Parallelized Latent Dirichlet Allocation Provides a Novel Interpretability of Mutation Signatures in Cancer Genomes


Abstract
:1. Introduction
2. Methods
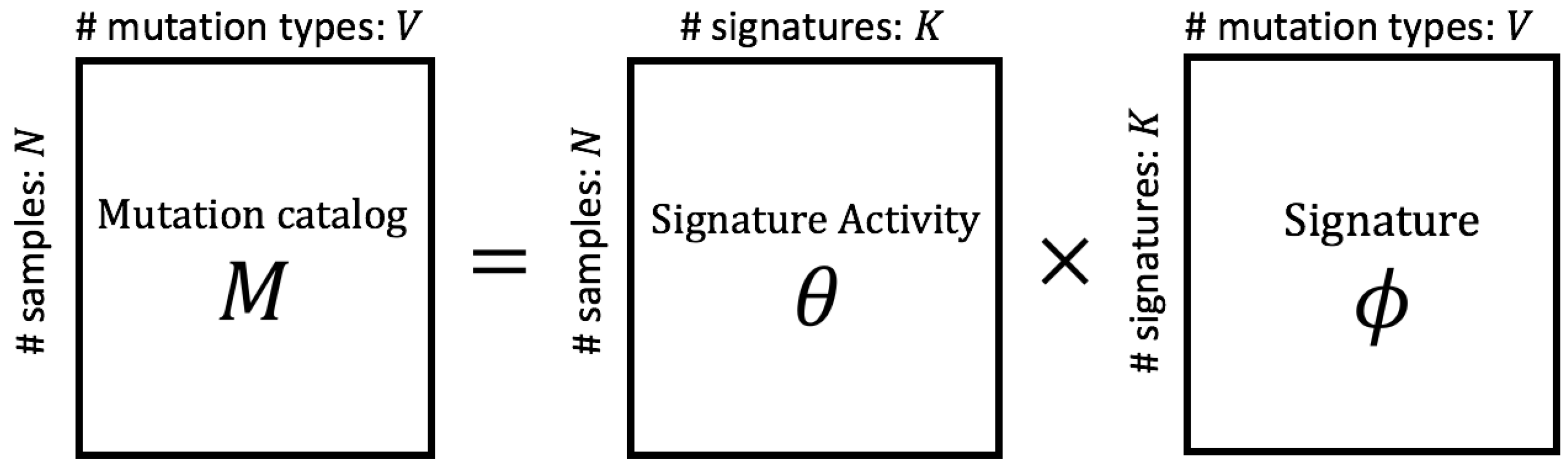
2.1. Modeling Mutation Catalogs
2.2. The Property of PLDA
2.3. Comparison with Other Similar Models
3. Result
3.1. Simulation with Artificial Data Following Generation Process of PLDA
3.2. Simulation with PCAWG Synthetic Data
3.3. Real Data Analysis
4. Discussion
4.1. Difficulty to Predict Signatures with Some Tumor Types
4.2. Some Important Signatures Could Not Be Extracted by PLDA
4.3. Perspective
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NMF | Non-negative Matrix Factorization |
| LDA | latent Dirichlet allocation |
| PLDA | Parallelized latent Dirichlet allocation |
| PCA | principal component analysis |
| t-SNE | t-distributed Stochastic Neighbor Embedding |
| SBS | Single Base Substitution |
| PCAWG | Pan Cancer Analysis of Whole Genomes |
| RR | Reconstruction Rate |
| CNS | Central Nervous System |
| HCC | Hepatocellular Carcinoma |
| Oligo | Oligodendroglioma |
| AdenoCa | Adeno Carcinoma |
References
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [PubMed] [Green Version]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res. Mol. Mech. Mutagen. 2005, 571, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S. Cancer mutation signatures, DNA damage mechanisms, and potential clinical implications. Genome Med. 2013, 5, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.D.; Seung, H.S. Algorithms for non-negative matrix factorization. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2001; pp. 556–562. [Google Scholar]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415. [Google Scholar]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helleday, T.; Eshtad, S.; Nik-Zainal, S. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014, 15, 585. [Google Scholar]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82. [Google Scholar] [CrossRef] [Green Version]
- Fischer, A.; Illingworth, C.J.; Campbell, P.J.; Mustonen, V. EMu: Probabilistic inference of mutational processes and their localization in the cancer genome. Genome Biol. 2013, 14, R39. [Google Scholar] [CrossRef] [Green Version]
- Rosales, R.A.; Drummond, R.D.; Valieris, R.; Dias-Neto, E.; da Silva, I.T. signeR: An empirical Bayesian approach to mutational signature discovery. Bioinformatics 2016, 33, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, Y.; Tremmel, G.; Miyano, S.; Stephens, M. A simple model-based approach to inferring and visualizing cancer mutation signatures. PLoS Genet. 2015, 11, e1005657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsutani, T.; Ueno, Y.; Fukunaga, T.; Hamada, M. Discovering novel mutation signatures by latent Dirichlet allocation with variational Bayes inference. Bioinformatics 2019, 35, 4543–4552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Owusu, M.; Harris, R.; Jackson, S.P.; Loizou, J.I.; Nik-Zainal, S. Validating the concept of mutational signatures with isogenic cell models. Nat. Commun. 2018, 9, 1744. [Google Scholar] [CrossRef] [Green Version]
- Haradhvala, N.; Kim, J.; Maruvka, Y.; Polak, P.; Rosebrock, D.; Livitz, D.; Hess, J.; Leshchiner, I.; Kamburov, A.; Mouw, K.; et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat. Commun. 2018, 9, 1746. [Google Scholar] [CrossRef] [Green Version]
- Tan, V.Y.; Févotte, C. Automatic relevance determination in nonnegative matrix factorization with the/spl beta/-divergence. IEEE Trans. Pattern Anal. Mach. Intell. 2012, 35, 1592–1605. [Google Scholar] [CrossRef]
- Mcauliffe, J.D.; Blei, D.M. Supervised topic models. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2008; pp. 121–128. [Google Scholar]
- Blei, D.M.; Ng, A.Y.; Jordan, M.I. Latent dirichlet allocation. J. Mach. Learn. Res. 2003, 3, 993–1022. [Google Scholar]
- Corduneanu, A.; Bishop, C.M. Variational Bayesian model selection for mixture distributions. In Artificial intelligence and Statistics; Morgan Kaufmann: Waltham, MA, USA, 2001; Volume 2001, pp. 27–34. [Google Scholar]
- Van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Nakatani, Y.; McLysaght, A. Genomes as documents of evolutionary history: A probabilistic macrosynteny model for the reconstruction of ancestral genomes. Bioinformatics 2017, 33, i369–i378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, S.; Nishijima, S.; Fukunaga, T.; Hattori, M.; Hamada, M. Revealing the microbial assemblage structure in the human gut microbiome using latent Dirichlet allocation. Microbiome 2020, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Boot, A.; Huang, M.N.; Ng, A.W.; Ho, S.C.; Lim, J.Q.; Kawakami, Y.; Chayama, K.; Teh, B.T.; Nakagawa, H.; Rozen, S.G. In-depth characterization of the cisplatin mutational signature in human cell lines and in esophageal and liver tumors. Genome Res. 2018, 28, 654–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.N.; Yu, W.; Teoh, W.W.; Ardin, M.; Jusakul, A.; Ng, A.W.T.; Boot, A.; Abedi-Ardekani, B.; Villar, S.; Myint, S.S.; et al. Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors. Genome Res. 2017, 27, 1475–1486. [Google Scholar] [CrossRef] [Green Version]
- Rustad, E.H.; Yellapantula, V.; Leongamornlert, D.; Bolli, N.; Ledergor, G.; Nadeu, F.; Angelopoulos, N.; Dawson, K.J.; Mitchell, T.J.; Osborne, R.J.; et al. Timing the initiation of multiple myeloma. Nat. Commun. 2020, 11, 1–14. [Google Scholar]
- Maura, F.; Degasperi, A.; Nadeu, F.; Leongamornlert, D.; Davies, H.; Moore, L.; Royo, R.; Ziccheddu, B.; Puente, X.S.; Avet-Loiseau, H.; et al. A practical guide for mutational signature analysis in hematological malignancies. Nat. Commun. 2019, 10, 1–12. [Google Scholar]
- Steuer, C.E.; Ramalingam, S.S. Tumor mutation burden: Leading immunotherapy to the era of precision medicine. J. Clin. Oncol. 2018, 36, 631–632. [Google Scholar]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular determinants of response to anti–programmed cell death (PD)-1 and anti–programmed death-ligand 1 (PD-L1) blockade in patients with non–small-cell lung cancer profiled with targeted next-generation sequencing. J. Clin. Oncol. 2018, 36, 633. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | SBS1 | SBS2 | SBS3 | SBS4 | SBS5 |
|---|---|---|---|---|---|
| 1st | ∘ | ∘ | × | × | × |
| 2nd | × | ∘ | ∘ | × | × |
| 3rd | × | × | ∘ | ∘ | × |
| 4th | × | × | × | ∘ | ∘ |
| 5th | ∘ | × | × | × | ∘ |
| Methods\ | 2 | 3 | 4 | 5 (True) | 6 | 7 | 8 | Total |
|---|---|---|---|---|---|---|---|---|
| PLDA (Proposed) | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 100 |
| SignatureAnalyzer | 0 | 0 | 0 | 100 | 0 | 0 | 0 | 100 |
| SigProfiler | 0 | 0 | 91 | 8 | 1 | 0 | 0 | 100 |
| Normal LDA | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 100 |
| Method | SBS1 | SBS2 | SBS3 | SBS4 | SBS5 |
|---|---|---|---|---|---|
| PLDA (Proposed) | 100 | 100 | 100 | 100 | 100 |
| SignatureAnalyzer | 100 | 100 | 100 | 100 | 100 |
| SigProfiler | 100 | 100 | 9 | 100 | 100 |
| Normal LDA | 100 | 100 | 98 | 0 | 0 |
| Method | Correct Matched | Duplicated | Not Matched | Total |
|---|---|---|---|---|
| PLDA (Proposed) | 500 | 0 | 0 | 500 |
| SignatureAnalyzer | 500 | 0 | 0 | 500 |
| SigProfiler | 409 | 0 | 1 | 410 |
| Normal LDA | 298 | 0 | 2 | 300 |
| Tumor Type | SBS1 | SBS2 | SBS3 | SBS4 | SBS5 | SBS6 | SBS7-a | SBS7-b | SBS7-c | SBS7-d |
|---|---|---|---|---|---|---|---|---|---|---|
| 1st | ∘ | ∘ | × | × | × | × | × | × | × | × |
| 2nd | × | ∘ | ∘ | × | × | × | × | × | × | × |
| 3rd | × | × | ∘ | ∘ | × | × | × | × | × | × |
| 4th | × | × | × | ∘ | ∘ | × | × | × | × | × |
| 5th | ∘ | × | × | × | ∘ | × | × | × | × | × |
| 6th | × | × | × | × | × | ∘ | × | × | × | × |
| 7th | × | × | × | × | × | × | ∘ | × | × | × |
| 8th | × | × | × | × | × | × | × | ∘ | × | × |
| 9th | × | × | × | × | × | × | × | × | ∘ | × |
| 10th | × | × | × | × | × | × | × | × | × | ∘ |
| Methods \ | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 (true) | 11 | 12 | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PLDA (Proposed) | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 72 | 26 | 1 | 0 | 100 |
| SignatureAnalyzer | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 93 | 7 | 0 | 0 | 100 |
| SigProfiler | 0 | 0 | 0 | 0 | 0 | 7 | 26 | 29 | 38 | 0 | 0 | 100 |
| Normal LDA | 0 | 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 100 |
| Method | SBS1 | SBS2 | SBS3 | SBS4 | SBS5 | SBS6 | SBS7-a | SBS7-b | SBS7-c | SBS7-d |
|---|---|---|---|---|---|---|---|---|---|---|
| PLDA (Proposed) | 100 | 100 | 95 | 100 | 96 | 79 | 22 | 98 | 100 | 100 |
| SignatureAnalyzer | 100 | 100 | 99 | 100 | 64 | 0 | 0 | 97 | 100 | 100 |
| SigProfiler | 100 | 100 | 46 | 100 | 0 | 0 | 0 | 35 | 100 | 100 |
| Normal LDA | 100 | 100 | 89 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Method | Correct Matched | Duplicated | Not Matched | Total |
|---|---|---|---|---|
| PLDA (Proposed) | 890 | 0 | 37 | 927 |
| SignatureAnalyzer | 760 | 0 | 147 | 907 |
| SigProfiler | 581 | 0 | 317 | 898 |
| Normal LDA | 289 | 0 | 11 | 300 |
| Predicted | Matched | Cosine Similarity | True | Matched | Cosine Similarity |
|---|---|---|---|---|---|
| 1 | SBS2 | 0.7780 | SBS1 | 15 | 1.0000 |
| 2 | SBS13 | 0.9987 | SBS13 | 2 | 0.9987 |
| 3 | SBS5 | 0.8729 | SBS15 | 13 | 0.9946 |
| 4 | SBS3 | 0.9155 | SBS17a | 14 | 0.9999 |
| 5 | SBS26 | 0.9691 | SBS17b | 7 | 1.0000 |
| 6 | SBS5 | 0.8792 | SBS18 | 9 | 0.9933 |
| 7 | SBS17b | 1.0000 | SBS2 | 17 | 0.9999 |
| 8 | SBS22 | 0.9858 | SBS21 | 5 | 0.7372 |
| 9 | SBS18 | 0.9933 | SBS22 | 8 | 0.9858 |
| 10 | SBS41 | 0.9856 | SBS26 | 5 | 0.9691 |
| 11 | SBS40 | 0.7277 | SBS28 | 11 | 0.7042 |
| 12 | SBS4 | 0.9308 | SBS29 | 9 | 0.8110 |
| 13 | SBS15 | 0.9946 | SBS3 | 4 | 0.9155 |
| 14 | SBS17a | 0.9999 | SBS30 | 6 | 0.7484 |
| 15 | SBS1 | 1.0000 | SBS4 | 12 | 0.9308 |
| 16 | SBS8 | 0.9320 | SBS40 | 16 | 0.7617 |
| 17 | SBS2 | 0.9999 | SBS41 | 10 | 0.9856 |
| 18 | SBS44 | 0.9446 | SBS44 | 18 | 0.9446 |
| 19 | SBS5 | 0.8762 | SBS5 | 6 | 0.8792 |
| - | - | - | SBS8 | 16 | 0.9320 |
| - | - | - | SBS9 | 11 | 0.7027 |
| Method | # Extracted (True:21) | Avg. Cosine Similarity | Reconstruction Rate |
|---|---|---|---|
| PLDA (proposed) | 16 | 0.9194 | 0.9895 |
| SigProfiler | 16 | 0.9387 | 0.9949 |
| SignatureAnalyzer | 19 | 0.9642 | 0.9018 |
| SBS1 | SBS2 | SBS3 | SBS4 | SBS5 | SBS6 | SBS7a | SBS7b | SBS7c | SBS7d |
| ∘ | ∘ | × | ∘ | × | ∘∘∘× | ∘ | |||
| SBS8 | SBS9 | SBS10a | SBS10b | SBS11 | SBS12 | SBS13 | SBS14 | SBS15 | SBS16 |
| ∘∘∘▵ | ∘ | ∘× | × | ∘ | ∘ | ∘ | ∘ | ||
| SBS17a | SBS17b | SBS18 | SBS19 | SBS20 | SBS21 | SBS22 | SBS23 | SBS24 | SBS25 |
| ∘ | ∘× | ∘ | ▵ | ∘× | ∘ | ||||
| SBS26 | SBS27 | SBS28 | SBS29 | SBS30 | SBS31 | SBS32 | SBS33 | SBS34 | SBS35 |
| ▵ | ∘∘ | × | ▵ | ||||||
| SBS36 | SBS37 | SBS38 | SBS39 | SBS40 | SBS41 | SBS42 | SBS43 | SBS44 | SBS45 |
| × | ▵ | ▵ | × | × | |||||
| SBS46 | SBS47 | SBS48 | SBS49 | SBS50 | SBS51 | SBS52 | SBS53 | SBS54 | SBS55 |
| × | |||||||||
| SBS56 | SBS57 | SBS58 | SBS59 | SBS60 | SBS84 | SBS85 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsutani, T.; Hamada, M. Parallelized Latent Dirichlet Allocation Provides a Novel Interpretability of Mutation Signatures in Cancer Genomes. Genes 2020, 11, 1127. https://doi.org/10.3390/genes11101127
Matsutani T, Hamada M. Parallelized Latent Dirichlet Allocation Provides a Novel Interpretability of Mutation Signatures in Cancer Genomes. Genes. 2020; 11(10):1127. https://doi.org/10.3390/genes11101127
Chicago/Turabian StyleMatsutani, Taro, and Michiaki Hamada. 2020. "Parallelized Latent Dirichlet Allocation Provides a Novel Interpretability of Mutation Signatures in Cancer Genomes" Genes 11, no. 10: 1127. https://doi.org/10.3390/genes11101127
APA StyleMatsutani, T., & Hamada, M. (2020). Parallelized Latent Dirichlet Allocation Provides a Novel Interpretability of Mutation Signatures in Cancer Genomes. Genes, 11(10), 1127. https://doi.org/10.3390/genes11101127