Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. X Inactivation
2.3. Targeted Next-Generation Sequencing
2.4. Variant Segregation and Additional Analysis
3. Results
3.1. Targeted Next-Generation Sequencing
3.2. Segregation Analysis and Genotype-Phenotype Correlation
3.2.1. Pathogenic/Likely Pathogenic Variants
Family ID0707
Family ID1122
Family ID1208
3.2.2. Variants of Unknown Significance
Family ID0216
Family ID1128
Family ID1402
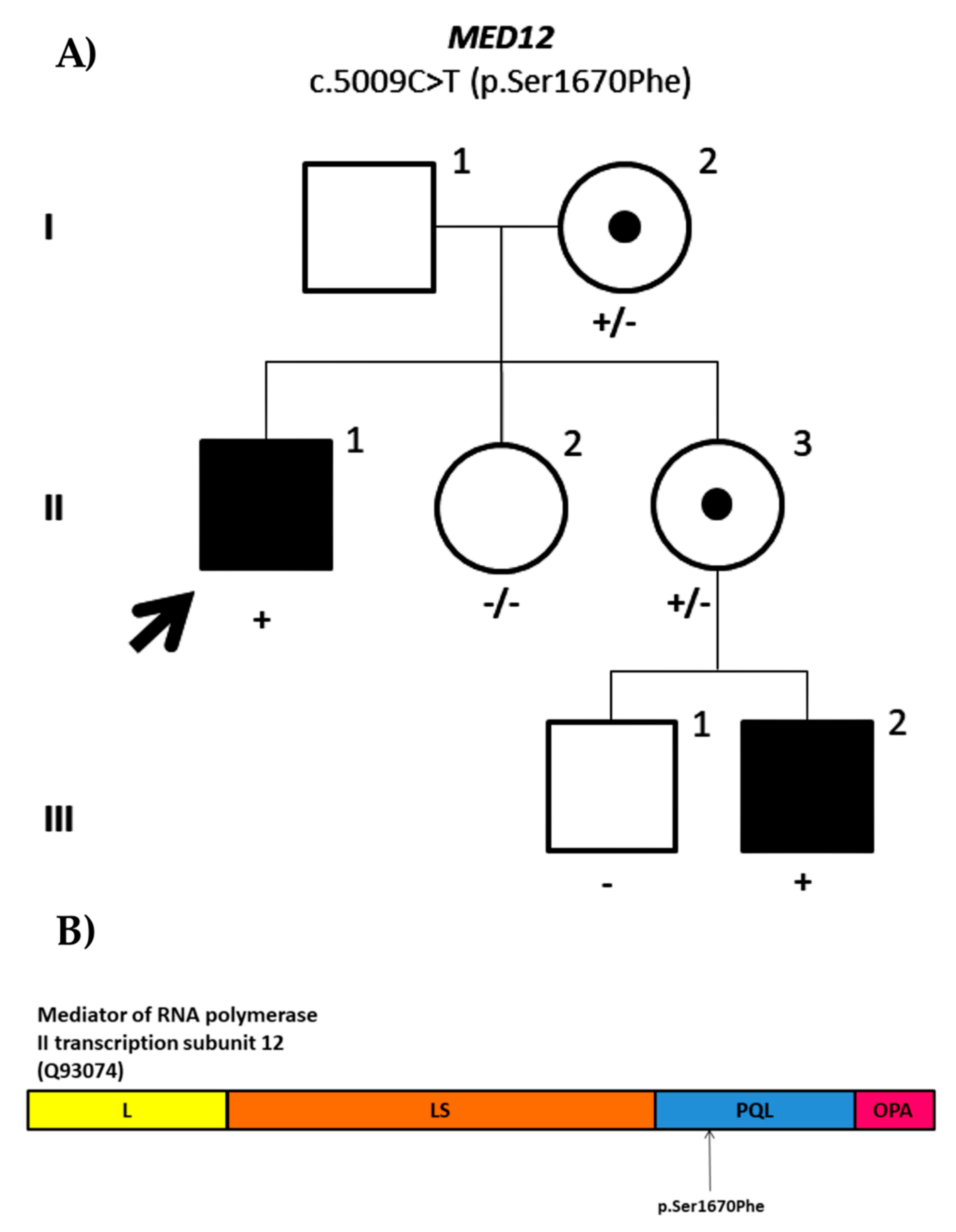
Family ID1405
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maulik, P.K.; Mascarenhas, M.N.; Mathers, C.D.; Dua, T.; Saxena, S. Prevalence of intellectual disability: A meta-analysis of population-based studies. Res. Dev. Disabil. 2011, 32, 419–436. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, K.; Milton, M.; Smith, G.; Ouellette-Kuntz, H. Systematic Review of the prevalence and incidence of intellectual disabilities: Current trends and issues. Curr. Dev. Disord. Rep. 2016, 3, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Schalock, R.L.; Borthwick-Duffy, S.A.; Bradley, V.J.; Buntinx, W.H.E.; Coulter, D.L.; Craig, E.M.; Gomez, S.C.; Lachapelle, Y.; Luckasson, R.; Reeve, A.; et al. Intellectual Disability: Definition, Classification, and Systems of Supports, 11th ed.; American Association on Intellectual and Developmental Disabilities: Washington, DC, USA, 2010; pp. 1–280. [Google Scholar]
- Stevenson, R.E.; Schwartz, C.E. Clinical and molecular contributions to the understanding of X-linked mental retardation. Cytogenet. Genome Res. 2002, 99, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Ropers, H.H.; Hamel, B.C.J. X-linked mental retardation. Nat. Rev. Genet. 2005, 6, 46–57. [Google Scholar] [CrossRef]
- Ropers, H.H. X-linked mental retardation: Many genes for a complex disorder. Curr. Opin. Genet. Dev. 2006, 16, 260–269. [Google Scholar] [CrossRef]
- Stevenson, R.E.; Schwartz, C.E. X-linked intellectual disability: Unique vulnerability of the male genome. Dev. Disabil. Res. Rev. 2009, 15, 361–368. [Google Scholar] [CrossRef]
- Lubs, H.A.; Stevenson, R.E.; Schwartz, C.E. Fragile X and X-linked intellectual disability: Four decades of discovery. Am. J. Hum. Genet. 2012, 90, 579–590. [Google Scholar] [CrossRef] [Green Version]
- Vissers, L.E.L.M.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2015, 17, 9–18. [Google Scholar] [CrossRef]
- Neri, G.; Schwartz, C.E.; Lubs, H.A.; Stevenson, R.E. X-linked intellectual disability update 2017. Am. J. Med. Genet. Part A 2018, 176, 1375–1388. [Google Scholar] [CrossRef] [Green Version]
- Crawford, D.C.; Acuña, J.M.; Sherman, S.L. FMR1 and the Fragile X Syndrome: Human Genome Epidemiology Review. Genet. Med. 2001, 3, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Tarpey, P.S.; Smith, R.; Pleasance, E.; Whibley, A.; Edkins, S.; Hardy, C.; O’Meara, S.; Latimer, C.; Dicks, E.; Menzies, A.; et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009, 41, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Tzschach, A.; Grasshoff, U.; Beck-Woedl, S.; Dufke, C.; Bauer, C.; Kehrer, M.; Evers, C.; Moog, U.; Oehl-Jaschkowitz, B.; Di Donato, N.; et al. Next-generation sequencing in X-linked intellectual disability. Eur. J. Hum. Genet. 2015, 23, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Haas, S.A.; Chelly, J.; Van Esch, H.; Raynaud, M.; De Brouwer, A.P.M.; Weinert, S.; Froyen, G.; Frints, S.G.M.; Laumonnier, F.; et al. X-exome sequencing of 405 unresolved families identifies seven novel intellectual disability genes. Mol. Psychiatry 2016, 21, 133–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niranjan, T.S.; Skinner, C.; May, M.; Turner, T.; Rose, R.; Stevenson, R.; Schwartz, C.E.; Wang, T. Affected kindred analysis of human X chromosome exomes to identify novel X-linked intellectual disability genes. PLoS ONE 2015, 10, 116454. [Google Scholar] [CrossRef] [Green Version]
- Philips, A.K.; Sirén, A.; Avela, K.; Somer, M.; Peippo, M.; Ahvenainen, M.; Doagu, F.; Arvio, M.; Kääriäinen, H.; Van Esch, H.; et al. X-exome sequencing in Finnish families with Intellectual Disability—Four novel mutations and two novel syndromic phenotypes. Orphanet J. Rare Dis. 2014, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Sanchis-Juan, A.; Bitsara, C.; Low, K.Y.; Carss, K.J.; French, C.E.; Spasic-Boskovic, O.; Jarvis, J.; Field, M.; Raymond, F.L.; Grozeva, D. Rare genetic variation in 135 families with family history suggestive of X-linked intellectual disability. Front. Genet. 2019, 10, 578. [Google Scholar] [CrossRef]
- Vissers, L.E.L.M.; De Ligt, J.; Gilissen, C.; Janssen, I.; Steehouwer, M.; De Vries, P.; Van Lier, B.; Arts, P.; Wieskamp, N.; Del Rosario, M.; et al. A de novo paradigm for mental retardation. Nat. Genet. 2010, 42, 1109. [Google Scholar] [CrossRef]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar]
- Ørstavik, K.H. X chromosome inactivation in clinical practice. Hum. Genet. 2009, 126, 363–373. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1082. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [Green Version]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Bodamer, O.A.; Bloesch, S.M.; Gregg, A.R.; Stockler-Ipsiroglu, S.; O’Brien, W.E. Analysis of guanidinoacetate and creatine by isotope dilution electrospray tandem mass spectrometry. Clin. Chim. Acta 2001, 308, 173–178. [Google Scholar] [CrossRef]
- Laumonnier, F.; Holbert, S.; Ronce, N.; Faravelli, F.; Lenzner, S.; Schwartz, C.E.; Lespinasse, J.; Van Esch, H.; Lacombe, D.; Goizet, C.; et al. Mutations in PHF8 are associated with X linked mental retardation and cleft lip/cleft palate. J. Med. Genet. 2005, 42, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Siderius, L.E.; Hamel, B.C.J.; Van Bokhoven, H.; De Jager, F.; Van Den Helm, B.; Kremer, H.; Heineman-de Boer, J.A.; Ropers, H.H.; Mariman, E.C.M. X-linked mental retardation associated with cleft lip/palate maps to Xp11.3-q21.3. Am. J. Med. Genet. 1999, 85, 216–220. [Google Scholar] [CrossRef]
- Koivisto, A.M.; Ala-Mello, S.; Lemmelä, S.; Komu, H.A.; Rautio, J.; Järvelä, I. Screening of mutations in the PHF8 gene and identification of a novel mutation in a Finnish family with XLMR and cleft lip/cleft palate. Clin. Genet. 2007, 72, 145–149. [Google Scholar] [CrossRef]
- Abidi, F.E.; Miano, M.G.; Murray, J.C.; Schwartz, C.E. A novel mutation in the PHF8 gene is associated with X-linked mental retardation with cleft lip/cleft palate. Clin. Genet. 2007, 72, 19–22. [Google Scholar] [CrossRef]
- Redin, C.; Gerard, B.; Lauer, J.; Herenger, Y.; Muller, J.; Quartier, A.; Masurel-Paulet, A.; Willems, M.; Lesca, G.; El-Chehadeh, S.; et al. Efficient strategy for the molecular diagnosis of intellectual disability using targeted high-throughput sequencing. J. Med. Genet. 2014, 51, 724–736. [Google Scholar] [CrossRef]
- Faundes, V.; Newman, W.G.; Bernardini, L.; Canham, N.; Clayton-Smith, J.; Dallapiccola, B.; Davies, S.J.; Demos, M.K.; Goldman, A.; Gill, H.; et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018, 102, 175–187. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Hu, H.; Kahrizi, K.; Musante, L.; Fattahi, Z.; Herwig, R.; Hosseini, M.; Oppitz, C.; Abedini, S.S.; Suckow, V.; Larti, F.; et al. Genetics of intellectual disability in consanguineous families Genetics of intellectual disability in consanguineous families. Mol. Psychiatry 2018, 24, 1027–1039. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, T.W.; Gerety, S.S.; Jones, W.D.; Van Kogelenberg, M.; King, D.A.; McRae, J.; Morley, K.I.; Parthiban, V.; Al-Turki, S.; Ambridge, K.; et al. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar]
- Salomons, G.S.; Van Dooren, S.J.M.; Verhoeven, N.M.; Cecil, K.M.; Ball, W.S.; Degrauw, T.J.; Jakobs, C. X-linked creatine-transporter gene (SLC6A8) defect: A new creatine-deficiency syndrome. Am. J. Hum. Genet. 2001, 68, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Rosenberg, E.H.; Almeida, L.S.; Wood, T.C.; Jakobs, C.; Stevenson, R.E.; Schwartz, C.E.; Salomons, G.S. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum. Genet. 2006, 119, 604–610. [Google Scholar] [CrossRef]
- Andrade, F. The arginine-creatine pathway is disturbed in children and adolescents with renal transplants (Pediatric Research (2008) 64 (218-222). Pediatr. Res. 2009, 65, 248. [Google Scholar]
- Shoubridge, C.; Tarpey, P.S.; Abidi, F.; Ramsden, S.L.; Rujirabanjerd, S.; Murphy, J.A.; Boyle, J.; Shaw, M.; Gardner, A.; Proos, A.; et al. Mutations in the guanine nucleotide exchange factor gene IQSEC2 cause nonsyndromic intellectual disability. Nat. Genet. 2010, 42, 486–488. [Google Scholar] [CrossRef] [Green Version]
- Rogers, E.J.; Jada, R.; Schragenheim-Rozales, K.; Sah, M.; Cortes, M.; Florence, M.; Levy, N.S.; Moss, R.; Walikonis, R.S.; Palty, R.; et al. An IQSEC2 mutation associated with intellectual disability and autism results in decreased surface AMPA receptors. Front. Mol. Neurosci. 2019, 12, 13. [Google Scholar] [CrossRef]
- Shoubridge, C.; Harvey, R.J.; Dudding-Byth, T. IQSEC2 mutation update and review of the female-specific phenotype spectrum including intellectual disability and epilepsy. Hum. Mutat. 2019, 40, 5–24. [Google Scholar] [CrossRef] [Green Version]
- Kalscheuer, V.M.; James, V.M.; Himelright, M.L.; Long, P.; Oegema, R.; Jensen, C.; Bienek, M.; Hu, H.; Haas, S.A.; Topf, M.; et al. Novel Missense Mutation A789V in IQSEC2 Underlies X-Linked Intellectual Disability in the MRX78 Family. Front. Mol. Neurosci. 2016, 8, 85. [Google Scholar] [CrossRef] [Green Version]
- Shoubridge, C.; Walikonis, R.S.; Gécz, J.; Harvey, R.J. Subtle functional defects in the Arf-specific guanine nucleotide exchange factor IQSEC2 cause non-syndromic X-linked intellectual disability. Small GTPases 2010, 1, 98–103. [Google Scholar] [CrossRef] [Green Version]
- Levy, N.S.; Umanah, G.K.E.; Rogers, E.J.; Jada, R.; Lache, O.; Levy, A.P. IQSEC2-Associated Intellectual Disability and Autism. Int. J. Mol. Sci. 2019, 20, 3038. [Google Scholar] [CrossRef] [Green Version]
- Petersen, A.; Brown, J.C.; Gerges, N.Z. BRAG1/IQSEC2 as a regulator of small GTPase-dependent trafficking. Small GTPases 2018, 11, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.R.; Wang, G.; Sheng, Y.; Conger, K.K.; Casanova, J.E.; Zhu, J.J. Arf6-GEF BRAG1 regulates JNK-mediated synaptic removal of glua1-containing AMPA receptors: A new mechanism for nonsyndromic X-linked mental disorder. J. Neurosci. 2012, 32, 11716–11726. [Google Scholar] [CrossRef] [Green Version]
- Gilfillan, G.D.; Selmer, K.K.; Roxrud, I.; Smith, R.; Eiklid, K.; Kroken, M.; Mattingsdal, M.; Egeland, T.; Stenmark, H.; Sjøholm, H.; et al. SLC9A6 Mutations Cause X-Linked Mental Retardation, Microcephaly, Epilepsy, and Ataxia, a Phenotype Mimicking Angelman Syndrome. Am. J. Hum. Genet. 2008, 82, 1003–1010. [Google Scholar] [CrossRef] [Green Version]
- Pescosolido, M.F.; Stein, D.M.; Schmidt, M.; Moufawad, C.; Achkar, E.; Sabbagh, M.; Rogg, J.M.; Tantravahi, U.; Mclean, R.L.; Liu, J.S.; et al. Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Ann. Neurol. 2014, 76, 581–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masurel-Paulet, A.; Piton, A.; Chancenotte, S.; Redin, C.; Thauvin-Robinet, C.; Henrenger, Y.; Minot, D.; Creppy, A.; Ruffier-Bourdet, M.; Thevenon, J.; et al. A new family with an SLC9A6 mutation expanding the phenotypic spectrum of Christianson syndrome. Am. J. Med. Genet. Part A 2016, 170, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Ilie, A.; Gao, A.Y.L.L.; Boucher, A.; Park, J.; Berghuis, A.M.; Hoffer, M.J.V.V.; Hilhorst-Hofstee, Y.; McKinney, R.A.; Orlowski, J. A potential gain-of-function variant of SLC9A6 leads to endosomal alkalinization and neuronal atrophy associated with Christianson Syndrome. Neurobiol. Dis. 2019, 121, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Lena, J.-C.; Mishra, S.; Riaz, H.A.; Schmidt, M.; Morrow, E.M. Functional Assessment In Vivo of the Mouse Homologue of the Human Ala-9-Ser NHE6 Variant. ENeuro 2019, 6. [Google Scholar] [CrossRef]
- Froyen, G.; Corbett, M.; Vandewalle, J.; Jarvela, I.; Lawrence, O.; Meldrum, C.; Bauters, M.; Govaerts, K.; Vandeleur, L.; Van Esch, H.; et al. Submicroscopic Duplications of the Hydroxysteroid Dehydrogenase HSD17B10 and the E3 Ubiquitin Ligase HUWE1 Are Associated with Mental Retardation. Am. J. Hum. Genet. 2008, 82, 432–443. [Google Scholar] [CrossRef] [Green Version]
- Bosshard, M.; Aprigliano, R.; Gattiker, C.; Palibrk, V.; Markkanen, E.; Hoff Backe, P.; Pellegrino, S.; Raymond, F.L.; Froyen, G.; Altmeyer, M.; et al. Impaired oxidative stress response characterizes HUWE1-promoted X-linked intellectual disability. Sci. Rep. 2017, 7, 15050. [Google Scholar] [CrossRef] [Green Version]
- Friez, M.J.; Brooks, S.S.; Stevenson, R.E.; Field, M.; Basehore, M.J.; Adès, L.C.; Sebold, C.; Mcgee, S.; Saxon, S.; Skinner, C.; et al. HUWE1 mutations in Juberg-Marsidi and Brooks syndromes: The results of an X-chromosome exome sequencing study. BMJ Open 2016, 6, 9537. [Google Scholar] [CrossRef] [Green Version]
- Moortgat, S.; Berland, S.; Aukrust, I.; Maystadt, I.; Baker, L.; Benoit, V.; Caro-Llopis, A.; Cooper, N.S.; Debray, F.G.; Faivre, L.; et al. HUWE1 variants cause dominant X-linked intellectual disability: A clinical study of 21 patients. Eur. J. Hum. Genet. 2018, 26, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Juberg, R.C.; Marsidi, I. A new form of X-linked mental retardation with growth retardation, deafness, and microgenitalism. Am. J. Hum. Genet. 1980, 32, 714–722. [Google Scholar]
- Brooks, S.S.; Wisniewski, K.; Brown, W.T. New X-linked mental retardation (XLMR) syndrome with distinct facial appearance and growth retardation. Am. J. Med. Genet. 1994, 51, 586–590. [Google Scholar] [CrossRef]
- Garcia, C.C.; Blair, H.J.; Seager, M.; Coulthard, A.; Tennant, S.; Buddles, M.; Curtis, A.; Goodship, J.A. Identification of a mutation in synapsin I, a synaptic vesicle protein, in a family with epilepsy. J. Med. Genet. 2004, 41, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Fassio, A.; Patry, L.; Congia, S.; Onofri, F.; Piton, A.; Gauthier, J.; Pozzi, D.; Messa, M.; Defranchi, E.; Fadda, M.; et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 2011, 20, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Risheg, H.; Graham, J.M.; Clark, R.D.; Rogers, R.C.; Opitz, J.M.; Moeschler, J.B.; Peiffer, A.P.; May, M.; Joseph, S.M.; Jones, J.R.; et al. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat. Genet. 2007, 39, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.E.; Tarpey, P.S.; Lubs, H.A.; Verloes, A.; May, M.M.; Risheg, H.; Friez, M.J.; Futreal, P.A.; Edkins, S.; Teague, J.; et al. The original Lujan syndrome family has a novel missense mutation (p.N1007S) in the MED12 gene. J. Med. Genet. 2007, 44, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silfhout, A.T.V.; De Vries, B.B.A.; Van Bon, B.W.M.; Hoischen, A.; Ruiterkamp-versteeg, M.; Gilissen, C.; Gao, F.; Van Zwam, M.; Harteveld, C.L.; Van Essen, A.J.; et al. Mutations in MED12 cause X-linked ohdo syndrome. Am. J. Hum. Genet. 2013, 92, 401–406. [Google Scholar]
- Charzewska, A.; Maiwald, R.; Kahrizi, K.; Oehl-Jaschkowitz, B.; Dufke, A.; Lemke, J.R.; Enders, H.; Najmabadi, H.; Tzschach, A.; Hachmann, W.; et al. The power of the Mediator complex-Expanding the genetic architecture and phenotypic spectrum of MED12 -related disorders. Clin. Genet. 2018, 94, 450–456. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, E.; Brussino, A.; Biamino, E.; Belligni, E.F.; Bruselles, A.; Ciolfi, A.; Caputo, V.; Pizzi, S.; Calcia, A.; Di Gregorio, E.; et al. Exome sequencing in children of women with skewed X-inactivation identifies atypical cases and complex phenotypes. Eur. J. Paediatr. Neurol. 2016, 21, 484–745. [Google Scholar] [CrossRef]
- Plenge, R.M.; Stevenson, R.A.; Lubs, H.A.; Schwartz, C.E.; Willard, H.F. Skewed X-Chromosome Inactivation Is a Common Feature of X-Linked Mental Retardation Disorders. Am. J. Hum. Genet. 2002, 71, 168–173. [Google Scholar] [CrossRef] [Green Version]
- Ziats, C.A.; Schwartz, C.E.; Gecz, J.; Shaw, M.; Field, M.J.; Stevenson, R.E.; Neri, G. X-linked intellectual disability: Phenotypic expression in carrier females. Clin. Genet. 2019. [Google Scholar] [CrossRef]
- Laumonnier, F.; Shoubridge, C.; Antar, C.; Nguyen, L.S.; Van Esch, H.; Kleefstra, T.; Briault, S.; Fryns, J.P.; Hamel, B.; Chelly, J.; et al. Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol. Psychiatry 2010, 15, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Tejada, M.I.; Villate, O.; Ibarluzea, N.; la Hoz, A.-B.D.; Martínez-Bouzas, C.; Beristain, E.; Martínez, F.; Friez, M.J.; Sobrino, B.; Barros, F. Molecular and clinical characterization of a novel nonsense variant in exon 1 of the UPF3B gene found in a large Spanish Basque family (MRX82). Front. Genet. 2019, 10, 1074. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X Linked | Siblings | |||
|---|---|---|---|---|
| Total | n = 47 | n = 14 | ||
| Age range | 2–63 | 2–24 | ||
| 0–10 | 26 | 55.32% | 9 | 64.29% |
| 10–20 | 10 | 21.28% | 4 | 28.57% |
| >20 | 11 | 23.40% | 1 | 7.14% |
| Intellectual disability | ||||
| Mild/Borderline | 16 | 34.04% | 5 | 35.71% |
| Moderate | 6 | 12.77% | 1 | 7.14% |
| Severe | 3 | 6.38% | 1 | 7.14% |
| Profound | 2 | 4.26% | 0 | |
| Unknown | 20 | 42.55% | 7 | 50% |
| Comorbidity | ||||
| Macrocephaly | 1 | 2.13% | 0 | |
| Microcephaly | 1 | 2.13% | 1 | 7.14% |
| Autism Spectrum Disorder | 14 | 29.79% | 4 | 28.57% |
| Hypotonia | 1 | 2.13% | 0 | |
| Epilepsy | 7 | 14.89% | 0 | |
| Behavioural disturbances | 5 | 10.64% | 2 | 14.29% |
| Previous studies | ||||
| Karyotype | 47 | 100% | 14 | 100% |
| Fragile-X | 47 | 100% | 14 | 100% |
| MLPA-X | 42 | 89.36% | 14 | 100% |
| array CGH | 15 | 31.91% | 14 | 100% |
| Patient ID | Gene | Variant | Inheritance 1 | Family History | GnomAD Allele Freq males | dbSNP | ClinVar | CADD 2 | InterVar 3 | InterVar (Manually Adjusted) |
|---|---|---|---|---|---|---|---|---|---|---|
| Pathogenic/likely pathogenic variants | ||||||||||
| ID0707 | PHF8 | NM_001184896.1: c.252C>A; p.Tyr84* | Maternal (97.04%) | Sib-pair | 36 | Pathogenic (PVS1, PM2, PP3) | Pathogenic (PVS1, PM2, PP3) | |||
| ID1204 | UPF3B | NM_080632.2: c.371-1G>C | Maternal (80.49%) | Sib-pair | Pathogenic (PVS1, PM2, PP3) | |||||
| ID1122 | SLC6A8 | NM_005629.3: c.1390_1392del GAT; p.Asp464del | Maternal (79.37%) | X-linked | Likely Pathogenic (PVS1, PM2) | Pathogenic (PS3, PM2, PM4, PP1, PP3) | ||||
| ID1208 | IQSEC2 | NM_001111125.2: c.128G>C; p.Arg43Pro | Maternal (53.7%) | X-linked | 26.2 | VUS (PM2) | Likely Pathogenic (PM2, PP1, PP2, PP3, PP4) | |||
| Variants of unknown significance | ||||||||||
| ID0216 | SLC9A6 | NM_001042537.1: c.316A>G; p.Met106Val | Maternal (57.09%) | X-linked | 23.1 | VUS (PM1, PM2, BP1) | VUS (PM1, PM2, PP1, BP1) | |||
| ID0919 | UPF3B | NM_080632.2: c.1118G>A; p.Arg373His | Maternal (uninformative) | X-linked | 2/66856 | rs146785878 | VUS | 26.7 | VUS (PM2, PP3) | |
| ID1010 | DLG3 | NM_021120.3: c.1424C>T; p.Ser475Leu | Maternal (91.97%) | Sib-pair | 1/75937 | rs953325312 | 31 | VUS (PM1, PM2, BP1) | ||
| ID1011 | NHS | NM_198270.3: c.1270A>G; p.Arg424Gly | Maternal (uninformative) | X-linked | 23.7 | VUS (PM2, BP1) | ||||
| ID1125 | FMR1 | NM_002024.5: c.1816C>T; p.Arg606Cys | Maternal (83.18%) | X-linked | 1/67871 | rs782778170 | 34 | VUS (PM1, PM2) | ||
| ID1128 | HUWE1 | NM_031407.6: c.1125G>T; p.Met375Ile | Maternal (86.55%) | Sib-pair | 0/41548 | rs1043071474 | 22.6 | VUS (PM1, PM2) | VUS (PM1, PM2) | |
| ID1205 | CASK | NM_003688.3: c.490G>A; p.Gly164Arg | Maternal (74.26%) | X-linked | 32 | VUS (PM1, PM2, PP3) | ||||
| ID1206 | HUWE1 | NM_031407.6: c.12209C>G; p.Ser4070Cys | Maternal (97.31%) | Sib-pair | 24 | VUS (PM1, PM2) | ||||
| ID1304 | CCDC22 | NM_014008.4: c.1388C>G; p.Ala463Gly | Maternal (54.33%) | X-linked | 1/73246 | rs782691732 | 26.9 | VUS (PM2) | ||
| ID1307 | PRPS1 | NM_002764.3: c.611G>A; p.Arg204His | Maternal (uninformative) | X-linked | rs1169615098 | 24 | VUS (PM1, PM2, PP2) | |||
| ID1402 | SYN1 | NM_006950.3: c.796G>A; p.Val266Met | Maternal (69.45%) | X-linked | rs1327735600 | 26.4 | VUS (PM1, PM2) | VUS (PM1, PM2, PP3) | ||
| ID1405 | MED12 | NM_005120.2: c.5009C>T; p.Ser1670Phe | Maternal (95.36%) | X-linked | 33 | VUS (PM2) | VUS (PM2, PP1, PP3) | |||
| Benign/likely benign variants | ||||||||||
| ID1208 | MAOA | NM_000240.3: c.617G>A; p.Arg206Gln | Maternal (53.7%) | X-linked | rs1218703391 | 31 | VUS (PM1, PM2, PP3) | Likely benign (PM1, PM2, PP3, BS2, BP5) | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibarluzea, N.; de la Hoz, A.B.; Villate, O.; Llano, I.; Ocio, I.; Martí, I.; Guitart, M.; Gabau, E.; Andrade, F.; Gener, B.; et al. Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability. Genes 2020, 11, 51. https://doi.org/10.3390/genes11010051
Ibarluzea N, de la Hoz AB, Villate O, Llano I, Ocio I, Martí I, Guitart M, Gabau E, Andrade F, Gener B, et al. Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability. Genes. 2020; 11(1):51. https://doi.org/10.3390/genes11010051
Chicago/Turabian StyleIbarluzea, Nekane, Ana Belén de la Hoz, Olatz Villate, Isabel Llano, Intzane Ocio, Itxaso Martí, Miriam Guitart, Elisabeth Gabau, Fernando Andrade, Blanca Gener, and et al. 2020. "Targeted Next-Generation Sequencing in Patients with Suggestive X-Linked Intellectual Disability" Genes 11, no. 1: 51. https://doi.org/10.3390/genes11010051