Molecular Strategies for RPGR Gene Therapy

 ,
,
Abstract
:1. Introduction
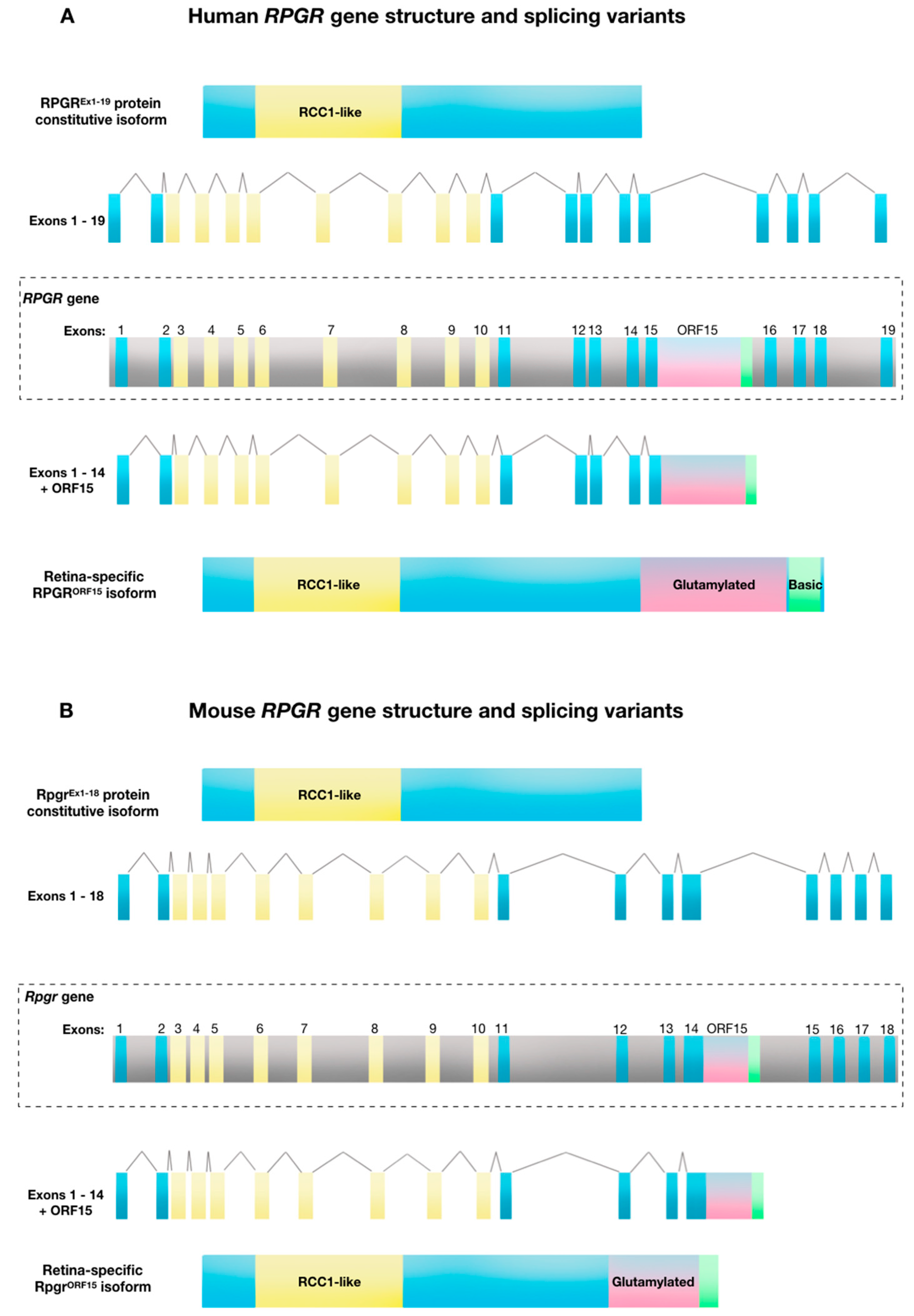
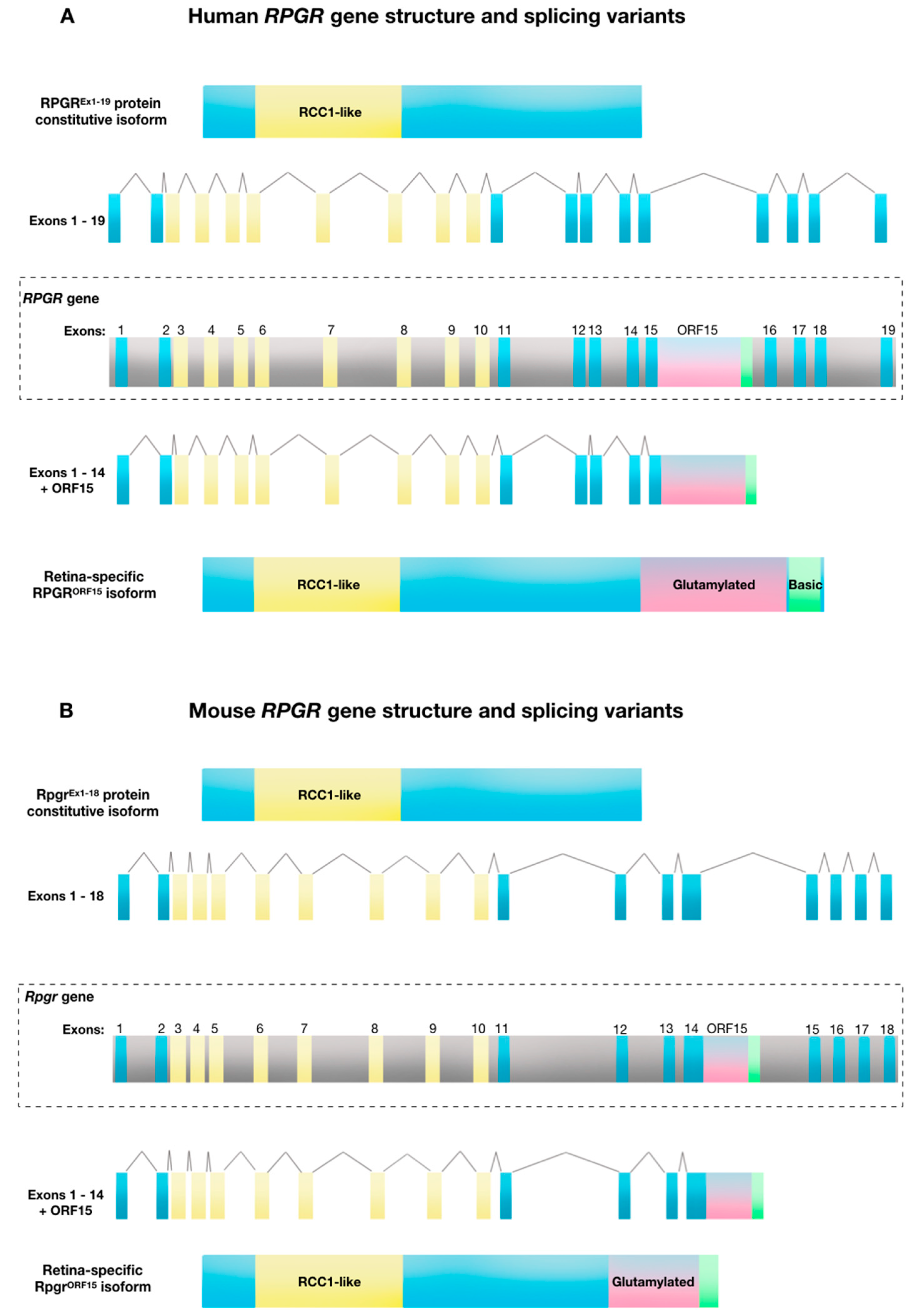
2. Structure and Function of Retinitis Pigmentosa GTPase Regulator (RPGR)
3. Molecular Mechanisms and Pathogenesis of RPGR-Related X-Linked Retinitis Pigmentosa (RP)
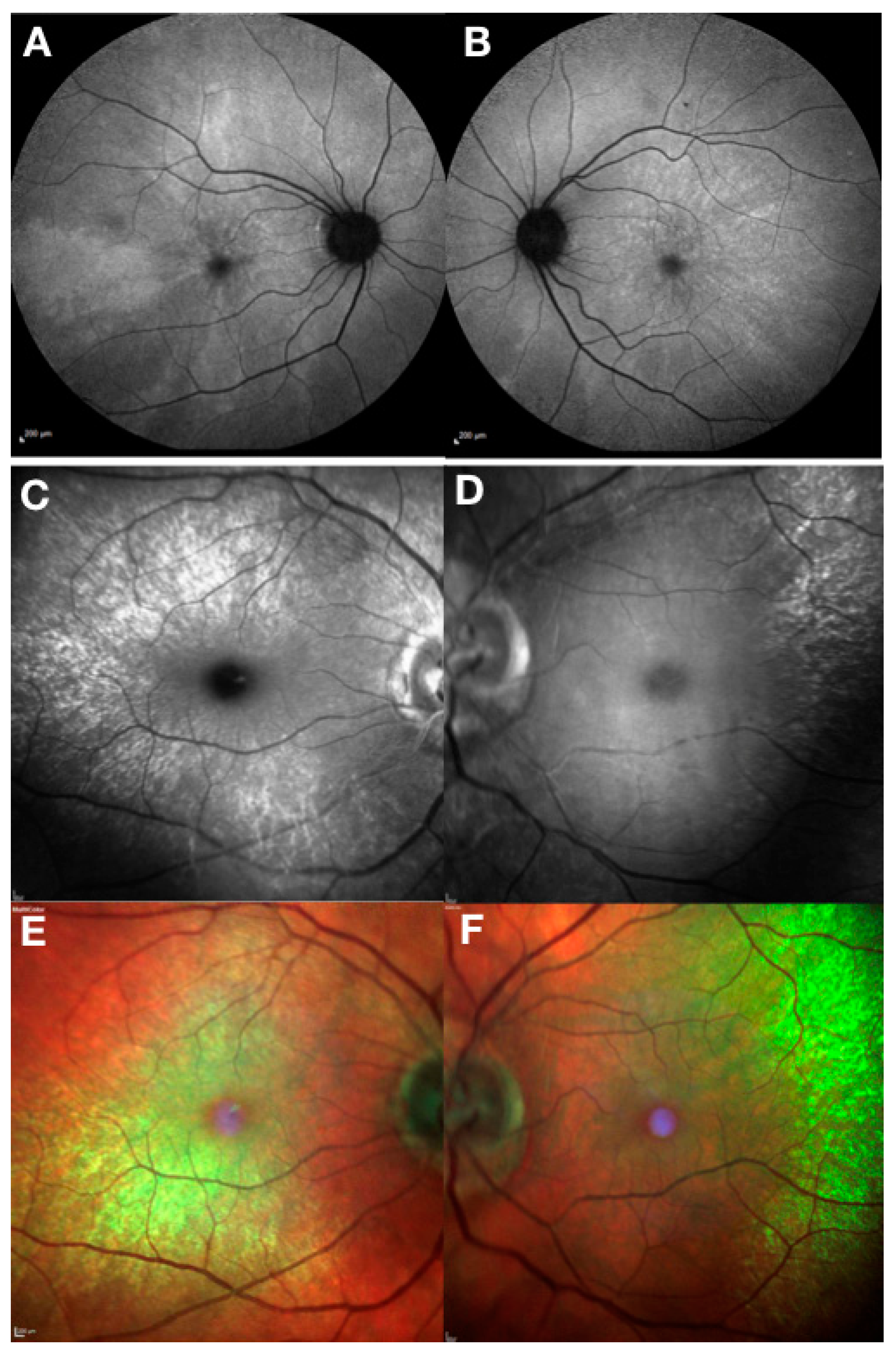
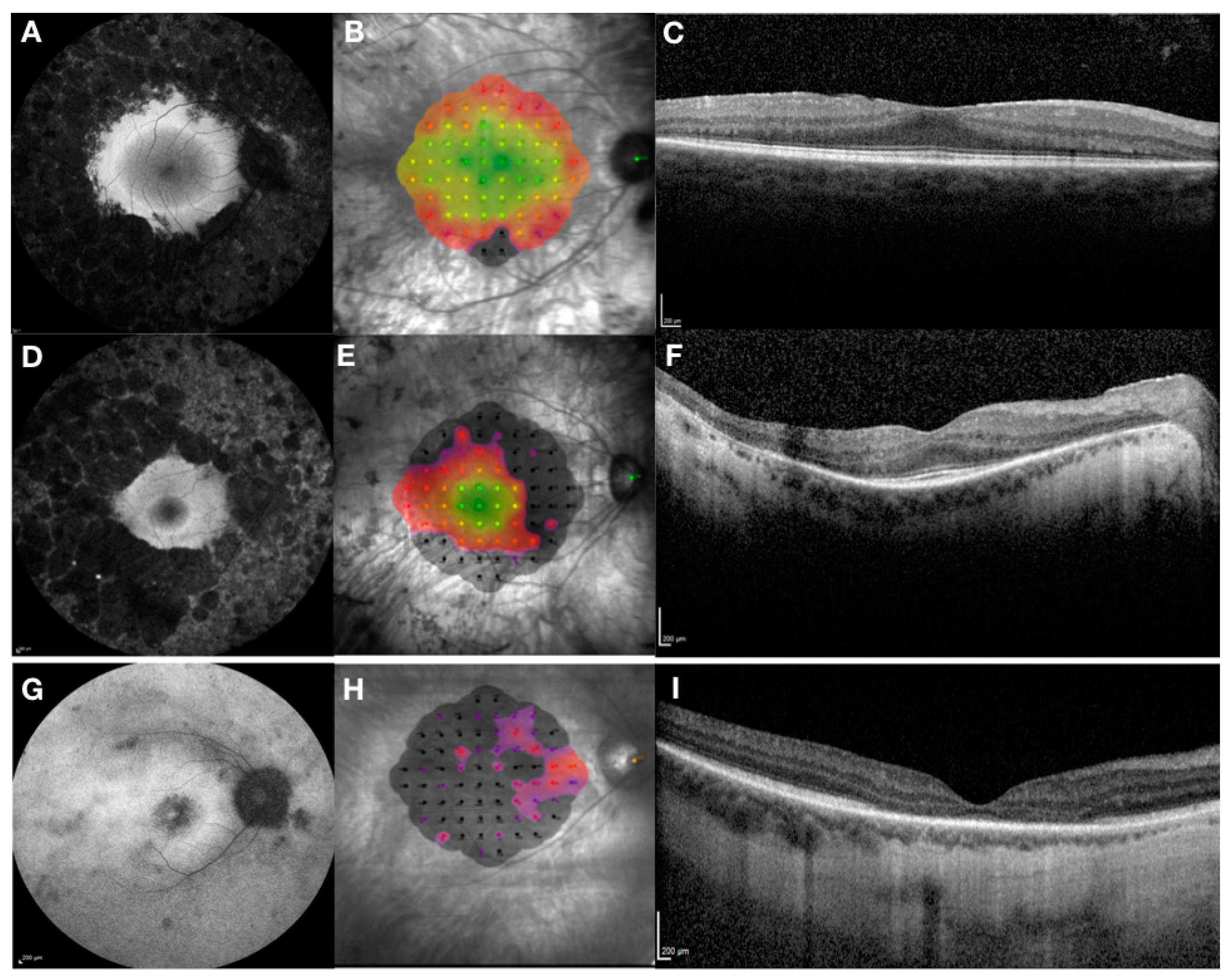
4. Clinical and Genetic Diagnosis of RPGR-Related X-Linked RP
5. Treatment Options for RPGR-Related X-Linked RP
6. Gene Therapy Clinical Trials for RPGR-Related X-Linked RP
7. Summary
Funding
Conflicts of Interest
References
- Tee, J.J.; Smith, A.J.; Hardcastle, A.J.; Michaelides, M. RPGR-associated retinopathy: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2016, 100, 1022–1027. [Google Scholar] [CrossRef]
- Talib, M.; van Schooneveld, M.J.; Thiadens, A.A.; Fiocco, M.; Wijnholds, J.; Florijn, R.J.; Schalij-Delfos, N.E.; van Genderen, M.M.; Putter, H.; Cremers, F.P.M.; et al. Clinical and genetic characteristics of male patients with RPGR-associated retinal dystrophies: A long-term follow-up study. Retina 2019, 39, 1186–1199. [Google Scholar] [CrossRef]
- Pelletier, V.; Jambou, M.; Delphin, N.; Zinovieva, E.; Stum, M.; Gigarel, N.; Dollfus, H.; Hamel, C.; Toutain, A.; Dufier, J.L.; et al. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: Genotype-phenotype correlations and impact on genetic counseling. Hum. Mutat. 2007, 28, 81–91. [Google Scholar] [CrossRef]
- Branham, K.; Othman, M.; Brumm, M.; Karoukis, A.J.; Atmaca-Sonmez, P.; Yashar, B.M.; Schwartz, S.B.; Stover, N.B.; Trzupek, K.; Wheaton, D.; et al. Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8232–8237. [Google Scholar] [CrossRef]
- Webb, T.R.; Parfitt, D.A.; Gardner, J.C.; Martinez, A.; Bevilacqua, D.; Davidson, A.E.; Zito, I.; Thiselton, D.L.; Ressa, J.H.; Apergi, M.; et al. Deep intronic mutation in OFD1, identified by targeted genomic next-generation sequencing, causes a severe form of X-linked retinitis pigmentosa (RP23). Hum. Mol. Genet. 2012, 21, 3647–3654. [Google Scholar] [CrossRef]
- Comander, J.; Weigel-DiFranco, C.; Sandberg, M.A.; Berson, E.L. Visual function in carriers of X-linked retinitis pigmentosa. Ophthalmology 2015, 122, 1899–1906. [Google Scholar] [CrossRef]
- Nanda, A.; Salvetti, A.P.; Clouston, P.; Downes, S.M.; MacLaren, R.E. Exploring the Variable Phenotypes of RPGR Carrier Females in Assessing their Potential for Retinal Gene Therapy. Genes 2018, 9, 643. [Google Scholar] [CrossRef]
- Wu, H.; Luo, J.; Yu, H.; Rattner, A.; Mo, A.; Wang, Y.; Smallwood, P.M.; Erlanger, B.; Wheelan, S.J.; Nathans, J. Cellular resolution maps of X chromosome inactivation: Implications for neural development, function, and disease. Neuron 2014, 81, 103–119. [Google Scholar] [CrossRef]
- Raghupathy, R.K.; Gautier, P.; Soares, D.C.; Wright, A.F.; Shu, X. Evolutionary characterization of the retinitis pigmentosa GTPase regulator gene. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6255–6264. [Google Scholar] [CrossRef]
- Vervoort, R.; Lennon, A.; Bird, A.C.; Tulloch, B.; Axton, R.; Miano, M.G.; Meindl, A.; Meitinger, T.; Ciccodicola, A.; Wright, A.F. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat. Genet. 2000, 25, 462–466. [Google Scholar] [CrossRef]
- Vervoort, R.; Wright, A.F. Mutations of RPGR in X-linked retinitis pigmentosa (RP3). Hum. Mutat. 2002, 19, 486–500. [Google Scholar] [CrossRef]
- Megaw, R.D.; Soares, D.C.; Wright, A.F. RPGR: Its role in photoreceptor physiology, human disease, and future therapies. Exp. Eye Res. 2015, 138, 32–41. [Google Scholar] [CrossRef]
- Pawlyk, B.S.; Bulgakov, O.V.; Sun, X.; Adamian, M.; Shu, X.; Smith, A.J.; Berson, E.L.; Ali, R.R.; Khani, S.; Wright, A.F.; et al. Photoreceptor rescue by an abbreviated human RPGR gene in a murine model of X-linked retinitis pigmentosa. Gene. Ther. 2015, 23, 196–204. [Google Scholar] [CrossRef]
- Fischer, M.D.; McClements, M.E.; Martinez-Fernandez De La Camara, C.; Bellingrath, J.S.; Dauletbekov, D.; Ramsden, S.C.; Hickey, D.G.; Barnard, A.R.; MacLaren, R.E. Codon-optimized RPGR improves stability and efficacy of AAV8 gene therapy in two mouse models of X-linked retinitis pigmentosa. Mol. Ther. 2017, 25, 1854–1865. [Google Scholar] [CrossRef]
- Beltran, W.A.; Cideciyan, A.V.; Boye, S.E.; Ye, G.J.; Iwabe, S.; Dufour, V.L.; Marinho, L.F.; Swider, M.; Kosyk, M.S.; Sha, J.; et al. Optimization of retinal gene therapy for X-linked retinitis pigmentosa due to RPGR mutations. Mol. Ther. 2017, 25, 1866–1880. [Google Scholar] [CrossRef]
- Hong, D.H.; Li, T. Complex expression pattern of RPGR reveals a role for purine-rich exonic splicing enhancers. Investig. Ophthalmol. Vis. Sci. 2002, 43, 3373–3382. [Google Scholar]
- Meindl, A.; Dry, K.; Herrmann, K.; Manson, E.; Ciccodicola, A.; Edgar, A.; Carvalho, M.R.; Achatz, H.; Hellebrand, H.; Lennon, A.; et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X–linked retinitis pigmentosa (RP3). Nat. Genet. 1996, 13, 35–42. [Google Scholar] [CrossRef]
- Kirschner, R.; Erturk, D.; Zeitz, C.; Sahin, S.; Ramser, J.; Cremers, F.P.; Ropers, H.H.; Berger, W. DNA sequence comparison of human and mouse retinitis pigmentosa GTPase regulator (RPGR) identifies tissue-specific exons and putative regulatory elements. Hum. Genet. 2001, 109, 271–278. [Google Scholar] [CrossRef]
- Hong, D.H.; Pawlyk, B.; Sokolov, M.; Strissel, K.J.; Yang, J.; Tulloch, B.; Wright, A.F.; Arshavsky, V.Y.; Li, T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2413–2421. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Zhao, H.; Ferreira, P.A. Species-specific subcellular localization of RPGR and RPGRIP isoforms: Implications for the phenotypic variability of congenital retinopathies among species. Hum. Mol. Genet. 2002, 11, 1899–1907. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.A.; Atkins, S.J.; Peranen, J.; Swaroop, A.; Khanna, H. Interaction of retinitis pigmentosa GTPase regulator (RPGR) with RAB8A GTPase: Implications for cilia dysfunction and photoreceptor degeneration. Hum. Mol. Genet. 2010, 19, 3591–3598. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.-H.; Zhang, K.; Chen, C.-K.; Frederick, J.M.; Prestwich, G.D.; Baehr, W. Photoreceptor cGMP phosphodiesterase delta subunit (PDEδ) functions as a prenyl-binding protein. J. Biol. Chem. 2004, 279, 407–413. [Google Scholar] [CrossRef]
- Khanna, H.; Hurd, T.W.; Lillo, C.; Shu, X.; Parapuram, S.K.; He, S.; Akimoto, M.; Wright, A.F.; Margolis, B.; Williams, D.S.; et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J. Biol. Chem. 2005, 280, 33580–33587. [Google Scholar] [CrossRef]
- Otto, E.A.; Loeys, B.; Khanna, H.; Hellemans, J.; Sudbrak, R.; Fan, S.; Muerb, U.; O’Toole, J.F.; Helou, J.; Attanasio, M.; et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior-Loken syndrome and interacts with RPGR and calmodulin. Nat. Genet. 2005, 37, 282–288. [Google Scholar] [CrossRef]
- Chang, B.; Khanna, H.; Hawes, N.; molecular, D.J.H. In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse. Hum. Mol. Genet. 2006, 15, 1847–1857. [Google Scholar] [CrossRef]
- Wright, R.N.; Hong, D.-H.; Perkins, B. RpgrORF15 Connects to the usher protein network through direct interactions with multiple whirlin isoforms. Investig. Opthalmol. Vis. Sci. 2012, 53, 1519. [Google Scholar] [CrossRef]
- Rao, K.N.; Anand, M.; Khanna, H. The carboxyl terminal mutational hotspot of the ciliary disease protein RPGRORF15 (retinitis pigmentosa GTPase regulator) is glutamylated in vivo. Biol. Open 2016, 5, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, K.; Gadadhar, S.; Souphron, J.; Magiera, M.M.; Janke, C. Molecular interactions between tubulin tails and glutamylases reveal determinants of glutamylation patterns. EMBO Rep. 2017, 18, 1013–1026. [Google Scholar] [CrossRef]
- Sun, X.; Park, J.H.; Gumerson, J.; Wu, Z.; Swaroop, A.; Qian, H.; Roll-Mecak, A.; Li, T. Loss of RPGR glutamylation underlies the pathogenic mechanism of retinal dystrophy caused by TTLL5 mutations. Proc. Natl. Acad. Sci. USA 2016, 113, E2925–E2934. [Google Scholar] [CrossRef]
- Shu, X.; McDowall, E.; Brown, A.F.; Wright, A.F. The human retinitis pigmentosa GTPase regulator gene variant database. Hum. Mutat. 2008, 29, 605–608. [Google Scholar] [CrossRef]
- Hosch, J.; Lorenz, B.; Stieger, K. RPGR: Role in the photoreceptor cilium, human retinal disease, and gene therapy. Ophthalmic Genet. 2011, 32, 1–11. [Google Scholar] [CrossRef]
- Iannaccone, A.; Breuer, D.K.; Wang, X.F.; Kuo, S.F.; Normando, E.M.; Filippova, E.; Baldi, A.; Hiriyanna, S.; MacDonald, C.B.; Baldi, F.; et al. Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation. J. Med. Genet. 2003, 40, e118. [Google Scholar] [CrossRef]
- Zito, I.; Downes, S.M.; Patel, R.J.; Cheetham, M.E.; Ebenezer, N.D.; Jenkins, S.A.; Bhattacharya, S.S.; Webster, A.R.; Holder, G.E.; Bird, A.C.; et al. RPGR mutation associated with retinitis pigmentosa, impaired hearing, and sinorespiratory infections. J. Med. Genet. 2003, 40, 609–615. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.G.; Fishman, G.A.; Kretzer, F.L. Abnormal axonemes in X-linked retinitis pigmentosa. Arch. Ophthal. 1988, 106, 362–368. [Google Scholar] [CrossRef]
- Hunter, D.G.; Fishman, G.A.; Mehta, R.S.; Kretzer, F.L. Abnormal sperm and photoreceptor axonemes in Usher’s syndrome. Arch. Ophthalmol. 1986, 104, 385–389. [Google Scholar] [CrossRef]
- He, S.; Parapuram, S.K.; Hurd, T.W.; Behnam, B.; Margolis, B.; Swaroop, A.; Khanna, H. Retinitis pigmentosa GTPase regulator (RPGR) protein isoforms in mammalian retina: Insights into X-linked retinitis pigmentosa and associated ciliopathies. Vision Res. 2008, 48, 366–376. [Google Scholar] [CrossRef]
- Jacobi, F.K.; Karra, D.; Broghammer, M.; Blin, N.; Pusch, C.M. Mutational risk in highly repetitive exon ORF15 of the RPGR multidisease gene is not associated with haplotype background. Int. J. Mol. Med. 2005, 16, 1175–1178. [Google Scholar] [CrossRef]
- Karra, D.; Jacobi, F.K.; Broghammer, M.; Blin, N.; Pusch, C.M. Population haplotypes of exon ORF15 of the retinitis pigmentosa GTPase regulator gene in Germany: Implications for screening for inherited retinal disorders. Mol. Diagn. Ther. 2006, 10, 115–123. [Google Scholar] [CrossRef]
- Sergouniotis, P.I.; Chakarova, C.; Murphy, C.; Becker, M.; Lenassi, E.; Arno, G.; Lek, M.; MacArthur, D.G.; Bhattacharya, S.S.; Moore, A.T.; et al. UCL-Exomes Consortium Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am. J. Hum. Genet. 2014, 94, 760–769. [Google Scholar] [CrossRef]
- Sandberg, M.A.; Rosner, B.; Weigel-DiFranco, C.; Dryja, T.P.; Berson, E.L. Disease course of patients with X-linked retinitis pigmentosa due to RPGR gene mutations. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1298–1304. [Google Scholar] [CrossRef]
- Huang, W.C.; Wright, A.F.; Roman, A.J.; Cideciyan, A.V.; Manson, F.D.; Gewaily, D.Y.; Schwartz, S.B.; Sadigh, S.; Limberis, M.P.; Bell, P.; et al. RPGR-associated retinal degeneration in human X-linked RP and a murine model. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5594–5608. [Google Scholar] [CrossRef]
- Mitamura, Y.; Mitamura-Aizawa, S.; Nagasawa, T.; Katome, T.; Eguchi, H.; Naito, T. Diagnostic imaging in patients with retinitis pigmentosa. J. Med. Investig. 2012, 59, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Tee, J.J.L.; Kalitzeos, A.; Webster, A.R.; Peto, T.; Michaelides, M. Quantitative analysis of hyperautofluorescent rings to characterize the natural history and progression in RPGR-associated retinopathy. Retina 2018, 38, 2401–2414. [Google Scholar] [CrossRef]
- Robson, A.G.; Michaelides, M.; Luong, V.A.; Holder, G.E.; Bird, A.C.; Webster, A.R.; Moore, A.T.; Fitzke, F.W. Functional correlates of fundus autofluorescence abnormalities in patients with RPGR or RIMS1 mutations causing cone or cone rod dystrophy. Br. J. Ophthalmol. 2008, 92, 95–102. [Google Scholar] [CrossRef]
- Zhang, Q.; Giacalone, J.C.; Searby, C.; Stone, E.M.; Tucker, B.A.; Sheffield, V.C. Disruption of RPGR protein interaction network is the common feature of RPGR missense variations that cause XLRP. Proc. Natl. Acad. Sci. USA 2019, 116, 1353–1360. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, D.R.; Hughbanks-Wheaton, D.K.; Pearson, N.S.; Fish, G.E.; Spencer, R.; Takacs, A.; Klein, M.; Locke, K.G.; Birch, D.G. Four-year placebo-controlled trial of docosahexaenoic acid in X-linked retinitis pigmentosa (DHAX trial): A randomized clinical trial. JAMA Ophthalmol. 2014, 132, 866–873. [Google Scholar] [CrossRef]
- Beltran, W.A.; Wen, R.; Acland, G.M.; Aguirre, G.D. Intravitreal injection of ciliary neurotrophic factor (CNTF) causes peripheral remodeling and does not prevent photoreceptor loss in canine RPGR mutant retina. Exp. Eye Res. 2007, 84, 753–771. [Google Scholar] [CrossRef] [Green Version]
- Ho, A.C.; Humayun, M.S.; Dorn, J.D.; da Cruz, L.; Dagnelie, G.; Handa, J.; Barale, P.O.; Sahel, J.A.; Stanga, P.E.; Hafezi, F.; et al. Long-term results from an epiretinal prosthesis to restore sight to the blind. Ophthalmology 2015, 122, 1547–1554. [Google Scholar] [CrossRef]
- Edwards, T.L.; Cottriall, C.L.; Xue, K.; Simunovic, M.P.; Ramsden, J.D.; Zrenner, E.; MacLaren, R.E. Assessment of the Electronic Retinal Implant α AMS in Restoring Vision to Blind Patients with End-Stage Retinitis Pigmentosa. Ophthalmology 2018, 125, 432–443. [Google Scholar] [CrossRef]
- Cehajic-Kapetanovic, J.; Eleftheriou, C.; Allen, A.E.; Milosavljevic, N.; Pienaar, A.; Bedford, R.; Davis, K.E.; Bishop, P.N.; Lucas, R.J. Restoration of Vision with Ectopic Expression of Human Rod Opsin. Curr. Biol. 2015, 25, 2111–2122. [Google Scholar] [CrossRef] [Green Version]
- Eleftheriou, C.G.; Cehajic-Kapetanovic, J.; Martial, F.P.; Milosavljevic, N.; Bedford, R.A.; Lucas, R.J. Meclofenamic acid improves the signal to noise ratio for visual responses produced by ectopic expression of human rod opsin. Mol. Vis. 2017, 23, 334–345. [Google Scholar]
- Hong, D.H.; Pawlyk, B.S.; Adamian, M.; Sandberg, M.A.; Li, T. A single, abbreviated RPGR-ORF15 variant reconstitutes RPGR function in vivo. Investig. Ophthalmol. Vis. Sci. 2005, 46, 435–441. [Google Scholar] [CrossRef]
- Deng, W.T.; Dyka, F.M.; Dinculescu, A.; Li, J.; Zhu, P.; Chiodo, V.A.; Boye, S.L.; Conlon, T.J.; Erger, K.; Cossette, T.; et al. Stability and Safety of an AAV Vector for Treating RPGR-ORF15 X-Linked Retinitis Pigmentosa. Hum. Gene. Ther. 2015, 26, 593–602. [Google Scholar] [CrossRef]
- Wu, Z.; Hiriyanna, S.; Qian, H.; Mookherjee, S.; Campos, M.M.; Gao, C.; Fariss, R.; Sieving, P.A.; Li, T.; Colosi, P.; et al. A long-term efficacy study of gene replacement therapy for RPGR-associated retinal degeneration. Hum. Mol. Genet. 2015, 24, 3956–3970. [Google Scholar] [CrossRef] [Green Version]
- Beltran, W.A.; Cideciyan, A.V.; Lewin, A.S.; Iwabe, S.; Khanna, H.; Sumaroka, A.; Chiodo, V.A.; Fajardo, D.S.; Román, A.J.; Deng, W.T.; et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2012, 109, 2132–2137. [Google Scholar] [CrossRef] [Green Version]
- Giacalone, J.C.; Andorf, J.L.; Zhang, Q.; Burnight, E.R.; Ochoa, D.; Reutzel, A.J.; Collins, M.M.; Sheffield, V.C.; Mullins, R.F.; Han, I.C.; et al. Development of a Molecularly Stable Gene Therapy Vector for the Treatment of RPGR-Associated X-Linked Retinitis Pigmentosa. Hum. Gene. Ther. 2019, 30, 967–974. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
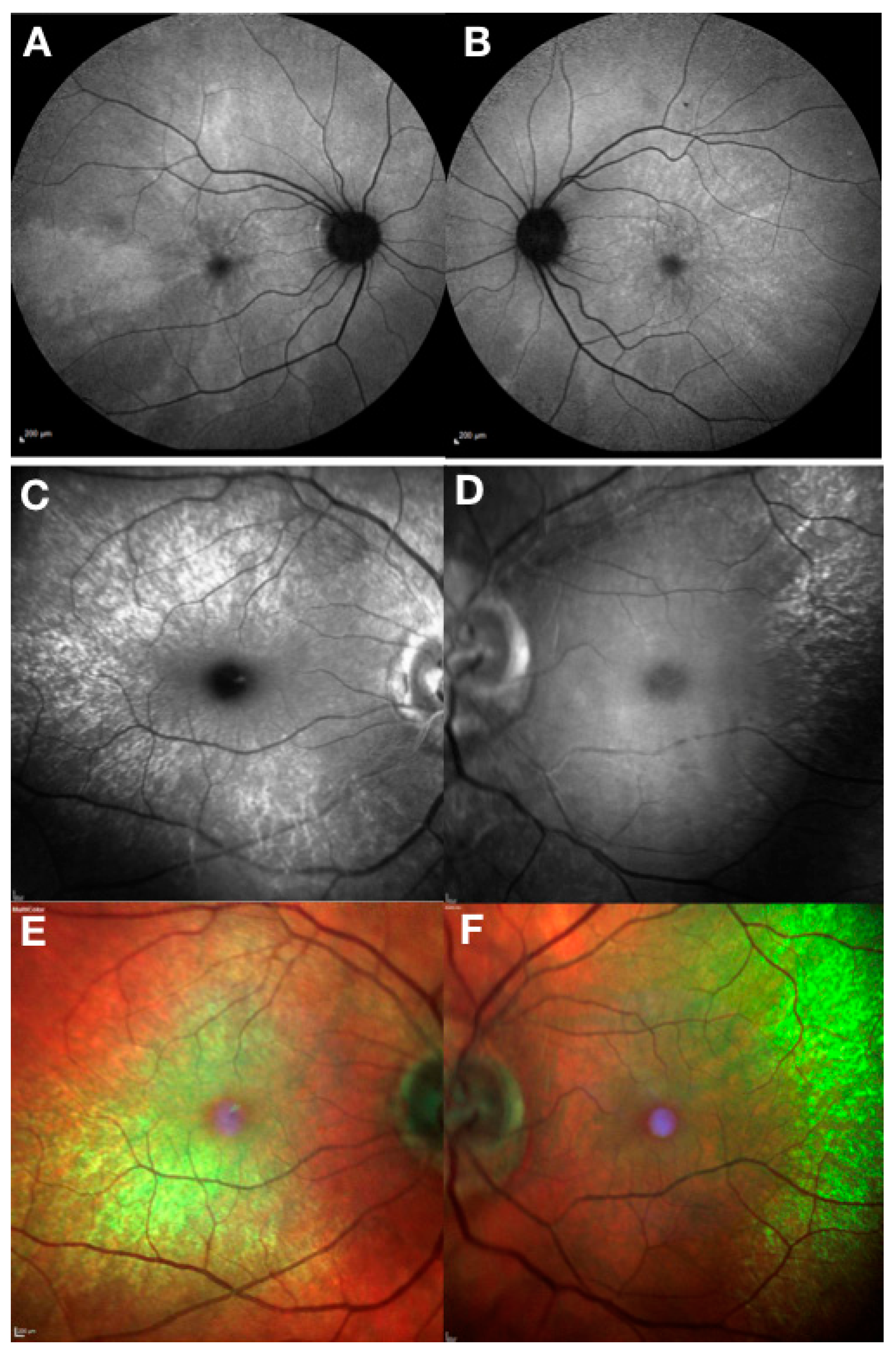{kind=link}
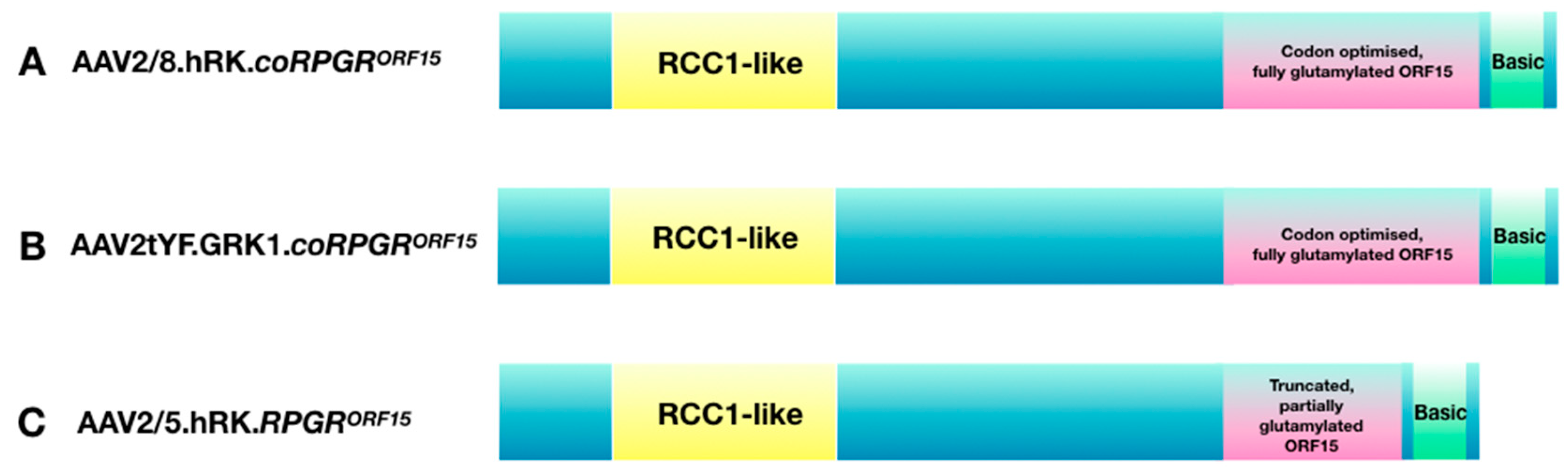{kind=link}
| DNA Sequence | Amino Acid Sequence | |||||
|---|---|---|---|---|---|---|
| Species | Coding Sequence Prior to ORF15 | Percentage of Purine Bases | Region with Homology to Human ORF15 * | Percentage of Purine Bases | ORF15 Amino Acid Length (Percentage Glu-Gly) * | Glutamylation Region (Percentage Glu-Gly) * |
| Homo sapiens | 1 to 14 | 54% | ORF15 | 89% | 567 (67%) | 351 (88%) |
| 1.7 kb | 1.7 kb | |||||
| Mus musculus | 1 to 14 | 57% | Intron 14 | 86% | 488 (60%) | 273 (84%) |
| 2.5 kb | 1.5 kb | |||||
| Canis lupus familiaris | 1 to 13 | 58% | Exon 14/Intron 14 | 88% | 522 (66%) | 331 (72%) |
| 2.5 kb | 1.5 kb | |||||
| Pan troglodytes | 1 to 14 | 54% | Exon 15/Intron 15 | 89% | 560 (66%) | 330 (88%) |
| 1.7 kb | 1.7 kb | |||||
| Gorilla gorilla gorilla | 1 to 14 | 54% | Exon 15/Intron 15 | 89% | 549 (66%) | 321 (88%) |
| 1.7 kb | 1.7 kb | |||||
| Macaca mulatta | 1 to 14 | 53% | Exon 15/Intron 15 | 89% | 549 (65%) | 323 (86%) |
| 1.7 kb | 1.7 kb | |||||
| Xenopus tropicalis | 1 to 13 | 57% | Exon 14/Intron 14/Exon 15 | 77% | 679 (45%) | 232 (82%) |
| 1.6 kb | 2.0 kb | |||||
| Clinical Trial (clinicaltrials.gov) | Intervention/Observation | Clinical Centre/s | Sponsor |
|---|---|---|---|
| Phase I/II/III NCT03116113 multicenter, open-label Part 1: non-randomised, dose-selection study 18 participants Part 2: dose expansion study (randomised to low dose, high dose, control) 63 participants Start date: March 2017 | Subretinal delivery of AAV8-hRK-coRPGRORF15 | Oxford, UK Manchester, UK Southampton, UK Florida, USA Oregon, USA Pennsylvania, USA | Nightstar Therapeutics (now Biogen Inc), UK |
| Phase I/II NCT03252847 Non-randomised, open-label, dose-escalation trial 36 participants Start date: July 2017 | Subretinal delivery of AAV2/5-hRK-RPGRORF15 | London, UK | MeiraGTx, UK |
| Phase I/II NCT03316560 Non-randomised, open-label, multicenter, dose-escalation trial 30 participants with RPGR ORF15 mutations Start date: April 2018 | Subretinal delivery of rAAV2tYF-GRK1-coRPGRORF15 | Colorado, USA Massachusetts, USA New York, USA North Carolina, USA Ohio, USA Oregon, USA Pennsylvania, USA Texas, USA | Applied Genetic Technologies Corporation (AGTC), USA |
| Prospective natural history study of XLRP with genetically confirmed mutation in RPGR 150 participants Start date: December 2017 | Observational study | Multiple centres in UK, Germany, Holland, France, USA | Nightstar Therapeutics (now Biogen Inc), UK |
| Prospective natural history study of XLRP NCT03349242 Start date: December 2017 | Observational study | Massachusetts, USA Michigan, USA | MeiraGTx, UK |
| Prospective natural history study of XLRP caused by RPGR-ORF15 mutations 45 participants NCT03314207 Start date: December 2017 | Observational study | New York, USA North Carolina, USA Ohio, USA Oregon, USA Texas, USA | Applied Genetic Technologies Corporation (AGTC), USA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cehajic Kapetanovic, J.; McClements, M.E.; Martinez-Fernandez de la Camara, C.; MacLaren, R.E. Molecular Strategies for RPGR Gene Therapy. Genes 2019, 10, 674. https://doi.org/10.3390/genes10090674
Cehajic Kapetanovic J, McClements ME, Martinez-Fernandez de la Camara C, MacLaren RE. Molecular Strategies for RPGR Gene Therapy. Genes. 2019; 10(9):674. https://doi.org/10.3390/genes10090674
Chicago/Turabian StyleCehajic Kapetanovic, Jasmina, Michelle E McClements, Cristina Martinez-Fernandez de la Camara, and Robert E MacLaren. 2019. "Molecular Strategies for RPGR Gene Therapy" Genes 10, no. 9: 674. https://doi.org/10.3390/genes10090674
APA StyleCehajic Kapetanovic, J., McClements, M. E., Martinez-Fernandez de la Camara, C., & MacLaren, R. E. (2019). Molecular Strategies for RPGR Gene Therapy. Genes, 10(9), 674. https://doi.org/10.3390/genes10090674