Yeast as a Tool for Deeper Understanding of Human Manganese-Related Diseases


Abstract
:1. Yeast as a Model to Study Human Pathologies
2. Manganese Homeostasis: From Yeast to Human
2.1. Biological Functions of Manganese
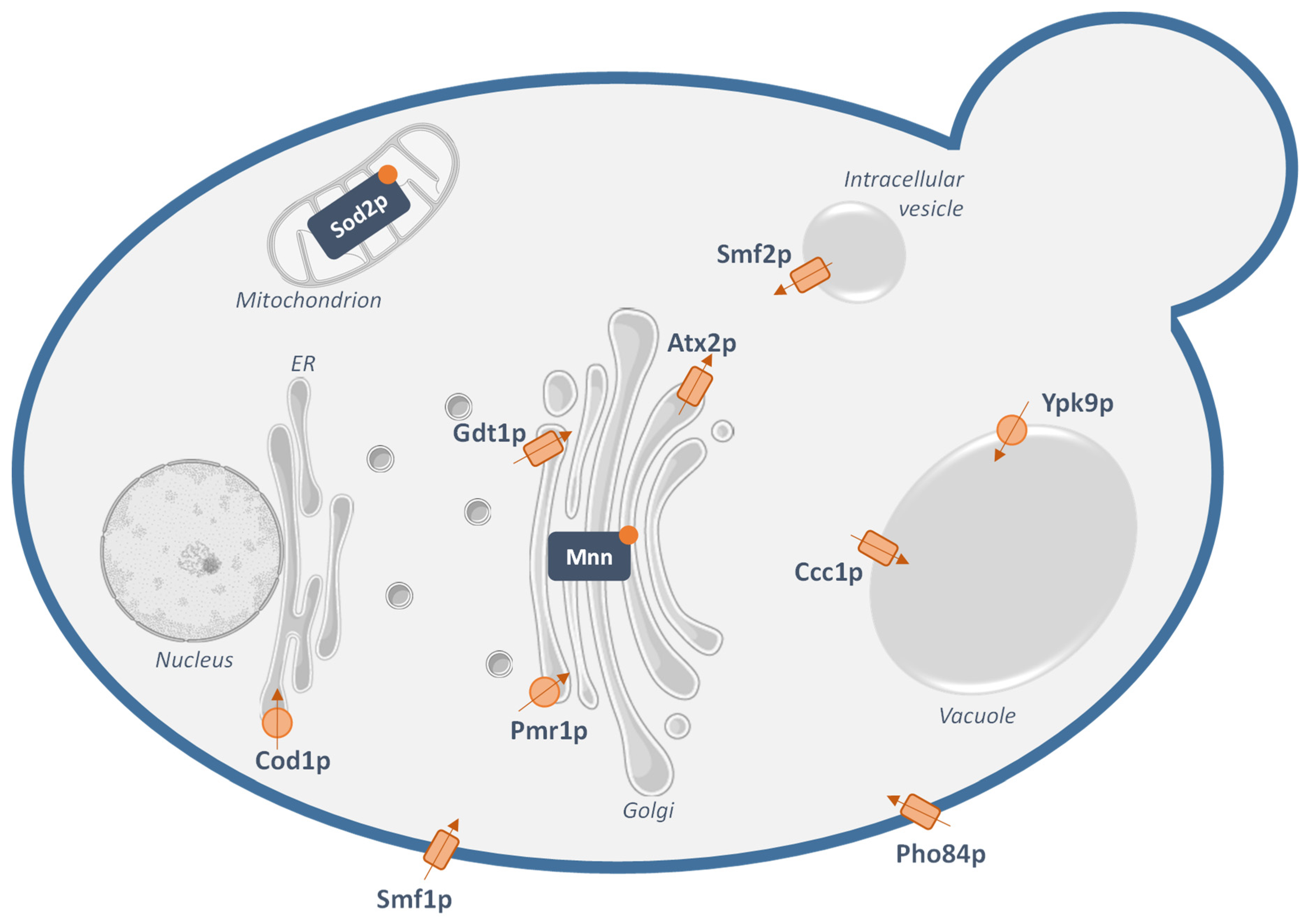
2.2. Manganese Transporters in Yeast
2.3. From Yeast to Human
3. Use of Yeast to Elucidate the Molecular Background of Manganese-Related Disorders
3.1. Molecular Mechanisms Behind Cellular Manganese Toxicity
3.2. Disorders Related to Altered Bioavailability of Mn2+ as Non-Enzymatic Antioxidant
3.3. Disorders Related to Altered Bioavailability of Mn2+ as a Cofactor
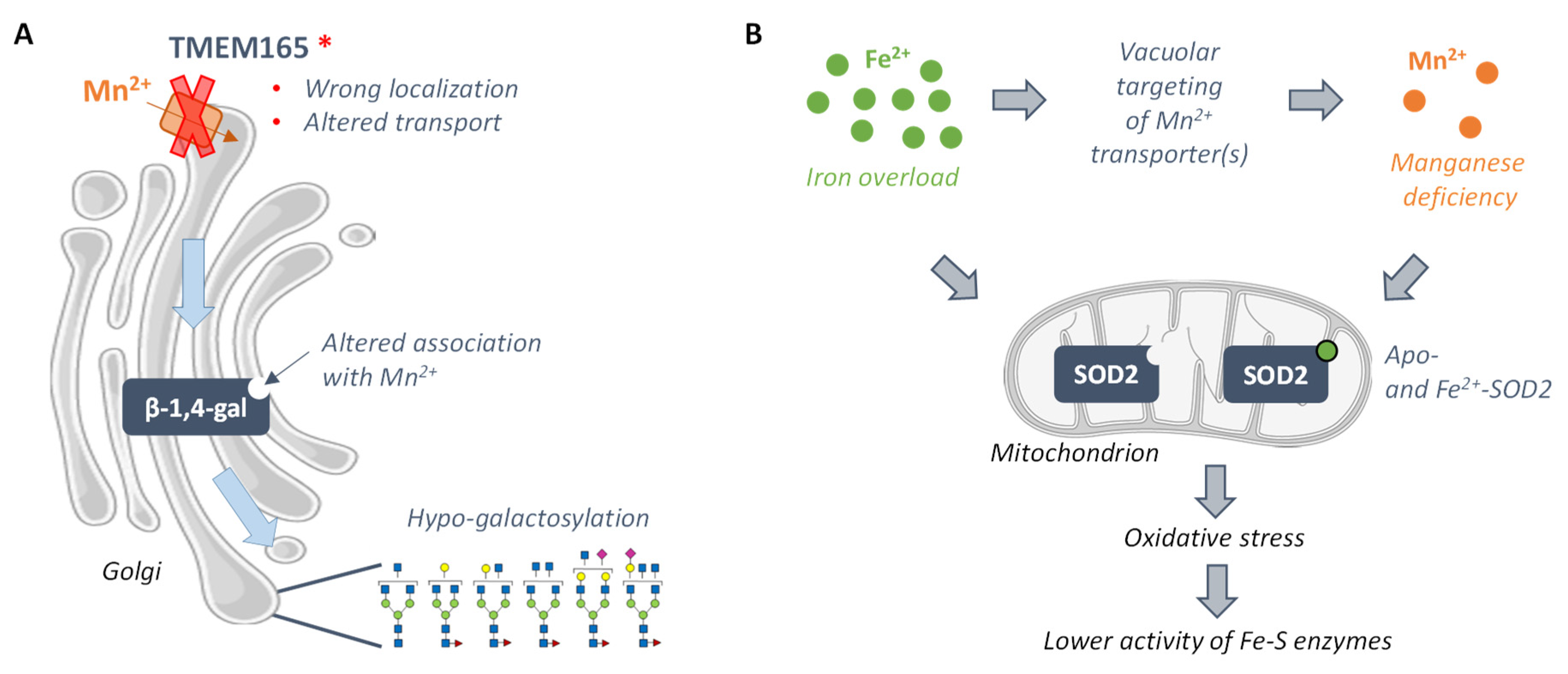
3.3.1. TMEM165-CDG and Mn2+-Dependent Glycosyltransferases
3.3.2. Friedreich Ataxia and the Mn2+-Dependent Superoxide Dismutase 2 SOD2
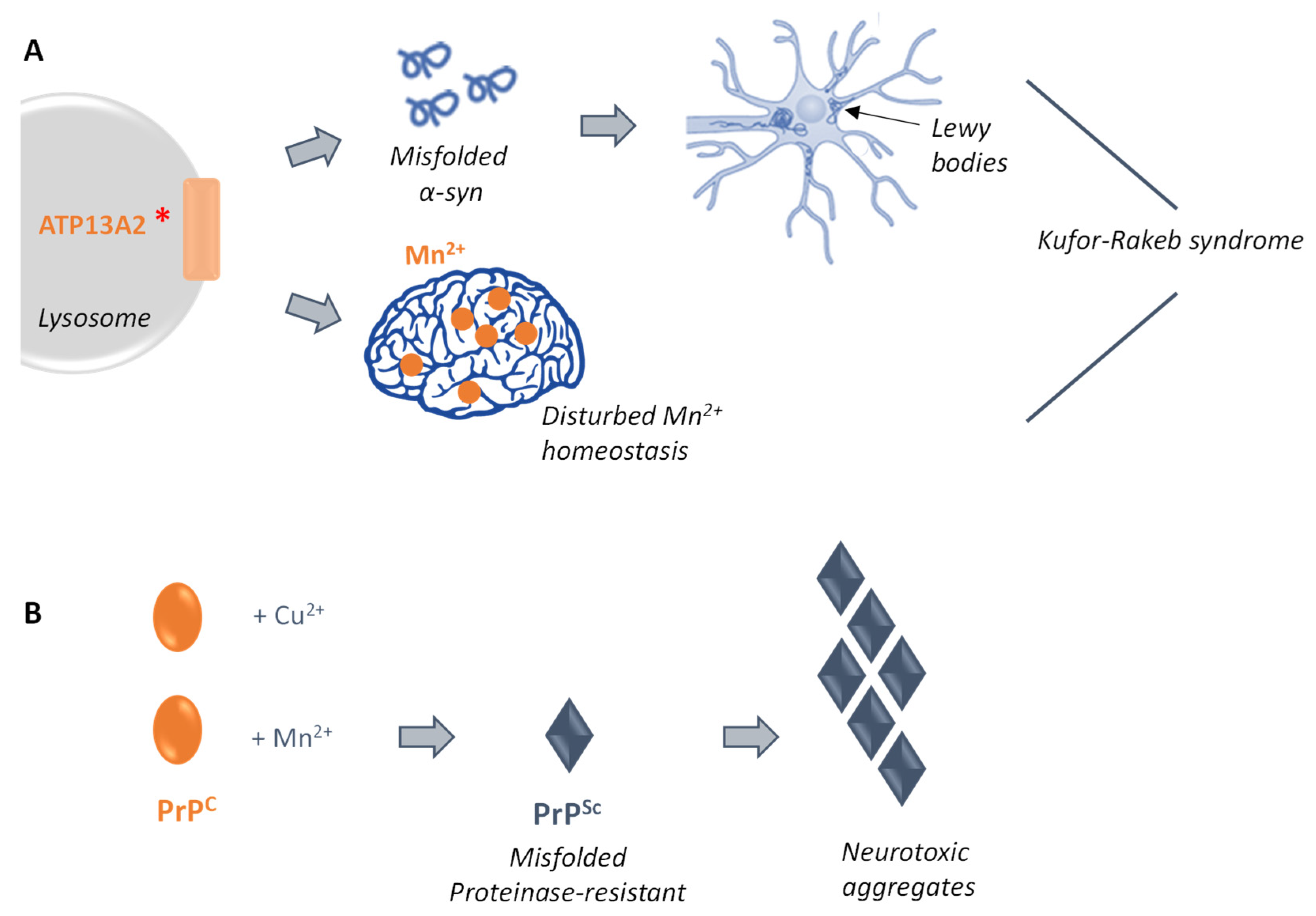
3.4. Manganese-Related Neurodegenerative Disorders
3.4.1. Ypk9p and Early Parkinson
3.4.2. Prion-Related Diseases
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gerngross, T.U. Advances in the production of human therapeutic proteins in yeasts and filamentous fungi. Nat. Biotechnol. 2004, 22, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 546–567. [Google Scholar] [CrossRef] [PubMed]
- Botstein, D.; Chervitz, S.A.; Cherry, M. Yeast as a Model Organism. Science 1997, 277, 1259–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.G.; Snyder, M. Yeast as a Model for Human Disease. Curr. Protoc. Hum. Genet. 2006, 48, 1–15. [Google Scholar] [CrossRef]
- Petranovic, D.; Nielsen, J. Can yeast systems biology contribute to the understanding of human disease? Trends Biotechnol. 2008, 26, 584–590. [Google Scholar] [CrossRef]
- Garber, K. Beyond the Nobel Prize: Cell cycle research offers new view of cancer. J. Natl. Cancer Inst. 2001, 93, 1766–1768. [Google Scholar] [CrossRef]
- Kornberg, R.D. The molecular basis of eukaryotic transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 12955–12961. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, E.H. Telomeres and Telomerase: The Means to the End (Nobel Lecture). Angew. Chem. Int. Ed. 2010, 49, 7405–7421. [Google Scholar] [CrossRef]
- Ferro-Novick, S.; Brose, N. Traffic control system within cells. Nature 2013, 504, 98. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Frake, R.A. Yoshinori Ohsumi’s Nobel Prize for mechanisms of autophagy: From basic yeast biology to therapeutic potential. J. R. Coll. Physicians Edinb. 2016, 46, 228–233. [Google Scholar] [CrossRef]
- Mager, W.H.; Winderickx, J. Yeast as a model for medical and medicinal research. Trends Pharmacol. Sci. 2005, 26, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Bassett, D.E., Jr.; Boguski, M.S.; Hieter, P. Yeast genes and human disease. Nature 1996, 379, 589–590. [Google Scholar] [CrossRef] [PubMed]
- Perocchi, F.; Mancera, E.; Steinmetz, L.M. Systematic screens for human disease genes, from yeast to human and back. Mol. Biosyst. 2008, 4, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Archibald, F.S.; Fridovich, I. The scavenging of superoxide radical by manganous complexes: In vitro. Arch. Biochem. Biophys. 1982, 214, 452–463. [Google Scholar] [CrossRef]
- Barnese, K.; Gralla, E.B.; Valentine, J.S.; Cabelli, D.E. Biologically relevant mechanism for catalytic superoxide removal by simple manganese compounds. Proc. Natl. Acad. Sci. USA 2012, 109, 6892–6897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, M.J.; Gaidamakova, E.K.; Matrosova, V.Y.; Vasilenko, A.; Zhai, M.; Venkateswaran, A.; Hess, M.; Omelchenko, M.V.; Kostandarithes, H.M.; Makarova, K.S.; et al. Accumulation of Mn (II) in Deinococcus radiodurans facilitates gamma-radiation resistance. Science 2004, 306, 1025–1028. [Google Scholar] [CrossRef] [PubMed]
- Archibald, F.S.; Fridovich, I. Manganese and defenses against oxygen toxicity in Lactobacillus plantarum. J. Bacteriol. 1981, 145, 442–451. [Google Scholar] [PubMed]
- Culotta, V.C.; Daly, M.J. Manganese complexes: Diverse metabolic routes to oxidative stress resistance in prokaryotes and yeast. Antioxid. Redox Signal. 2013, 19, 933–944. [Google Scholar] [CrossRef]
- Jensen, A.N.; Jensen, L.T. Manganese Transport, Trafficking and Function in Invertebrates. Toxicology 2015, 22. [Google Scholar] [CrossRef]
- Reddi, A.R.; Jensen, L.T.; Culotta, V.C. Manganese homeostasis in Saccharomyces cerevisiae. Chem. Rev. 2009, 109, 4722–4732. [Google Scholar] [CrossRef]
- Culotta, V.C.; Yang, M.; Hall, M.D. Manganese transport and trafficking: Lessons learned from Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Nelson, H.; Nelson, N. The family of SMF metal ion transporters in yeast cells. J. Biol. Chem. 2000, 275, 33388–33394. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.T.; Ajua-Alemanji, M.; Culotta, V.C. The Saccharomyces cerevisiae high affinity phosphate transporter encoded by PHO84 also functions in manganese homeostasis. J. Biol. Chem. 2003, 278, 42036–42040. [Google Scholar] [CrossRef] [PubMed]
- Luk, E.E.; Culotta, V.C. Manganese superoxide dismutase in Saccharomyces cerevisiae acquires its metal co-factor through a pathway involving the Nramp metal transporter, Smf2p. J. Biol. Chem. 2001, 276, 47556–47562. [Google Scholar] [CrossRef] [PubMed]
- Durr, G.; Strayle, J.; Plemper, R.; Elbs, S.; Klee, S.K.; Catty, P.; Wolf, D.H.; Rudolph, H.K. The medial-Golgi ion pump Pmr1 supplies the yeast secretory pathway with Ca2+ and Mn2+ required for glycosylation, sorting, and endoplasmic reticulum-associated protein degradation. Mol. Biol. Cell 1998, 9, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Lapinskas, P.J.; Cunningham, K.W.; Liu, X.F.; Fink, G.R.; Culotta, V.C. Mutations in PMR1 suppress oxidative damage in yeast cells lacking superoxide dismutase. Mol. Cell. Biol. 1995, 15, 1382–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thines, L.; Deschamps, A.; Sengottaiyan, P.; Savel, O.; Stribny, J.; Morsomme, P. The yeast protein Gdt1p transports Mn2+ ions and thereby regulates manganese homeostasis in the Golgi. J. Biol. Chem. 2018, 293, 8048–8055. [Google Scholar] [CrossRef]
- Lin, S.J.; Culotta, V.C. Suppression of oxidative damage by Saccharomyces cerevisiae ATX2, which encodes a manganese-trafficking protein that localizes to Golgi-like vesicles. Mol. Cell. Biol. 1996, 16, 6303–6312. [Google Scholar] [CrossRef] [Green Version]
- Cohen, Y.; Megyeri, M.; Chen, O.C.; Condomitti, G.; Riezman, I.; Loizides-Mangold, U.; Abdul-Sada, A.; Rimon, N.; Riezman, H.; Platt, F.M.; et al. The yeast p5 type ATPase, spf1, regulates manganese transport into the endoplasmic reticulum. PLoS ONE 2013, 8, e85519. [Google Scholar] [CrossRef]
- Lapinskas, P.J.; Lin, S.-J.; Culotta, V.C. The role of the Saccharomyces cerevisiae CCC1 gene in the homeostasis of manganese ions. Mol. Microbiol. 1996, 21, 519–528. [Google Scholar] [CrossRef]
- Chesi, A.; Kilaru, A.; Fang, X.; Cooper, A.A.; Gitler, A.D. The role of the Parkinson’s disease gene PARK9 in essential cellular pathways and the manganese homeostasis network in yeast. PLoS ONE 2012, 7, e34178. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, P.A.; Moore, M.J. Labile Low-Molecular-Mass Metal Complexes in Mitochondria: Trials and Tribulations of a Burgeoning Field. Biochemistry 2016, 55, 4140–4153. [Google Scholar] [CrossRef] [PubMed]
- Au, C.; Benedetto, A.; Aschner, M. Manganese transport in eukaryotes: The role of DMT1. Neurotoxicology 2008, 29, 569–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courville, P.; Chaloupka, R.; Cellier, M.F. Recent progress in structure-function analyses of Nramp proton-dependent metal-ion transporters. Biochem. Cell Biol. 2006, 84, 960–978. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Mohamalawari, D.; Rao, R. A novel isoform of the secretory pathway Ca2+,Mn2+-ATPase, hSPCA2, has unusual properties and is expressed in the brain. J. Biol. Chem. 2005, 280, 11608–11614. [Google Scholar] [CrossRef] [PubMed]
- Vanoevelen, J.; Dode, L.; Van Baelen, K.; Fairclough, R.J.; Missiaen, L.; Raeymaekers, L.; Wuytack, F. The secretory pathway Ca2+/Mn2+-ATPase 2 is a Golgi-localized pump with high affinity for Ca2+ ions. J. Biol. Chem. 2005, 280, 22800–22808. [Google Scholar] [CrossRef] [PubMed]
- Demaegd, D.; Foulquier, F.; Colinet, A.S.; Gremillon, L.; Legrand, D.; Mariot, P.; Peiter, E.; Van Schaftingen, E.; Matthijs, G.; Morsomme, P. Newly characterized Golgi-localized family of proteins is involved in calcium and pH homeostasis in yeast and human cells. Proc. Natl. Acad. Sci. USA 2013, 110, 6859–6864. [Google Scholar] [CrossRef] [Green Version]
- Tan, J.; Zhang, T.; Jiang, L.; Chi, J.; Hu, D.; Pan, Q.; Wang, D.; Zhang, Z. Regulation of intracellular manganese homeostasis by Kufor-Rakeb syndrome-associated ATP13A2 protein. J. Biol. Chem. 2011, 286, 29654–29662. [Google Scholar] [CrossRef]
- Brown, S.; Taylor, N.L. Could mitochondrial dysfunction play a role in manganese toxicity? Environ. Toxicol. Pharmacol. 1999, 7, 49–57. [Google Scholar] [CrossRef]
- Putrament, A.; Baranowska, H.; Ejchart, A.; Jachymczyk, W. Manganese mutagenesis in yeast. VI. Mn2+ uptake, mitDNA replication and ER induction: Comparison with other divalent cations. Mol. Gen. Genet. 1977, 151, 69–76. [Google Scholar] [CrossRef]
- Putrament, A.; Branowska, H.; Ejchart, A.; Prazmo, W. Manganese mutagenesis in yeast. A practical application of manganese for the induction of mitochondrial antibiotic-resistant mutations. J. Gen. Microbiol. 1975, 90, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.A.; Li, Z.; Sridhar, S.; Khoshbouei, H. The effect of manganese on dopamine toxicity and dopamine transporter (DAT) in control and DAT transfected HEK cells. Neurotoxicology 2013, 35, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Zhao, Q.; Slavkovich, V.; Aschner, M.; Graziano, J.H. Alteration of iron homeostasis following chronic exposure to manganese in rats1. Brain Res. 1999, 833, 125–132. [Google Scholar] [CrossRef]
- Zheng, W.; Ren, S.; Graziano, J.H. Manganese inhibits mitochondrial aconitase: A mechanism of manganese neurotoxicity. Brain Res. 1998, 799, 334–342. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macmillan-Crow, L.A.; Cruthirds, D.L. Manganese superoxide dismutase in disease. Free Radic. Res. 2001, 34, 325–336. [Google Scholar] [CrossRef]
- Wessling-Resnick, M. Nramp1 and Other Transporters Involved in Metal Withholding during Infection. J. Biol. Chem. 2015, 290, 18984–18990. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.C.; Kosman, D.J. Intracellular Mn (II)-associated superoxide scavenging activity protects Cu,Zn superoxide dismutase-deficient Saccharomyces cerevisiae against dioxygen stress. J. Biol. Chem. 1989, 264, 12172–12178. [Google Scholar]
- Liu, X.F.; Elashvili, I.; Gralla, E.B.; Valentine, J.S.; Lapinskas, P.; Culotta, V.C. Yeast lacking superoxide dismutase. Isolation of genetic suppressors. J. Biol. Chem. 1992, 267, 18298–18302. [Google Scholar]
- Reddi, A.R.; Culotta, V.C. Regulation of manganese antioxidants by nutrient sensing pathways in Saccharomyces cerevisiae. Genetics 2011, 189, 1261–1270. [Google Scholar] [CrossRef]
- McNaughton, R.L.; Reddi, A.R.; Clement, M.H.S.; Sharma, A.; Barnese, K.; Rosenfeld, L.; Gralla, E.B.; Valentine, J.S.; Culotta, V.C.; Hoffman, B.M. Probing in vivo Mn2+ speciation and oxidative stress resistance in yeast cells with electron-nuclear double resonance spectroscopy. Proc. Natl. Acad. Sci. USA 2010, 107, 15335–15339. [Google Scholar] [CrossRef]
- Péanne, R.; de Lonlay, P.; Foulquier, F.; Kornak, U.; Lefeber, D.J.; Morava, E.; Pérez, B.; Seta, N.; Thiel, C.; Van Schaftingen, E.; et al. Congenital disorders of glycosylation (CDG): Quo vadis? Eur. J. Med. Genet. 2018, 61, 643–663. [Google Scholar] [CrossRef]
- Foulquier, F.; Amyere, M.; Jaeken, J.; Zeevaert, R.; Schollen, E.; Race, V.; Bammens, R.; Morelle, W.; Rosnoblet, C.; Legrand, D.; et al. TMEM165 deficiency causes a congenital disorder of glycosylation. Am. J. Hum. Genet. 2012, 91, 15–26. [Google Scholar] [CrossRef]
- Demaegd, D.; Colinet, A.S.; Deschamps, A.; Morsomme, P. Molecular evolution of a novel family of putative calcium transporters. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Colinet, A.S.; Sengottaiyan, P.; Deschamps, A.; Colsoul, M.L.; Thines, L.; Demaegd, D.; Duchêne, M.C.; Foulquier, F.; Hols, P.; Morsomme, P. Yeast Gdt1 is a Golgi-localized calcium transporter required for stress-induced calcium signaling and protein glycosylation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Dulary, E.; Yu, S.-Y.; Houdou, M.; de Bettignies, G.; Decool, V.; Potelle, S.; Duvet, S.; Krzewinski-Recchi, M.-A.; Garat, A.; Matthijs, G.; et al. Investigating the function of Gdt1p in yeast Golgi glycosylation. Biochim. Biophys. Acta Gen. Subj. 2017. [Google Scholar] [CrossRef]
- Potelle, S.; Morelle, W.; Dulary, E.; Duvet, S.; Vicogne, D.; Spriet, C.; Krzewinski-Recchi, M.A.; Morsomme, P.; Jaeken, J.; Matthijs, G.; et al. Glycosylation abnormalities in Gdt1p/TMEM165 deficient cells result from a defect in Golgi manganese homeostasis. Hum. Mol. Genet. 2016, 25, 1489–1500. [Google Scholar] [CrossRef] [Green Version]
- Morelle, W.; Potelle, S.; Witters, P.; Wong, S.; Climer, L.; Lupashin, V.; Matthijs, G.; Gadomski, T.; Jaeken, J.; Cassiman, D.; et al. Galactose Supplementation in Patients With TMEM165-CDG Rescues the Glycosylation Defects. J. Clin. Endocrinol. Metab. 2017, 102, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Rosnoblet, C.; Legrand, D.; Demaegd, D.; Hacine-Gherbi, H.; de Bettignies, G.; Bammens, R.; Borrego, C.; Duvet, S.; Morsomme, P.; Matthijs, G.; et al. Impact of disease-causing mutations on TMEM165 subcellular localization, a recently identified protein involved in CDG-II. Hum. Mol. Genet. 2013, 22, 2914–2928. [Google Scholar] [CrossRef] [Green Version]
- Colinet, A.S.; Thines, L.; Deschamps, A.; Flémal, G.; Demaegd, D.; Morsomme, P. Acidic and uncharged polar residues in the consensus motifs of the yeast Ca2+ transporter Gdt1p are required for calcium transport. Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Rötig, A.; Sidi, D.; Munnich, A.; Rustin, P. Molecular insights into Friedreich’s ataxia and antioxidant-based therapies. Trends Mol. Med. 2002, 8, 221–224. [Google Scholar] [CrossRef]
- Gerber, J.; Muhlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [Green Version]
- Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of [2Fe-2S] clusters in ISU-type proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef]
- Zhang, Y.; Lyver, E.R.; Knight, S.A.; Lesuisse, E.; Dancis, A. Frataxin and mitochondrial carrier proteins, Mrs3p and Mrs4p, cooperate in providing iron for heme synthesis. J. Biol. Chem. 2005, 280, 19794–19807. [Google Scholar] [CrossRef]
- O’Neill, H.A.; Gakh, O.; Park, S.; Cui, J.; Mooney, S.M.; Sampson, M.; Ferreira, G.C.; Isaya, G. Assembly of human frataxin is a mechanism for detoxifying redox-active iron. Biochemistry 2005, 44, 537–545. [Google Scholar] [CrossRef]
- Rötig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef]
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef]
- Foury, F.; Cazzalini, O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 1997, 411, 373–377. [Google Scholar] [CrossRef]
- Patel, P.I.; Isaya, G. Friedreich ataxia: From GAA triplet-repeat expansion to frataxin deficiency. Am. J. Hum. Genet. 2001, 69, 15–24. [Google Scholar] [CrossRef]
- Irazusta, V.; Cabiscol, E.; Reverter-Branchat, G.; Ros, J.; Tamarit, J. Manganese is the link between frataxin and iron-sulfur deficiency in the yeast model of Friedreich ataxia. J. Biol. Chem. 2006, 281, 12227–12232. [Google Scholar] [CrossRef]
- Irazusta, V.; Obis, E.; Moreno-Cermeño, A.; Cabiscol, E.; Ros, J.; Tamarit, J. Yeast frataxin mutants display decreased superoxide dismutase activity crucial to promote protein oxidative damage. Free Radic. Biol. Med. 2010, 48, 411–420. [Google Scholar] [CrossRef]
- Jiralerspong, S.; Ge, B.; Hudson, T.J.; Pandolfo, M. Manganese superoxide dismutase induction by iron is impaired in Friedreich ataxia cells. FEBS Lett. 2001, 509, 101–105. [Google Scholar] [CrossRef]
- Chantrel-Groussard, K.; Geromel, V.; Puccio, H.; Koenig, M.; Munnich, A.; Rotig, A.; Rustin, P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 2001, 10, 2061–2067. [Google Scholar] [CrossRef]
- Moreno-Cermeno, A.; Obis, E.; Belli, G.; Cabiscol, E.; Ros, J.; Tamarit, J. Frataxin depletion in yeast triggers up-regulation of iron transport systems before affecting iron-sulfur enzyme activities. J. Biol. Chem. 2010, 285, 41653–41664. [Google Scholar] [CrossRef]
- Markesbery, W.R.; Ehmann, W.D.; Alauddin, M.; Hossain, T.I. Brain trace element concentrations in aging. Neurobiol. Aging 1984, 5, 19–28. [Google Scholar] [CrossRef]
- Bowman, A.B.; Kwakye, G.F.; Herrero Hernández, E.; Aschner, M. Role of manganese in neurodegenerative diseases. J. Trace Elem. Med. Biol. 2011, 25, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef]
- Usenovic, M.; Tresse, E.; Mazzulli, J.R.; Taylor, J.P.; Krainc, D. Deficiency of ATP13A2 leads to lysosomal dysfunction, α-synuclein accumulation, and neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Van Veen, S.; Sørensen, D.M.; Holemans, T.; Holen, H.W.; Palmgren, M.G.; Vangheluwe, P. Cellular function and pathological role of ATP13A2 and related P-type transport ATPases in Parkinson’s disease and other neurological disorders. Front. Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.-C.; et al. α-Synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308. [Google Scholar] [CrossRef]
- Schmidt, K.; Wolfe, D.M.; Stiller, B.; Pearce, D.A. Cd2+, Mn2+, Ni2+ and Se2+ toxicity to Saccharomyces cerevisiae lacking YPK9p the orthologue of human ATP13A2. Biochem. Biophys. Res. Commun. 2009, 383, 198–202. [Google Scholar] [CrossRef]
- Outeiro, T.F.; Lindquist, S. Yeast Cells Provide Insight into α-Synuclein Biology and Pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef]
- Verina, T.; Schneider, J.S.; Guilarte, T.R. Manganese exposure induces α-synuclein aggregation in the frontal cortex of non-human primates. Toxicol. Lett. 2013, 217, 177–183. [Google Scholar] [CrossRef]
- Crossgrove, J.; Zheng, W. Manganese toxicity upon overexposure. NMR Biomed. 2004, 17, 544–553. [Google Scholar] [CrossRef]
- Prusiner, S. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef]
- Come, J.H.; Fraser, P.E.; Lansbury, P.T. A kinetic model for amyloid formation in the prion diseases: Importance of seeding. Proc. Natl. Acad. Sci. USA 1993, 90, 5959–5963. [Google Scholar] [CrossRef]
- Imran, M.; Mahmood, S. An overview of human prion diseases. Virol. J. 2011, 8, 559. [Google Scholar] [CrossRef]
- Brown, D.R. Prion protein expression aids cellular uptake and veratridine-induced release of copper. J. Neurosci. Res. 1999, 58, 717–725. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef]
- Brown, D.R.; Besinger, A. Prion protein expression and superoxide dismutase activity. Biochem. J. 1998, 334, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.; Marken, F.; Salter, S.; Brown, D.R. Thermodynamic and voltammetric characterization of the metal binding to the prion protein: Insights into pH dependence and redox chemistry. Biochemistry 2009, 48, 2610–2619. [Google Scholar] [CrossRef]
- Hornshaw, M.P.; McDermott, J.R.; Candy, J.M.; Lakey, J.H. Copper binding to the N-terminal tandem repeat region of mammalian and avian prion protein: Structural studies using synthetic peptides. Biochem. Biophys. Res. Commun. 1995, 214, 993–999. [Google Scholar] [CrossRef]
- Brown, D.R.; Hafiz, F.; Glasssmith, L.L.; Wong, B.S.; Jones, I.M.; Clive, C.; Haswell, S.J. Consequences of manganese replacement of copper for prion protein function and proteinase resistance. EMBO J. 2000, 19, 1180–1186. [Google Scholar] [CrossRef] [Green Version]
- Brazier, M.W.; Davies, P.; Player, E.; Marken, F.; Viles, J.H.; Brown, D.R. Manganese binding to the prion protein. J. Biol. Chem. 2008, 283, 12831–12839. [Google Scholar] [CrossRef]
- Treiber, C.; Simons, A.; Multhaup, G. Effect of copper and manganese on the de novo generation of protease-resistant prion protein in yeast cells. Biochemistry 2006, 45, 6674–6680. [Google Scholar] [CrossRef]
- Brown, D.R. Prions and manganese: A maddening beast. Metallomics 2011, 3, 229–238. [Google Scholar] [CrossRef]
- Suhre, M.H.; Hess, S.; Golser, A.V.; Scheibel, T. Influence of divalent copper, manganese and zinc ions on fibril nucleation and elongation of the amyloid-like yeast prion determinant Sup35p-NM. J. Inorg. Biochem. 2009, 103, 1711–1720. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Human Transporter | Yeast Mn2+-Transporting Ortholog | Function in Human Cells |
|---|---|---|
| DMT1 [33] | Smf1p and Smf2p | Plasma membrane metal importer |
| Involved in dietary manganese intake | ||
| NRAMP1 [20,34] | Smf1p and Smf2p | Plasma membrane metal importer |
| Mostly expressed in the phagosome of macrophages where it limits the bioavailability of metals to invading microbes | ||
| SPCA1 and SPCA2 [35,36] | Pmr1p | Golgi-localized Ca2+ and Mn2+ P-type ATPases |
| Involved in manganese detoxification and Golgi Mn2+ supply | ||
| TMEM165 [27,37] | Gdt1p | Golgi-localized Ca2+ and Mn2+ secondary transporter |
| Putatively involved in manganese detoxification and Golgi Mn2+ supply | ||
| ATP13A2 [38] | Ypk9p | Putative lysosomal Mn2+ transporter |
| Involved in manganese sequestration in the lysosome | ||
| ATP13A1 [29] | Cod1p | Mn2+ P-type ATPase at the endoplasmic reticulum |
| Supplies the secretory pathway with manganese |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thines, L.; Deschamps, A.; Stribny, J.; Morsomme, P. Yeast as a Tool for Deeper Understanding of Human Manganese-Related Diseases. Genes 2019, 10, 545. https://doi.org/10.3390/genes10070545
Thines L, Deschamps A, Stribny J, Morsomme P. Yeast as a Tool for Deeper Understanding of Human Manganese-Related Diseases. Genes. 2019; 10(7):545. https://doi.org/10.3390/genes10070545
Chicago/Turabian StyleThines, Louise, Antoine Deschamps, Jiri Stribny, and Pierre Morsomme. 2019. "Yeast as a Tool for Deeper Understanding of Human Manganese-Related Diseases" Genes 10, no. 7: 545. https://doi.org/10.3390/genes10070545
APA StyleThines, L., Deschamps, A., Stribny, J., & Morsomme, P. (2019). Yeast as a Tool for Deeper Understanding of Human Manganese-Related Diseases. Genes, 10(7), 545. https://doi.org/10.3390/genes10070545