Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks

 , , ,
, , , 
Abstract
:1. Introduction
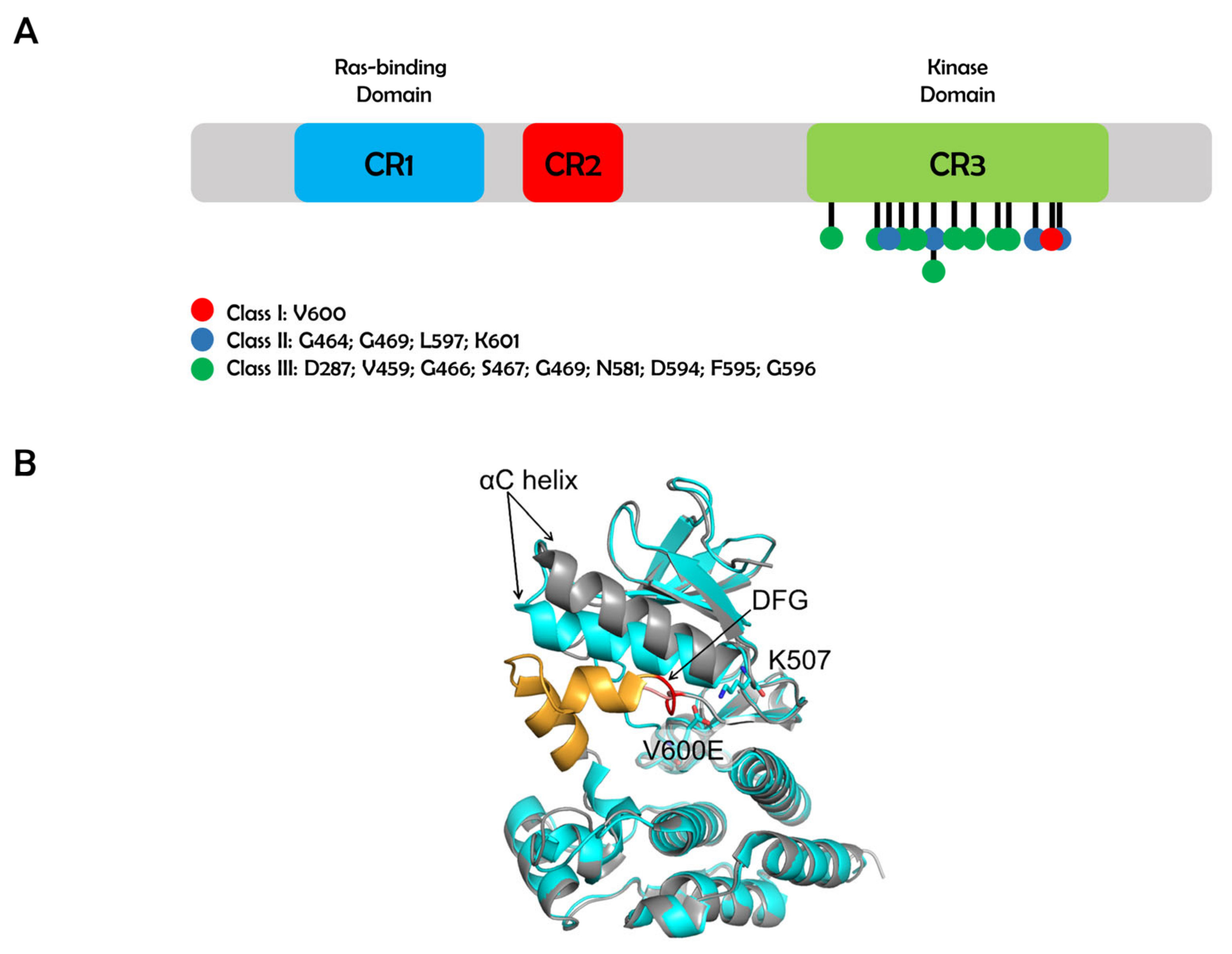
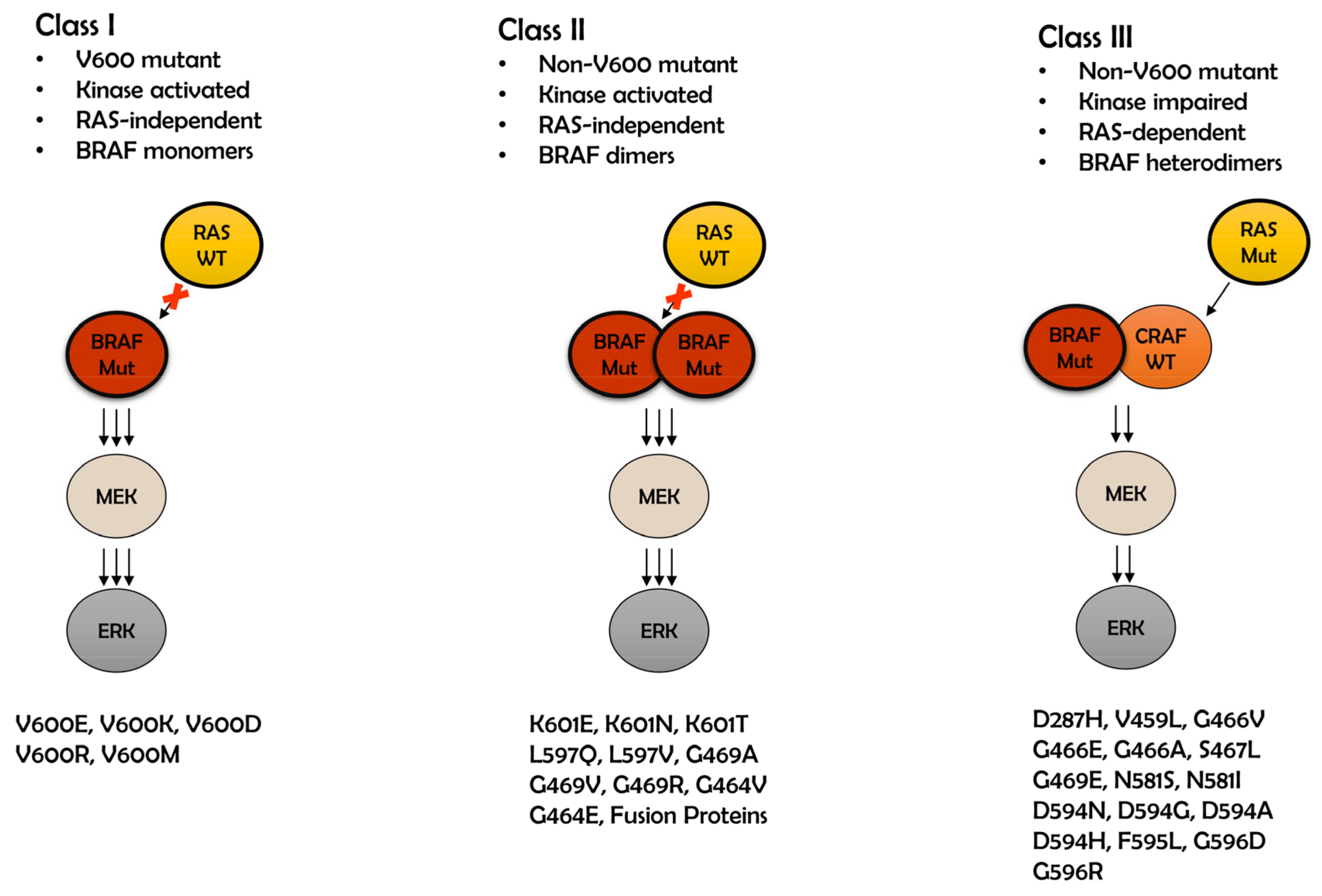
2. Class I BRAF Mutations
3. Class II BRAF Mutations
4. Class III BRAF Mutations
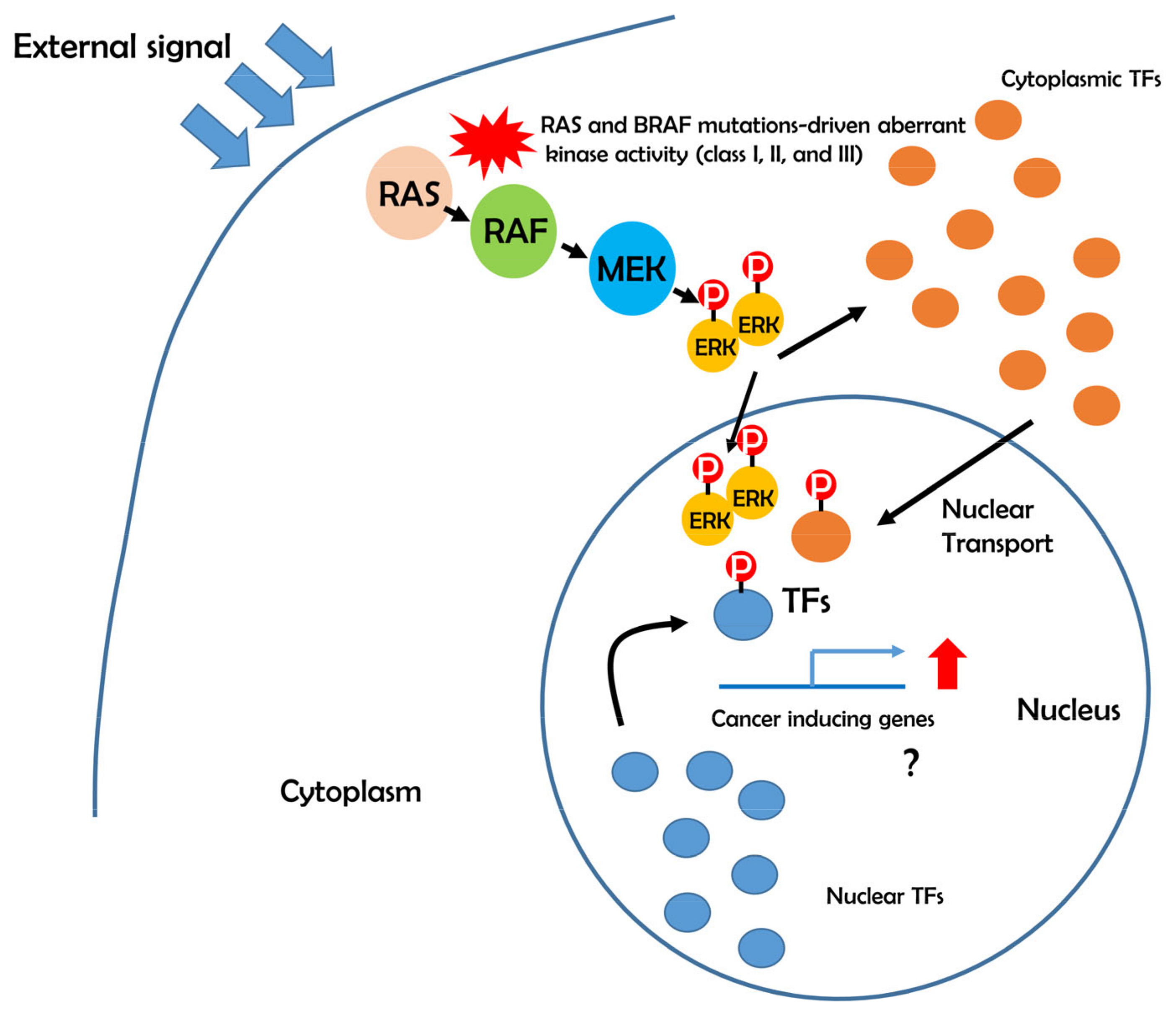
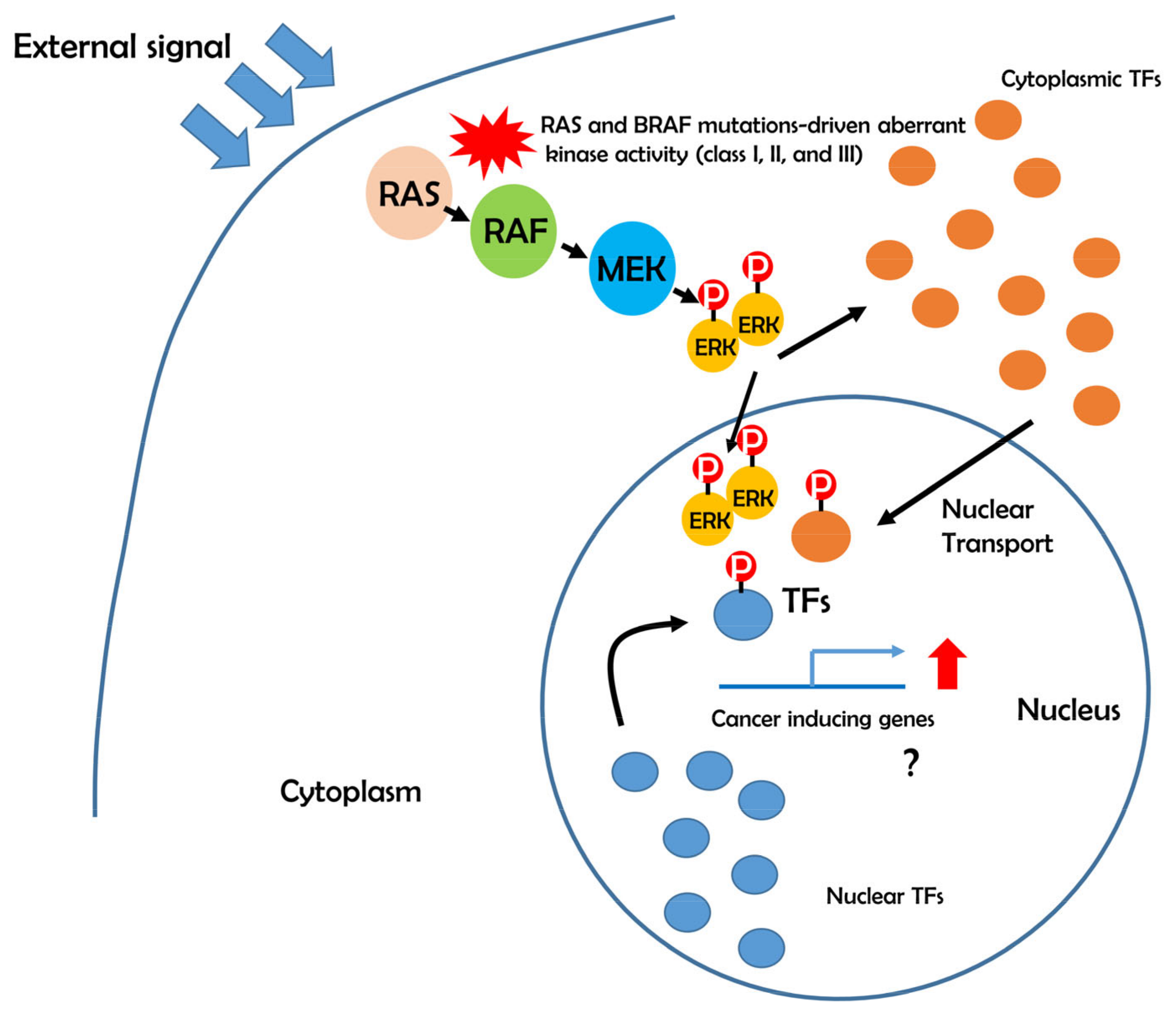
5. Possible Role of BRAF-ERK-TFs in Cancer Development
6. Aberrant Transcriptional Networks in BRAF Mutations
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kondoh, K.; Nishida, E. Regulation of MAP Kinases by MAP Kinase Phosphatases. Biochim. Biophys. Acta 2007, 1773, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.J.; Cobb, M.H. Mitogen-Activated Protein Kinase Pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Whitmarsh, A.J. Regulation of Gene Transcription by Mitogen-Activated Protein Kinase Signaling Pathways. Biochim. Biophys. Acta 2007, 1773, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.O.; Blenis, J. MAPK Signal Specificity: The Right Place at the Right Time. Trends Biochem. Sci. 2006, 31, 268–275. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Garnett, M.J.; Marais, R. Guilty as Charged: B-RAF is a Human Oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Zheng, G.; Tseng, L.H.; Chen, G.; Haley, L.; Illei, P.; Gocke, C.D.; Eshleman, J.R.; Lin, M.T. Clinical Detection and Categorization of Uncommon and Concomitant Mutations Involving BRAF. BMC Cancer 2015, 15, 779. [Google Scholar] [CrossRef] [Green Version]
- Aramini, J.M.; Vorobiev, S.M.; Tuberty, L.M.; Janjua, H.; Campbell, E.T.; Seetharaman, J.; Su, M.; Huang, Y.J.; Acton, T.B.; Xiao, R.; et al. The RAS-Binding Domain of Human BRAF Protein Serine/Threonine Kinase Exhibits Allosteric Conformational Changes upon Binding HRAS. Structure 2015, 23, 1382–1393. [Google Scholar] [CrossRef] [Green Version]
- Mandala, M.; Voit, C. Targeting BRAF in Melanoma: Biological and Clinical Challenges. Crit. Rev. Oncol. Hematol. 2013, 87, 239–255. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. RAF Protein-serine/threonine Kinases: Structure and Regulation. Biochem. Biophys. Res. Commun. 2010, 399, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Wenglowsky, S.; Ren, L.; Ahrendt, K.A.; Laird, E.R.; Aliagas, I.; Alicke, B.; Buckmelter, A.J.; Choo, E.F.; Dinkel, V.; Feng, B.; et al. Pyrazolopyridine Inhibitors of B-Raf(V600E). Part 1: The Development of Selective, Orally Bioavailable, and Efficacious Inhibitors. ACS Med. Chem. Lett. 2011, 2, 342–347. [Google Scholar] [CrossRef] [Green Version]
- Mason, C.S.; Springer, C.J.; Cooper, R.G.; Superti-Furga, G.; Marshall, C.J.; Marais, R. Serine and Tyrosine Phosphorylations Cooperate in Raf-1, but Not B-Raf Activation. EMBO J. 1999, 18, 2137–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.A.; Salajegheh, A.; Smith, R.A.; Lam, A.K. B-Raf Mutation: A Key Player in Molecular Biology of Cancer. Exp. Mol. Pathol. 2013, 95, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haling, J.R.; Sudhamsu, J.; Yen, I.; Sideris, S.; Sandoval, W.; Phung, W.; Bravo, B.J.; Giannetti, A.M.; Peck, A.; Masselot, A.; et al. Structure of the BRAF-MEK Complex Reveals a Kinase Activity Independent Role for BRAF in MAPK Signaling. Cancer. Cell 2014, 26, 402–413. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Rawson, S.; Li, K.; Kim, B.W.; Ficarro, S.B.; Pino, G.G.; Sharif, H.; Marto, J.A.; Jeon, H.; Eck, M.J. Architecture of Autoinhibited and Active BRAF-MEK1-14-3-3 Complexes. Nature 2019, 575, 545–550. [Google Scholar] [CrossRef]
- Cremolini, C.; Di Bartolomeo, M.; Amatu, A.; Antoniotti, C.; Moretto, R.; Berenato, R.; Perrone, F.; Tamborini, E.; Aprile, G.; Lonardi, S.; et al. BRAF Codons 594 and 596 Mutations Identify a New Molecular Subtype of Metastatic Colorectal Cancer at Favorable Prognosis. Ann. Oncol. 2015, 26, 2092–2097. [Google Scholar] [CrossRef]
- Wu, X.; Yan, J.; Dai, J.; Ma, M.; Tang, H.; Yu, J.; Xu, T.; Yu, H.; Si, L.; Chi, Z.; et al. Mutations in BRAF Codons 594 and 596 Predict Good Prognosis in Melanoma. Oncol. Lett. 2017, 14, 3601–3605. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.M.; Ashby, C.; Tytarenko, R.; Deshpande, S.; Wang, Y.; Sawyer, J.; Tian, E.; Johnson, S.; Rutherford, M.W.; Wardell, C.P.; et al. BRAF and DIS3 Mutations Associate with Adverse Outcome in a Long-Term Follow-Up of Patients with Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2422–2432. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of Novel Mutational Drivers Reveals Oncogene Dependencies in Multiple Myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with Class 3 BRAF Mutants are Sensitive to the Inhibition of Activated RAS. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Zhang, H.; Ding, H.; Qian, J.; Lizaso, A.; Lin, J.; Han-Zhang, H.; Xiang, J.; Li, Y.; Zhu, H. The Association between BRAF Mutation Class and Clinical Features in BRAF-Mutant Chinese Non-Small Cell Lung Cancer Patients. J. Transl. Med. 2019, 17, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF Gene in Human Cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Houben, R.; Becker, J.C.; Kappel, A.; Terheyden, P.; Brocker, E.B.; Goetz, R.; Rapp, U.R. Constitutive Activation of the Ras-Raf Signaling Pathway in Metastatic Melanoma is Associated with Poor Prognosis. J. Carcinog. 2004, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Ikenoue, T.; Hikiba, Y.; Kanai, F.; Tanaka, Y.; Imamura, J.; Imamura, T.; Ohta, M.; Ijichi, H.; Tateishi, K.; Kawakami, T.; et al. Functional Analysis of Mutations within the Kinase Activation Segment of B-Raf in Human Colorectal Tumors. Cancer Res. 2003, 63, 8132–8137. [Google Scholar]
- Lin, K.L.; Wang, O.C.; Zhang, X.H.; Dai, X.X.; Hu, X.Q.; Qu, J.M. The BRAF Mutation is Predictive of Aggressive Clinicopathological Characteristics in Papillary Thyroid Microcarcinoma. Ann. Surg. Oncol. 2010, 17, 3294–3300. [Google Scholar] [CrossRef]
- Naoki, K.; Chen, T.H.; Richards, W.G.; Sugarbaker, D.J.; Meyerson, M. Missense Mutations of the BRAF Gene in Human Lung Adenocarcinoma. Cancer Res. 2002, 62, 7001–7003. [Google Scholar]
- Sadlecki, P.; Walentowicz, P.; Bodnar, M.; Marszalek, A.; Grabiec, M.; Walentowicz-Sadlecka, M. Determination of BRAF V600E (VE1) Protein Expression and BRAF Gene Mutation Status in Codon 600 in Borderline and Low-Grade Ovarian Cancers. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E Mutation in Multiple Myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef] [Green Version]
- Wimmer, R.; Baccarini, M. Partner Exchange: Protein-Protein Interactions in the Raf Pathway. Trends Biochem. Sci. 2010, 35, 660–668. [Google Scholar] [CrossRef]
- Cantwell-Dorris, E.R.; O’Leary, J.J.; Sheils, O.M. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol. Cancer Ther. 2011, 10, 385–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Therrien, M. Regulation of RAF Protein Kinases in ERK Signalling. Nat. Rev. Mol. Cell Biol. 2015, 16, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Schirripa, M.; Biason, P.; Lonardi, S.; Pella, N.; Pino, M.S.; Urbano, F.; Antoniotti, C.; Cremolini, C.; Corallo, S.; Pietrantonio, F.; et al. Class 1, 2, and 3 BRAF-Mutated Metastatic Colorectal Cancer: A Detailed Clinical, Pathologic, and Molecular Characterization. Clin. Cancer Res. 2019, 25, 3954–3961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, S.; De Falco, V.; Tamburrino, A.; Barbi, F.; Tavano, M.; Avenia, N.; Santeusanio, F.; Santoro, M.; Macchiarulo, A.; Puxeddu, E. Insights into the Molecular Function of the Inactivating Mutations of B-Raf Involving the DFG Motif. Biochim. Biophys. Acta 2009, 1793, 1634–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hari, S.B.; Merritt, E.A.; Maly, D.J. Sequence Determinants of a Specific Inactive Protein Kinase Conformation. Chem. Biol. 2013, 20, 806–815. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, R.S.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L., Jr.; Levy, R.M. Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.N.; Lowe, E.D.; Noble, M.E.; Owen, D.J. The Eleventh Datta Lecture. the Structural Basis for Substrate Recognition and Control by Protein Kinases. FEBS Lett. 1998, 430, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Garnett, M.J.; Rana, S.; Paterson, H.; Barford, D.; Marais, R. Wild-Type and Mutant B-RAF Activate C-RAF through Distinct Mechanisms Involving Heterodimerization. Mol. Cell 2005, 20, 963–969. [Google Scholar] [CrossRef]
- Heidorn, S.J.; Milagre, C.; Whittaker, S.; Nourry, A.; Niculescu-Duvas, I.; Dhomen, N.; Hussain, J.; Reis-Filho, J.S.; Springer, C.J.; Pritchard, C.; et al. Kinase-Dead BRAF and Oncogenic RAS Cooperate to Drive Tumor Progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef]
- Scholl, F.A.; Dumesic, P.A.; Khavari, P.A. Effects of Active MEK1 Expression in Vivo. Cancer Lett. 2005, 230, 1–5. [Google Scholar] [CrossRef]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int. J. Mol. Sci. 2019, 20, 1194. [Google Scholar] [CrossRef] [Green Version]
- Pulverer, B.J.; Fisher, C.; Vousden, K.; Littlewood, T.; Evan, G.; Woodgett, J.R. Site-Specific Modulation of c-Myc Cotransformation by Residues Phosphorylated in Vivo. Oncogene 1994, 9, 59–70. [Google Scholar]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-Dependent Phosphorylation Pathways Regulate Myc Protein Stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Ai, J.; Shen, A.; Chen, Y.; Wang, X.; Peng, X.; Chen, H.; Shen, Y.; Huang, M.; Ding, J.; et al. C-Myc Alteration Determines the Therapeutic Response to FGFR Inhibitors. Clin. Cancer Res. 2017, 23, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Lan, H.Y. Diverse Roles of TGF-beta/Smads in Renal Fibrosis and Inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in Cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Guo, Y.; Cui, W.; Pei, Y.; Xu, D. Platelets Promote Invasion and Induce Epithelial to Mesenchymal Transition in Ovarian Cancer Cells by TGF-Beta Signaling Pathway. Gynecol. Oncol. 2019, 153, 639–650. [Google Scholar] [CrossRef]
- Wang, X.L.; Huang, C. Difference of TGF-beta/Smads Signaling Pathway in Epithelial-Mesenchymal Transition of Normal Colonic Epithelial Cells Induced by Tumor-Associated Fibroblasts and Colon Cancer Cells. Mol. Biol. Rep. 2019, 46, 2749–2759. [Google Scholar] [CrossRef]
- Zipper, L.M.; Mulcahy, R.T. Erk Activation is Required for Nrf2 Nuclear Localization during Pyrrolidine Dithiocarbamate Induction of Glutamate Cysteine Ligase Modulatory Gene Expression in HepG2 Cells. Toxicol. Sci. 2003, 73, 124–134. [Google Scholar] [CrossRef]
- Cho, S.J.; Ryu, J.H.; Surh, Y.J. Ajoene, a Major Organosulfide found in Crushed Garlic, Induces NAD(P)H:Quinone Oxidoreductase Expression through Nuclear Factor E2-Related Factor-2 Activation in Human Breast Epithelial Cells. J. Cancer Prev. 2019, 24, 112–122. [Google Scholar] [CrossRef]
- Jang, H.J.; Hong, E.M.; Kim, M.; Kim, J.H.; Jang, J.; Park, S.W.; Byun, H.W.; Koh, D.H.; Choi, M.H.; Kae, S.H.; et al. Simvastatin Induces Heme Oxygenase-1 Via NF-E2-Related Factor 2 (Nrf2) Activation through ERK and PI3K/Akt Pathway in Colon Cancer. Oncotarget 2016, 7, 46219–46229. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Gou, X.; Cai, P.; Xu, C.; Cao, L.; Zhao, Z.; Huang, M.; Jin, J. Sesamin Enhances Nrf2-Mediated Protective Defense Against Oxidative Stress and Inflammation in Colitis Via AKT and ERK Activation. Oxidative Med. Cell. Longev. 2019, 2019, 2432416. [Google Scholar] [CrossRef]
- Bucolo, C.; Drago, F.; Maisto, R.; Romano, G.L.; D’Agata, V.; Maugeri, G.; Giunta, S. Curcumin Prevents High Glucose Damage in Retinal Pigment Epithelial Cells through ERK1/2-Mediated Activation of the Nrf2/HO-1 Pathway. J. Cell. Physiol. 2019, 234, 17295–17304. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [Green Version]
- Huilgol, D.; Venkataramani, P.; Nandi, S.; Bhattacharjee, S. Transcription Factors that Govern Development and Disease: An Achilles Heel in Cancer. Genes 2019, 10, 794. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.; Sanchez-Ferras, O.; Bouchard, M. GATA Transcription Factors in Development and Disease. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Bonner, A.E.; Lemon, W.J.; You, M. Gene Expression Signatures Identify Novel Regulatory Pathways during Murine Lung Development: Implications for Lung Tumorigenesis. J. Med. Genet. 2003, 40, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Li, X.; Liu, H.; Jiang, R.; Yang, M.; Zhang, M.; Wang, Y.; Zhao, Y.; Li, H. GATA6-Upregulating Autophagy Promotes TKI Resistance in Nonsmall Cell Lung Cancer. Cancer Biol. Ther. 2019, 20, 1206–1212. [Google Scholar] [CrossRef]
- Sun, Z.; Yan, B. Multiple Roles and Regulatory Mechanisms of the Transcription Factor GATA6 in Human Cancers. Clin. Genet. 2020, 97, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Halmos, B.; Basseres, D.S.; Monti, S.; D’Alo, F.; Dayaram, T.; Ferenczi, K.; Wouters, B.J.; Huettner, C.S.; Golub, T.R.; Tenen, D.G. A Transcriptional Profiling Study of CCAAT/enhancer Binding Protein Targets Identifies Hepatocyte Nuclear Factor 3 Beta as a Novel Tumor Suppressor in Lung Cancer. Cancer Res. 2004, 64, 4137–4147. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Shibai, Y.; Mitsushita, J.; Shang, W.H.; Hirose, K.; Kamata, T. Oncogenic Ras Upregulates NADPH Oxidase 1 Gene Expression through MEK-ERK-Dependent Phosphorylation of GATA-6. Oncogene 2008, 27, 4921–4932. [Google Scholar] [CrossRef] [Green Version]
- Sekiya, S.; Suzuki, A. Direct Conversion of Mouse Fibroblasts to Hepatocyte-Like Cells by Defined Factors. Nature 2011, 475, 390–393. [Google Scholar] [CrossRef]
- Huang, P.; He, Z.; Ji, S.; Sun, H.; Xiang, D.; Liu, C.; Hu, Y.; Wang, X.; Hui, L. Induction of Functional Hepatocyte-Like Cells from Mouse Fibroblasts by Defined Factors. Nature 2011, 475, 386–389. [Google Scholar] [CrossRef]
- Enane, F.O.; Shuen, W.H.; Gu, X.; Quteba, E.; Przychodzen, B.; Makishima, H.; Bodo, J.; Ng, J.; Chee, C.L.; Ba, R.; et al. GATA4 Loss of Function in Liver Cancer Impedes Precursor to Hepatocyte Transition. J. Clin. Investig. 2017, 127, 3527–3542. [Google Scholar] [CrossRef] [Green Version]
- Fan, T.T.; Hu, P.F.; Wang, J.; Wei, J.; Zhang, Q.; Ning, B.F.; Yin, C.; Zhang, X.; Xie, W.F.; Chen, Y.X.; et al. Regression Effect of Hepatocyte Nuclear Factor 4alpha on Liver Cirrhosis in Rats. J. Dig. Dis. 2013, 14, 318–327. [Google Scholar] [CrossRef]
- Ning, B.F.; Ding, J.; Yin, C.; Zhong, W.; Wu, K.; Zeng, X.; Yang, W.; Chen, Y.X.; Zhang, J.P.; Zhang, X.; et al. Hepatocyte Nuclear Factor 4 Alpha Suppresses the Development of Hepatocellular Carcinoma. Cancer Res. 2010, 70, 7640–7651. [Google Scholar] [CrossRef] [Green Version]
- Smallridge, R.C.; Chindris, A.M.; Asmann, Y.W.; Casler, J.D.; Serie, D.J.; Reddi, H.V.; Cradic, K.W.; Rivera, M.; Grebe, S.K.; Necela, B.M.; et al. RNA Sequencing Identifies Multiple Fusion Transcripts, Differentially Expressed Genes, and Reduced Expression of Immune Function Genes in BRAF (V600E) Mutant Vs BRAF Wild-Type Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2014, 99, E338–E347. [Google Scholar] [CrossRef]
- Qu, Y.; Yang, Q.; Liu, J.; Shi, B.; Ji, M.; Li, G.; Hou, P. C-Myc is Required for BRAF(V600E)-Induced Epigenetic Silencing by H3K27me3 in Tumorigenesis. Theranostics 2017, 7, 2092–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; Correa, A.J.; LoPresti, J.S.; Epstein, A.L. BRAF V600E in Papillary Thyroid Carcinoma is Associated with Increased Programmed Death Ligand 1 Expression and Suppressive Immune Cell Infiltration. Thyroid 2014, 24, 1385–1393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flockhart, R.J.; Webster, D.E.; Qu, K.; Mascarenhas, N.; Kovalski, J.; Kretz, M.; Khavari, P.A. BRAFV600E Remodels the Melanocyte Transcriptome and Induces BANCR to Regulate Melanoma Cell Migration. Genome Res. 2012, 22, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochi, M.; Hinoi, T.; Niitsu, H.; Miguchi, M.; Saito, Y.; Sada, H.; Sentani, K.; Sakamoto, N.; Oue, N.; Tashiro, H.; et al. Oncogenic Mutation in RAS-RAF Axis Leads to Increased Expression of GREB1, Resulting in Tumor Proliferation in Colorectal Cancer. Cancer Sci. 2020, 111, 3540–3549. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.M.; Gu, J.; Zhang, Y.S.; Wei, W.J.; Qu, N.; Wen, D.; Liao, T.; Shi, R.L.; Zhang, L.; Ji, Q.H.; et al. Aberrant Hypermethylation of the HOXD10 Gene in Papillary Thyroid Cancer with BRAFV600E Mutation. Oncol. Rep. 2018, 39, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Rusinek, D.; Swierniak, M.; Chmielik, E.; Kowal, M.; Kowalska, M.; Cyplinska, R.; Czarniecka, A.; Piglowski, W.; Korfanty, J.; Chekan, M.; et al. BRAFV600E-Associated Gene Expression Profile: Early Changes in the Transcriptome, Based on a Transgenic Mouse Model of Papillary Thyroid Carcinoma. PLoS ONE 2015, 10, e0143688. [Google Scholar] [CrossRef] [Green Version]
- Barros-Filho, M.C.; de Mello, J.B.H.; Marchi, F.A.; Pinto, C.A.L.; da Silva, I.C.; Damasceno, P.K.F.; Soares, M.B.P.; Kowalski, L.P.; Rogatto, S.R. GADD45B Transcript is a Prognostic Marker in Papillary Thyroid Carcinoma Patients Treated with Total Thyroidectomy and Radioiodine Therapy. Front. Endocrinol. 2020, 11, 269. [Google Scholar] [CrossRef]
- Rusinek, D.; Pfeifer, A.; Cieslicka, M.; Kowalska, M.; Pawlaczek, A.; Krajewska, J.; Szpak-Ulczok, S.; Tyszkiewicz, T.; Halczok, M.; Czarniecka, A.; et al. TERT Promoter Mutations and their Impact on Gene Expression Profile in Papillary Thyroid Carcinoma. Cancers 2020, 12, 1597. [Google Scholar] [CrossRef]
- Wongchenko, M.J.; McArthur, G.A.; Dreno, B.; Larkin, J.; Ascierto, P.A.; Sosman, J.; Andries, L.; Kockx, M.; Hurst, S.D.; Caro, I.; et al. Gene Expression Profiling in BRAF-Mutated Melanoma Reveals Patient Subgroups with Poor Outcomes to Vemurafenib that may be Overcome by Cobimetinib Plus Vemurafenib. Clin. Cancer Res. 2017, 23, 5238–5245. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Mutation | Diagnosis | References |
|---|---|---|
| D594A | Metastatic colorectal cancer | [18] |
| D594E | Melanoma Multiple myeloma | [19] [20] |
| D594G | Metastatic colorectal cancer Non-small cell lung cancers Multiple myeloma | [18] [8] [20] |
| D594H | Non-small cell lung cancers | [8] |
| D594N | Metastatic colorectal cancer Non-small cell lung cancers Multiple myeloma | [18] [8] [20,21] |
| Mutation ID | Genomic DNA Change | Location of Mutations | Clinical Significance | Donors Affected | Cancer Type |
|---|---|---|---|---|---|
| CLASS I | |||||
| MU62030 | chr7: g.140453136 A > T | V600E | Pathogenic | 814 | Bladder Urothelial Cancer Chronic Lymphocytic Leukemia Colon Adenocarcinoma Colorectal Cancer Brain Glioblastoma Multiforme Head and Neck Squamous Cell Carcinoma Kidney Renal Papillary Cell Carcinoma Brain Lower Grade Glioma Liver Cancer Lung Adenocarcinoma Malignant Lymphoma Skin Cancer Pediatric Brain Cancer Rectum Adenocarcinoma Skin Adenocarcinoma Skin Cutaneous Melanoma Thyroid Cancer Head and Neck Thyroid Carcinoma |
| MU32987175 | chr7: g.140453137 C > T | V600M | Likely pathogenic | 54 | Skin Cancer Skin Adenocarcinoma Skin Cutaneous Melanoma |
| MU40909253 | chr7: g.140453136 AC > TT | V600K | Pathogenic | 17 | Skin Cancer |
| MU44780501 | chr7: g.140453136 AC > CT | V600R | Pathogenic | 2 | Skin Cancer |
| MU44927644 | chr7: g.140453135CA > GT | V600D | - | 1 | Skin Cancer |
| CLASS II | |||||
| MU1846052 | chr7: g.140453134 T > C | K601E | Pathogenic | 16 | Chronic Lymphocytic Leukemia Lymphoid Neoplasm Diffuse Large B-Cell Lymphoma Skin Cancer Prostate Adenocarcinoma Skin Cutaneous Melanoma Gastric Adenocarcinoma Head and Neck Thyroid Carcinoma Uterine Corpus Endometrial Carcinoma |
| MU161538 | chr7: g.140481402 C > G | G469A | Pathogenic | 14 | Bladder Cancer Bladder Urothelial Cancer Chronic Lymphocytic Leukemia Colon Adenocarcinoma Early Onset Prostate Cancer Lung Adenocarcinoma Lung Squamous Cell Carcinoma Oral Cancer Prostate Adenocarcinoma Skin Cutaneous Melanoma Gastric Adenocarcinoma Skin Adenocarcinoma |
| MU86259478 | chr7: g.140481402 C > A | G469V | Pathogenic | 6 | Lung Adenocarcinoma Malignant Lymphoma |
| MU1299736 | chr7: g.140481403 C > T | G469R | Likely pathogenic | 6 | Lung Squamous Cell Carcinoma Skin Cancer Skin Cutaneous Melanoma |
| MU1334968 | chr7: g.140481417 C > A | G464V | Pathogenic/Likely pathogenic | 3 | Biliary Tract Cancer Lung Adenocarcinoma Lung Squamous Cell Carcinoma |
| MU4410750 | chr7: g.140453145 A > T | L597Q | Pathogenic/Likely pathogenic | 2 | Chronic Lymphocytic Leukemia Skin Cutaneous Melanoma |
| MU50026 | chr7: g.140453132 T > A | K601N | Likely pathogenic | 2 | Chronic Lymphocytic Leukemia Blood Cancer - T-Cell and Nk-Cell Lymphoma |
| MU44221302 | chr7: g.140453146 G > C | L597V | Pathogenic | 2 | Biliary Tract Cancer Colon Adenocarcinoma |
| MU129883017 | chr7: g.140481417 C > T | G464E | Pathogenic/Likely pathogenic | 1 | Uterine Corpus Endometrial Carcinoma |
| MU6236086 | chr7: g.140453133 T > G | K601T | Pathogenic/Likely pathogenic | 1 | Gastric Adenocarcinoma |
| CLASS III | |||||
| MU126831 | chr7: g.140453155 C > T | D594N | Likely pathogenic | 13 | Colon Adenocarcinoma Colorectal Cancer Head and Neck Squamous Cell Carcinoma Liver Cancer Lung Adenocarcinoma Lung Squamous Cell Carcinoma Malignant Lymphoma Skin Cutaneous Melanoma Gastric Adenocarcinoma |
| MU831694 | chr7: g.140453193 T > C | N581S | Likely pathogenic | 12 | Biliary Tract Cancer Chronic Lymphocytic Leukemia Kidney Renal Papillary Cell Carcinoma Liver Cancer Lung Adenocarcinoma Ovarian Serous Cystadenocarcinoma Skin Cutaneous Melanoma |
| MU168532 | chr7: g.140481411 C > A | G466V | Pathogenic/Likely pathogenic | 9 | Colon Adenocarcinoma Esophageal Adenocarcinoma Lung Adenocarcinoma Lung Squamous Cell Carcinoma Skin Cutaneous Melanoma |
| MU50763 | chr7: g.140453154 T > C | D594G | Pathogenic | 9 | Bladder Cancer Chronic Lymphocytic Leukemia Colorectal Cancer Brain Lower Grade Glioma Malignant Lymphoma Pancreatic Cancer Pancreatic Cancer Endocrine Neoplasms |
| MU4440100 | chr7: g.140481411 C > T | G466E | Likely pathogenic | 8 | Breast Er+ and Her2- Cancer Head and Neck Squamous Cell Carcinoma Skin Cancer Skin Cutaneous Melanoma |
| MU4420958 | chr7: g.140481408 G > A | S467L | - | 5 | Skin Cancer Skin Cutaneous Melanoma |
| MU1661062 | chr7: g.140481402 C > T | G469E | Pathogenic | 4 | Skin Cancer Oral Cancer Skin Cutaneous Melanoma |
| MU51987727 | chr7: g.140481411 C > G | G466A | Likely pathogenic | 2 | Lung Adenocarcinoma Skin Cancer |
| MU4622596 | chr7: g.140453149 C > G | G596R | Pathogenic/Likely pathogenic | 2 | Bladder Cancer Lung Adenocarcinoma |
| MU30632423 | chr7: g.140453154 T > G | D594A | Likely pathogenic | 2 | Colorectal Cancer Liver Cancer |
| MU63537540 | chr7: g.140453155 C > G | D594H | Pathogenic/Likely pathogenic | 2 | Colorectal Cancer Lung Adenocarcinoma |
| MU591874 | chr7: g.140453148 C > T | G596D | Likely pathogenic | 1 | Brain Glioblastoma Multiforme |
| MU4622598 | chr7: g.140453152 A > G | F595L | Pathogenic/Likely pathogenic | 1 | Bladder Cancer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. https://doi.org/10.3390/genes11111342
Śmiech M, Leszczyński P, Kono H, Wardell C, Taniguchi H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes. 2020; 11(11):1342. https://doi.org/10.3390/genes11111342
Chicago/Turabian StyleŚmiech, Magdalena, Paweł Leszczyński, Hidetoshi Kono, Christopher Wardell, and Hiroaki Taniguchi. 2020. "Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks" Genes 11, no. 11: 1342. https://doi.org/10.3390/genes11111342
APA StyleŚmiech, M., Leszczyński, P., Kono, H., Wardell, C., & Taniguchi, H. (2020). Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes, 11(11), 1342. https://doi.org/10.3390/genes11111342