Construction of High-Density Genetic Map and Identification of QTLs Associated with Seed Vigor after Exposure to Artificial Aging Conditions in Sweet Corn Using SLAF-seq

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Plant Population
2.2. Artificial Aging Treatment
2.3. Experimental Design and Phenotypic Evaluation
2.4. SLAF Library Construction
2.5. Grouping and Genotyping of Sequence Data
2.6. Linkage Map Construction
2.7. QTL Analysis
3. Results
3.1. Analysis of SLAF-seq Data
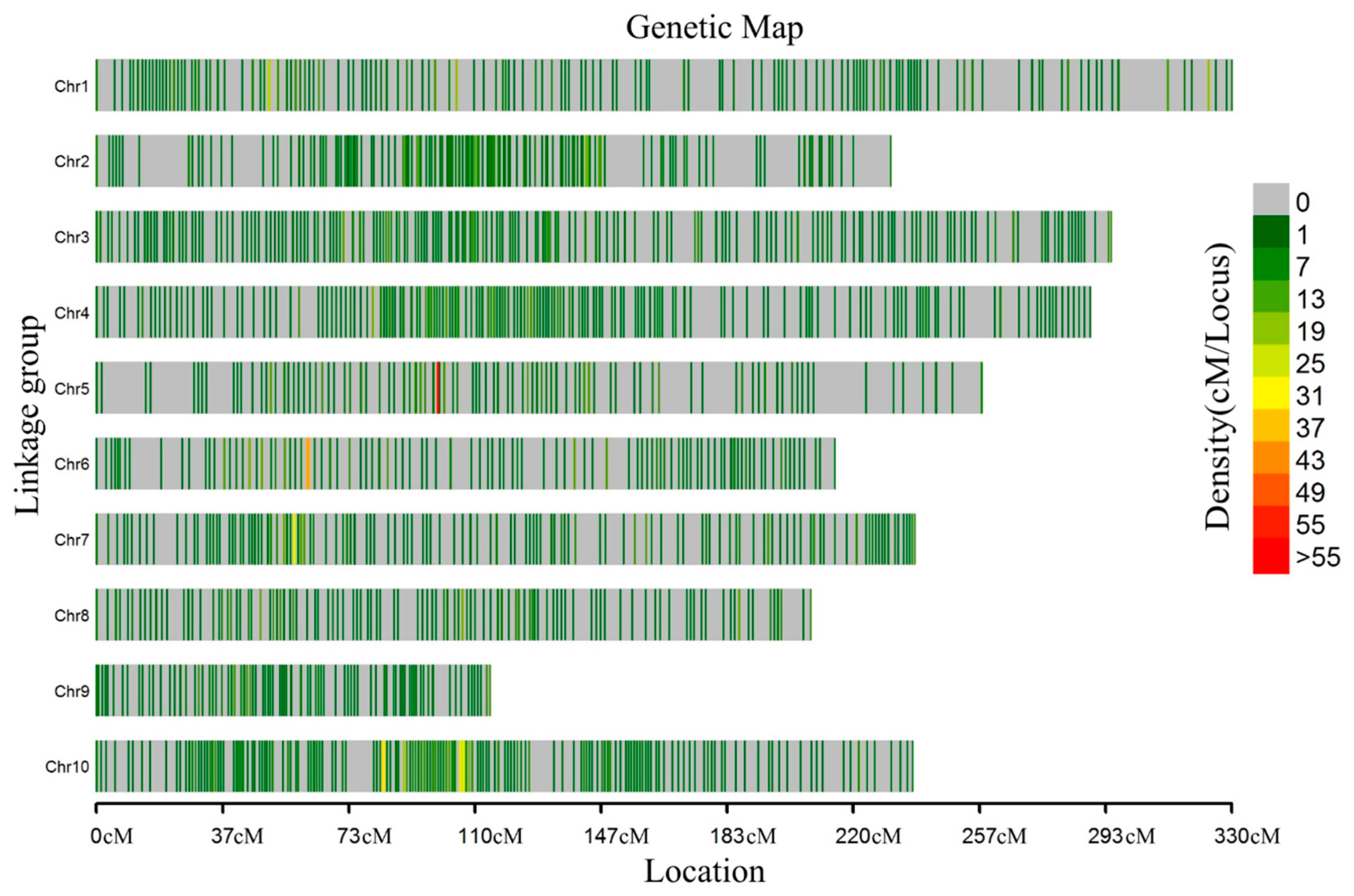
3.2. High-Density Genetic Map Construction
3.3. Quality and Accuracy of the Map
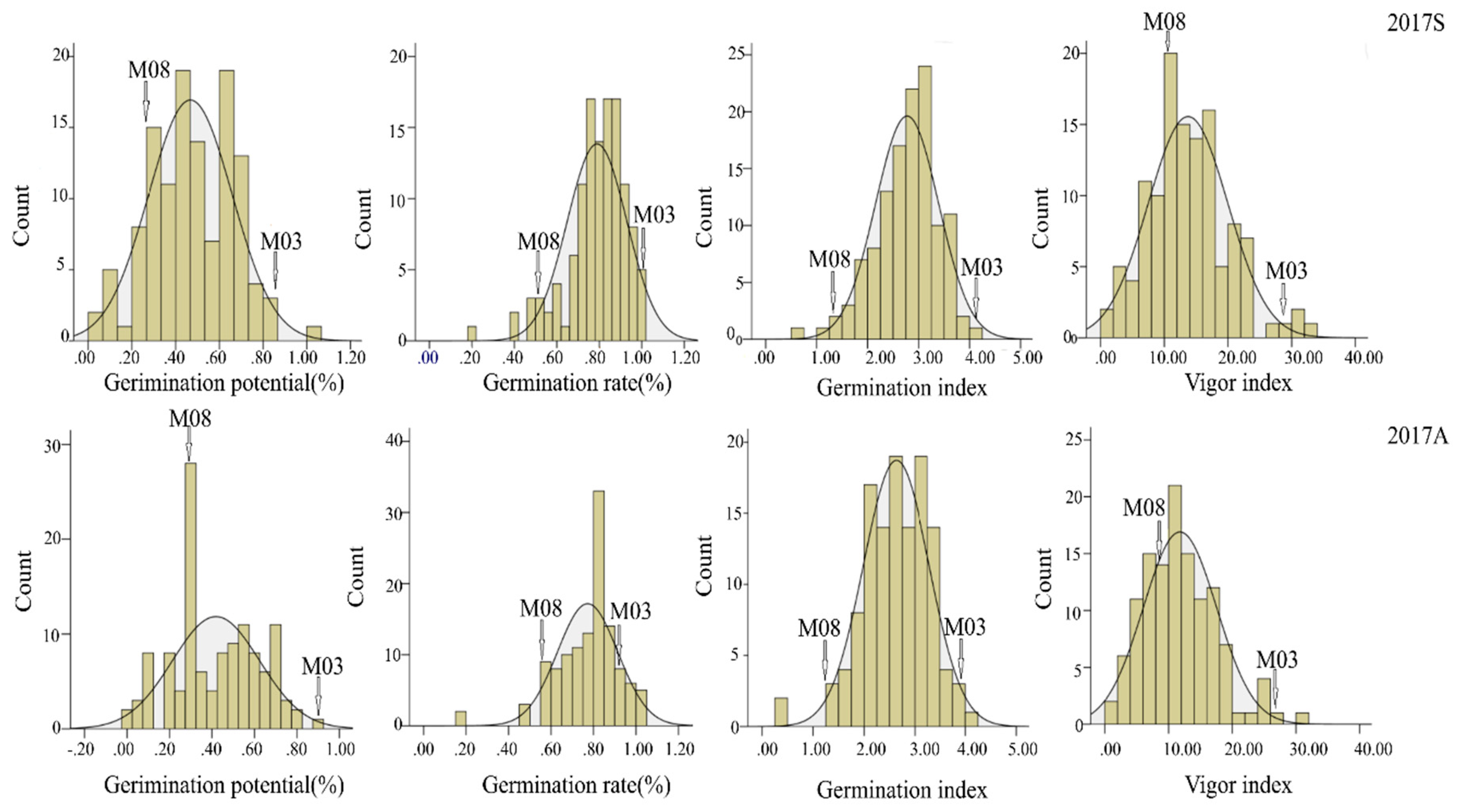
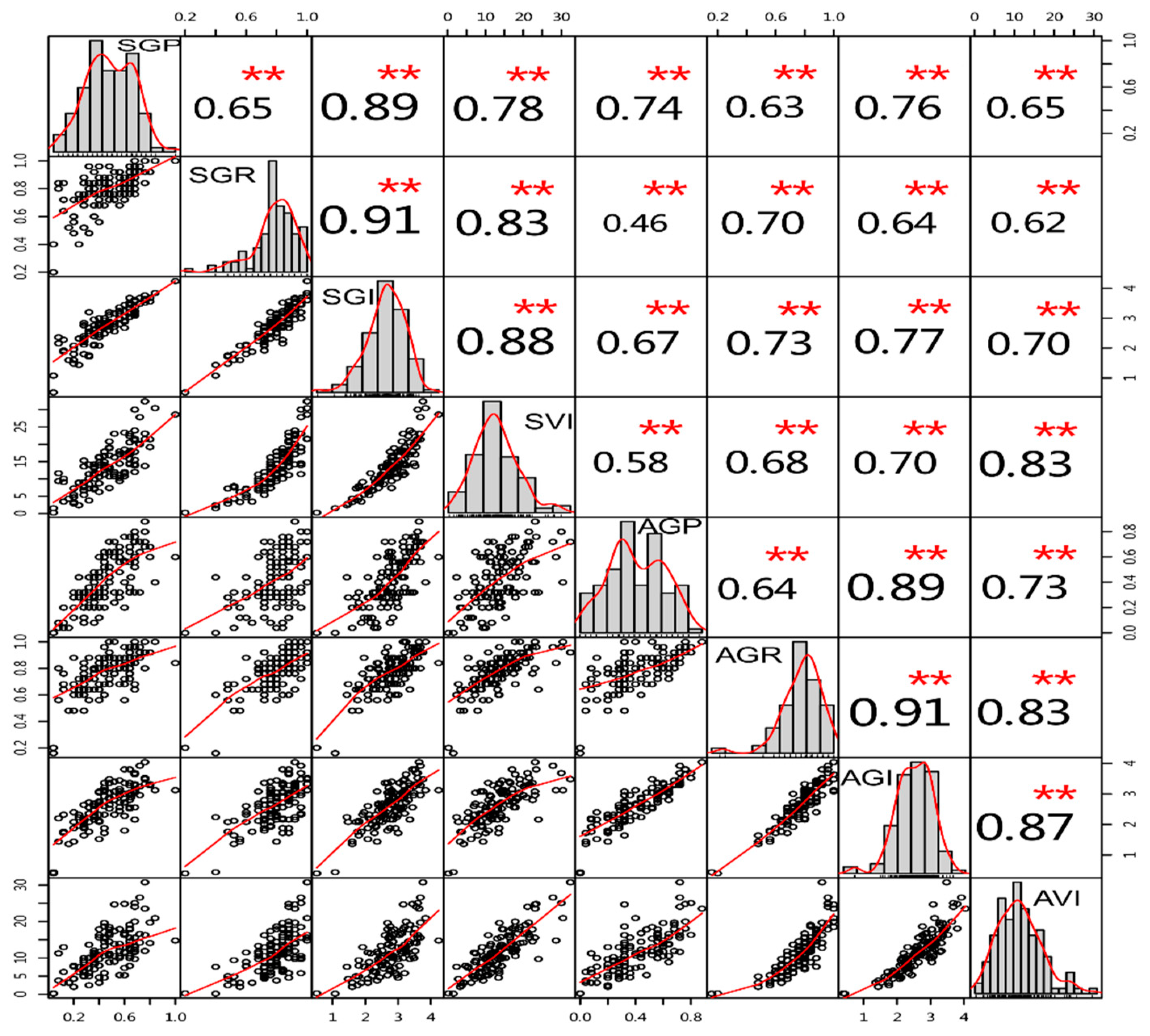
3.4. Phenotypic Analysis
3.5. QTL Analysis of Traits Associated with Seed Vigor in the BC4F3 Population
3.6. The Candidate Gene Analysis
4. Discussion
4.1. Characteristics of the SLAF-seq Method
4.2. QTL Mapping of Seed Vigor Related Traits Compared with Other Studies
4.3. Candidate Genes Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luo, J.; Wang, Z.; Tan, J.; Hu, J.; Yin, Y. Development situation and countermeasures of Guangdong sweet corn industry in 2013. Guangdong Agric. Sci. 2014, 5, 42–45. [Google Scholar] [CrossRef]
- Liu, D.; Ma, C.; Hong, W.; Huang, L.; Liu, M.; Zeng, H.; Deng, D.; Xin, H.; Song, J.; Xu, C.; et al. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS ONE 2014, 9, e98855. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Fan, S.; Liu, J.; Wang, Q.; Li, M.; Jiang, X.; Liu, Z.; Yin, Y. Identification of QTLs for seed storability in rice under natural aging conditions using two RILs with the same parent shennong 265. J. Integr. Agric. 2017, 16, 1084–1092. [Google Scholar] [CrossRef]
- Li, C.; Shao, G.; Wang, L.; Wang, Q.; Mao, X.; Wang, X.; Zhang, X.; Liu, S. QTL identification and fine mapping for seed storability in rice (Oryza sativa L.). Euphytica 2017, 213, 127–137. [Google Scholar] [CrossRef]
- Soltani, A. Genetic variation for and interrelationships among seed vigor traits in wheat from the Caspian Sea coast of Iran. Seed Sci. Technol. 2011, 29, 653–662. [Google Scholar]
- Landjeva, S.; Lohwasser, U.; Börner, A. Genetic mapping within the wheat D genome reveals QTL for germination, seed vigour and longevity, and early seedling growth. Euphytica 2010, 171, 129–143. [Google Scholar] [CrossRef]
- Mano, Y.; Takeda, K. Mapping quantitative trait loci for salt tolerance at germination and the seedling stage in barley (Hordeum vulgare L.). Euphytica 1997, 94, 263–272. [Google Scholar] [CrossRef]
- Styer, R.; Cantliffe, D.J. Dependence of seed vigor during germination on carbohydrate source in endosperm mutants of maize. Plant Physiol. 1984, 76, 196–200. [Google Scholar] [CrossRef] [Green Version]
- Tekrony, D.M.; Hunter, J.L. Effect of seed maturation and genotype on seed vigor in maize. Crop Sci. 1995, 35, 857–862. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Yu, Z.; Wang, Y.; Yang, Y.; Liu, Z.; Jiang, J.; Song, M.; Wu, Y. Superior storage stability in low lipoxygenase maize varieties. J. Stored Prod. Res. 2007, 43, 530–534. [Google Scholar] [CrossRef]
- Zhang, Z.; Shang, H.; Shi, Y.; Huang, L.; Li, J.; Ge, Q.; Gong, J.; Liu, A.; Chen, T.; Wang, D.; et al. Construction of a high-density genetic map by specific locus amplified fragment sequencing (SLAF-seq) and its application to Quantitative Trait Loci (QTL) analysis for boll weight in upland cotton (Gossypium hirsutum.). BMC Plant Biol. 2016, 16, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale DE NOVO SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huai, D.; Zhang, Z.; Cheng, K.; Kang, Y.; Wan, L.; Yan, L.; Jiang, H.; Lei, Y.; Liao, B. Development of a high-density genetic map based on specific length amplified fragment sequencing and its application in quantitative trait loci analysis for yield-related traits in cultivated peanut. Front. Plant Sci. 2018, 9, 827. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sun, Y.; Yang, P.; Cai, X.; Yang, L.; Ma, J.; Ou, Y.; Liu, T.; Ali, L.; Liu, D.; et al. A high density SLAF-seq SNP genetic map and QTL for seed size, oil and protein content in upland cotton. BMC Genomics. 2019, 20, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.; Liu, Y.; Liang, C.; Wang, W.; Li, C.; Guo, Y.; Ma, J.; Yu, Y.; Fan, L.; Yao, Y.; et al. Construction of a high-density genetic linkage map and QTL mapping of oleic acid content and three agronomic traits in sunflower (Helianthus annuus L.) using specific-locus amplified fragment sequencing (SLAF-seq). Breed. Sci. 2019, 18051. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Lin, S.; Yano, M.; Nagamine, T. Mapping quantitative trait loci controlling seed longevity in rice (Oryza sativa L.). Theor. Appl. Genet. 2002, 104, 981–986. [Google Scholar] [CrossRef]
- Nagel, M.; Vogel, H.; Landjeva, S.; Buck-Sorlin, G.; Lohwasser, U.; Scholz, U.; Borner, A. Seed conservation in ex situ genebanks genetic studies on longevity in barley. Euphytica 2009, 170, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.M.; Nagel, M.; Neumann, K.; Kobiljski, B.; Lohwasser, U.; Borner, A. Genetic studies of seed longevity in hexaploid wheat using segregation and association mapping approaches. Euphytica 2012, 186, 1–13. [Google Scholar] [CrossRef]
- Nagel, M.; Rosenhauer, M.; Willner, E.; Snowdon, R.J.; Friedt, W.; Borner, A. Seed longevity in oilseed rape (Brassica napus L.) genetic variation and QTL mapping. Plant. Genet. Res. 2011, 9, 260–263. [Google Scholar] [CrossRef]
- Bentsink, L.; Alonso-Blanco, C.; Vreugdenhil, D.; Tesnier, K.; Groot, S.; Koornneef, M. Genetic analysis of seed-soluble oligosaccharides in relation to seed storability of Arabidopsis. Plant Physiol. 2000, 124, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Fujino, K.; Sekiguchi, H.; Matsuda, Y.; Sugimoto, K.; Ono, K.; Masahiro, Y. Molecular identification of a major quantitative trait locus, qLTG3–1, controlling low-temperature germinability in rice. Proc. Natl. Acad. Sci. USA 2008, 105, 12623–12628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, K.; Matsuda, Y. Genome-wide analysis of genes targeted by qLTG3-1 controlling low-temperature germinability in rice. Plant Mol. Biol. 2010, 72, 137. [Google Scholar] [CrossRef] [PubMed]
- Fujino, K.; Sekiguchi, H. Origins of functional nucleotide polymorphisms in a major quantitative trait locus, qLTG3-1, controlling low-temperature germinability in rice. Plant Mol. Biol. 2011, 75, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Keizer, P.; van Eeuwijk, F.; Smeekens, S.; Bentsink, L. Natural variation for seed longevity and seed dormancy are negatively correlated in Arabidopsis. Plant Physiol. 2012, 160, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Ku, L.; Zhang, Z.; Zhang, J.; Guo, S.; Liu, H.; Zhao, R.; Ren, Z.; Zhang, L.; Su, H.; et al. QTLs for seed vigor-related traits identified in maize seeds germinated under artificial aging conditions. PLoS ONE 2014, 9, e92535. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Zhang, Z.; Fu, Z.; Liu, Z.; Hu, Y.; Tang, J. Comparative QTL analysis of maize seed artificial aging between an immortalized F2 population and its corresponding RILs. Crop. J. 2016, 4, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Hu, J.; Huang, X.; Wang, X.; Guan, Y.; Wang, Z. Relationship between changes of kernel nutritive components and seed vigor during development stages of F1 seeds of sh2 sweet corn. J. Zhejiang Univ. Sci. B 2008, 9, 964–968. [Google Scholar] [CrossRef]
- Saghai-Maroof, M.A.; Soliman, K.M.; Jorgensen, R.A.; Allard, R.W. Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc. Natl. Acad. Sci. USA 1984, 81, 8014–8018. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, Q.; Cheng, T.; Yang, W.; Pan, H.; Zhong, J.; Huang, L.; Liu, E. High-density genetic map construction and identification of a locus controlling weeping trait in an ornamental woody plant (Prunus mume Sieb. et Zucc). DNA Res. 2015, 22, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Van, O.; Stam, P.; Visser, R.; Eck, H. SMOOTH: A statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theor. Appl. Genet. 2005, 112, 187–194. [Google Scholar] [CrossRef]
- Liu, L.; Lai, Y.; Cheng, J.; Wang, L.; Du, L.; Wang, Z.; Zhang, H. Dynamic quantitative trait locus analysis of seed vigor at three maturity stages in rice. PLoS ONE 2014, 9, e11573212. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Feng, Q.; Qian, Q.; Zhao, Q.; Wang, L.; Wang, A.; Guan, J.; Fan, D.; Weng, Q.; Huang, T.; et al. High-throughput genotyping by whole-genome resequencing. Genome Res. 2009, 16, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.; Hu, Z. Mapping quantitative trait Loci using distorted markers. Int. J. Plant Genom. 2009, 410825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vision, T.J.; Brown, D.G.; Shmoys, D.B.; Durrett, R.T.; Tanksley, S.D. Selective mapping: A strategy for optimizing the construction of high-density linkage maps. Genetics 2000, 155, 407–420. [Google Scholar] [PubMed]
- Kosambi, D.D. The estimation of map distance from recombination values. Ann. Eugen. 1944, 12, 172–175. [Google Scholar] [CrossRef]
- Yang, J.; Hu, C.; Hu, H.; Yu, R.; Xia, Z.; Ye, X.; Zhu, J. QTLNetwork: Mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics 2008, 24, 721–723. [Google Scholar] [CrossRef] [Green Version]
- Stuber, C.W.; Edwards, M.D.; Wendel, J.F. Molecular marker facilitated investigations of quantitative trait loci in maize: II. Factors influencing yield and its component traits. Crop. Sci. 1987, 27, 639–648. [Google Scholar] [CrossRef] [Green Version]
- TeKrony, D.M.; Egli, D.B.; Wickham, D.A. Corn seed vigor effect on notillage field performance. I. Field emergence. Crop. Sci. 1989, 29, 1523–1528. [Google Scholar] [CrossRef]
- DeVries, M.; Goggi, A.S.; Moore, K.J. Determining seed performance of frost damaged maize seed lots. Crop Sci. 2007, 47, 2089–2097. [Google Scholar] [CrossRef]
- Hund, A.; Fracheboud, Y.; Soldati, A.; Frascaroli, E.; Salvi, S.; Stamp, P. QTL controlling root and shoot traits of maize seedlings under cold stress. Theor. Appl. Genet. 2004, 109, 618–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, Z.; Xie, H.; Hu, Y.; Liu, Z.; Duan, L.; Xu, S. Identification of QTLs for maize seed vigor at three stages of seed maturity using a RIL population. Euphytica 2011, 178, 127–135. [Google Scholar] [CrossRef]
- Colville, L.; Bradley, E.L.; Lloyd, A.S.; Pritchard, H.W.; Castle, L.; Kranner, I. Volatile fingerprints of seeds of four species indicate the involvement of alcoholic fermentation, lipid peroxidation, and Maillard reactions in seed deterioration during ageing and desiccation stress. J. Exp. Bot. 2012, 63, 6519–6530. [Google Scholar] [CrossRef] [PubMed]
- Dixon, D.P.; Lapthorn, A.; Edwards, R. Plant glutathione transferases. Genome Biol. 2002. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H. Change of physiological characteristics and seed vigor of waxy corn during ageing course. J. Maize. Sci. 2015, 23, 93–96. (In Chinese) [Google Scholar] [CrossRef]
- Ding, Y.; Li, Y.; Cai, Y.; Feng, K.; Zhao, Y.; Zhang, H. Effects of artificial accelerated aging and repairing process on seed vigor and physiological characteristics of wheat seeds. Seed 2016, 35, 13–16. [Google Scholar] [CrossRef]
- Roxas, V.; Smith, R.; Allen, E.; Allen, R. Overexpression of glutathione S-transferase/glutathioneperoxidase enhances the growth of transgenic tobacco seedlings during stress. Nat. Biotechnol. 1997, 15, 988. [Google Scholar] [CrossRef]
- Cummins, I.; Cole, D.J.; Edwards, R. A role for glutathione transferases functioning as glutathione peroxidases in resistance to multiple herbicides in black-grass. Plant J. 1999, 18, 285–292. [Google Scholar] [CrossRef]
- Uhrig, J.; Huang, L.; Barghahn, S.; Willmer, M.; Thurow, C.; Gatz, C. CC-type glutaredoxins recruit the transcriptional co-repressor TOPLESS to TGA-dependent target promoters in Arabidopsis thaliana. BBA-Gene. Regul. Mech. 2017, 1860, 218–226. [Google Scholar] [CrossRef]
- Connolly, J.D.; Hill, R.A. Dictionary of Terpenoids; Chapman and Hall: London, UK, 2013. [Google Scholar]
- Harborne, J.B.; Turner, B.L. Plant Chemosystematics; Academic Press: London, UK, 1984. [Google Scholar]
- Tholl, D. Biosynthesis and biological functions of terpenoids in plants. In Biotechnology of Isoprenoids; Springer: Cham, Switzerland, 2015; pp. 63–106. [Google Scholar] [CrossRef]
- Loreto, F.; Dicke, M.; Schnitzler, J.P.; Turlings, T. Plant volatiles and the environment. Plant Cell Environ. 2014, 37, 1905–1908. [Google Scholar] [CrossRef]
- Yadav, V.; Mallappa, C.; Gangappa, S.; Bhatia, S.; Chattopadhyay, S. A basic helix-loop-helix transcription factor in Arabidopsis, MYC2, acts as a repressor of blue light-mediated photomorphogenic growth. Plant Cell 2005, 17, 1953–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombrecht, B.; Xue, G.; Sprague, S.; Kirkegaard, J.; Ross, J.; Reid, J.; Fitt, G.; Sewelam, N.; Schenk, N.; Manners, J.M.; et al. MYC2 differentially modulates diverse jasmonate-dependent functions in Arabidopsis. Plant Cell 2007, 19, 2225–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, G.; Xue, X.; Mao, Y.; Wang, L.; Chen, X. Arabidopsis MYC2 interacts with DELLA proteins in regulating sesquiterpene synthase gene expression. Plant Cell 2012, 24, 2635–2648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esterhuysen, N.; Murray, S.L. Producing pathogen-resistant plants: An investigation towards functional genomics of maize Terpene synthase 11. S. Afr. J. Bot. 2007, 100, 333. [Google Scholar] [CrossRef]
- Chen, X.; Chen, H.; Yuan, J.; Köllner, T.G.; Chen, Y.; Guo, Y.; Zhuang, X.; Chen, X.; Zhang, Y.; Fu, J.; et al. The rice terpene synthase gene OsTPS19 functions as an (S)-limonene synthase in planta, and its overexpression leads to enhanced resistance to the blast fungus Magnaporthe oryzae. Plant. Biotechnol. J. 2018, 16, 1778–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Reads | GC Percentage (%) 1 | Q30 Percentage (%) 2 | Marker Number | Total Depth | Average Depth |
|---|---|---|---|---|---|---|
| M03 | 12,759,218 | 45.42 | 85.74 | 135,956 | 4,995,576 | 36.74 |
| M08 | 12,276,986 | 45.58 | 85.02 | 90,506 | 4,305,095 | 47.57 |
| Offspring | 1,370,055 | 45.15 | 86.39 | 98,023 | 536,053 | 5.47 |
| Chr 1 | Marker Number | Total cM 2 | Average cM 3 | Max Gap 4 | Gap < 5 cM |
|---|---|---|---|---|---|
| Chr1 | 475 | 329.92 | 0.69 | 14.2 | 95.14% |
| Chr2 | 353 | 230.88 | 0.65 | 14.0 | 90.86% |
| Chr3 | 464 | 294.87 | 0.64 | 12.2 | 92.74% |
| Chr4 | 433 | 288.80 | 0.67 | 8.98 | 97.24% |
| Chr5 | 405 | 257.25 | 0.64 | 15.28 | 98.61% |
| Chr6 | 343 | 214.57 | 0.63 | 9.33 | 99.26% |
| Chr7 | 381 | 237.86 | 0.62 | 7.03 | 98.49% |
| Chr8 | 325 | 207.57 | 0.64 | 6.32 | 97.31% |
| Chr9 | 179 | 114.32 | 0.64 | 4.97 | 96.84% |
| Chr10 | 518 | 237.22 | 0.46 | 8.1 | 97.94% |
| Total | 3876 | 2413.25 | 0.62 | 15.28 | 96.44% |
| Season | Trait | Parents (Mean ± SD) | BC4F3 Population | Skewness | Kurtosis | CV (%) | h2 | ||
|---|---|---|---|---|---|---|---|---|---|
| M03 | M08 | Range | Mean ± SD | ||||||
| 2017S | GP | 0.82 ± 0.23 | 0.36 ± 0.18 | 0.04–1.00 | 0.47 ± 0.19 | −0.06 | −0.42 | 41.02 | 0.72 |
| GR | 0.92 ± 0.13 | 0.55 ± 0.11 | 0.20–1.00 | 0.79 ± 0.14 | −1.25 | 2.33 | 17.81 | 0.64 | |
| GI | 3.86 ± 0.11 | 1.32 ± 0.54 | 0.52–4.24 | 2.78 ± 0.62 | −0.65 | 0.84 | 22.32 | 0.66 | |
| VI | 26.47 ± 5.39 | 11.44 ± 4.76 | 0.24–32.41 | 13.73 ± 6.26 | 0.48 | 0.39 | 45.64 | 0.73 | |
| 2017A | GP | 0.79 ± 0.16 | 0.36 ± 0.17 | 0.00–0.88 | 0.42 ± 0.21 | 0.01 | −0.80 | 49.20 | 0.73 |
| GR | 0.85 ± 0.20 | 0.53 ± 0.15 | 0.16–1.00 | 0.77 ± 0.14 | −1.28 | 3.63 | 18.42 | 075 | |
| GI | 3.43 ± 0.82 | 1.37 ± 0.56 | 0.40–4.04 | 2.63 ± 0.65 | −0.56 | 0.94 | 24.61 | 0.74 | |
| VI | 24.26 ± 5.27 | 8.92 ± 3.26 | 0.19–30.76 | 11.79 ± 5.76 | 0.58 | 0.49 | 18.91 | 0.67 | |
| Season | Trait | QTL | Chr 1 | QTL Region (cM) | Marker Interval | LOD 2 | ADD 3 | R2 (%) 4 |
|---|---|---|---|---|---|---|---|---|
| 2017S | GP | qGP6 | 6 | 38.800–38.810 | Marker1435–Marker1452 | 3.01 | 0.15 | 1.69 |
| qGP7-1 | 7 | 0.000–0.040 | Marker2623–Marker2627 | 2.40 | −0.09 | 0.62 | ||
| GR | qGR10 | 10 | 0.000–1.400 | Marker393–Marker398 | 3.24 | −0.20 | 5.53 | |
| qGR6 | 6 | 113.746–113.786 | Marker1652–Marker1656 | 3.55 | −0.13 | 2.38 | ||
| GI | qGI7-1 | 7 | 0.000–0.040 | Marker2623–Marker2627 | 4.24 | −0.46 | 1.49 | |
| VI | qVI7-1 | 7 | 0.000–0.040 | Marker2623–Marker2627 | 3.90 | −4.04 | 1.15 | |
| 2017A | GR | qGR10 | 10 | 0.000–1.400 | Marker393–Marker398 | 5.85 | −0.31 | 13.14 |
| GI | qGI10 | 10 | 0.000–1.400 | Marker393–Marker398 | 3.96 | −1.12 | 8.07 | |
| VI | qVI1 | 1 | 282.283–282.343 | Marker8370–Marker8371 | 2.16 | 6.08 | 3.03 | |
| qVI3-1 | 3 | 120.160–122.540 | Marker4631–Marker4636 | 2.28 | −4.57 | 1.71 | ||
| qVI3-2 | 3 | 220.090–221.731 | Marker4866–Marker4867 | 2.50 | −2.48 | 0.50 | ||
| qVI3-3 | 3 | 36.250–36.280 | Marker4245–Marker4248 | 2.06 | 3.04 | 0.76 | ||
| qVI3-4 | 3 | 81.440–81.470 | Marker4749–Marker4752 | 2.27 | −4.63 | 1.76 | ||
| qVI5-1 | 5 | 243.962–243.972 | Marker4126–Marker4127 | 2.05 | 6.01 | 2.96 | ||
| qVI9 | 9 | 56.586–56.596 | Marker1052–Marker1053 | 2.17 | 6.06 | 3.02 | ||
| qVI4 | 4 | 206.290–207.931 | Marker6830–Marker6834 | 2.53 | 4.93 | 2.02 | ||
| qVI5-2 | 5 | 90.936–90.946 | Marker3585–Marker3588 | 2.95 | 5.62 | 2.63 | ||
| qVI7-2 | 7 | 3.395–3.405 | Marker2628–Marker2631 | 3.00 | −2.73 | 0.62 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Feng, F.; Zhu, Y.; Xie, F.; Yang, J.; Gong, J.; Liu, Y.; Zhu, W.; Gao, T.; Chen, D.; et al. Construction of High-Density Genetic Map and Identification of QTLs Associated with Seed Vigor after Exposure to Artificial Aging Conditions in Sweet Corn Using SLAF-seq. Genes 2020, 11, 37. https://doi.org/10.3390/genes11010037
Wu X, Feng F, Zhu Y, Xie F, Yang J, Gong J, Liu Y, Zhu W, Gao T, Chen D, et al. Construction of High-Density Genetic Map and Identification of QTLs Associated with Seed Vigor after Exposure to Artificial Aging Conditions in Sweet Corn Using SLAF-seq. Genes. 2020; 11(1):37. https://doi.org/10.3390/genes11010037
Chicago/Turabian StyleWu, Xiaming, Faqiang Feng, Yuzhong Zhu, Fugui Xie, Jing Yang, Jie Gong, Yu Liu, Wei Zhu, Tianle Gao, Danyi Chen, and et al. 2020. "Construction of High-Density Genetic Map and Identification of QTLs Associated with Seed Vigor after Exposure to Artificial Aging Conditions in Sweet Corn Using SLAF-seq" Genes 11, no. 1: 37. https://doi.org/10.3390/genes11010037
APA StyleWu, X., Feng, F., Zhu, Y., Xie, F., Yang, J., Gong, J., Liu, Y., Zhu, W., Gao, T., Chen, D., Li, X., & Huang, J. (2020). Construction of High-Density Genetic Map and Identification of QTLs Associated with Seed Vigor after Exposure to Artificial Aging Conditions in Sweet Corn Using SLAF-seq. Genes, 11(1), 37. https://doi.org/10.3390/genes11010037