South China Indigenous Pigs. Genes. 2019; 10(5):346. https://doi.org/10.3390/genes10050346
Chicago/Turabian StyleDiao, Shuqi, Shuwen Huang, Zitao Chen, Jinyan Teng, Yunlong Ma, Xiaolong Yuan, Zanmou Chen, Hao Zhang, Jiaqi Li, and Zhe Zhang. 2019. "Genome-Wide Signatures of Selection Detection in Three South China Indigenous Pigs" Genes 10, no. 5: 346. https://doi.org/10.3390/genes10050346
APA StyleDiao, S., Huang, S., Chen, Z., Teng, J., Ma, Y., Yuan, X., Chen, Z., Zhang, H., Li, J., & Zhang, Z. (2019). Genome-Wide Signatures of Selection Detection in Three South China Indigenous Pigs. Genes, 10(5), 346. https://doi.org/10.3390/genes10050346