Identification of ADPKD-Related Genes and Pathways in Cells Overexpressing PKD2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Cell Culture and Transfection
2.3. Western Blot Analysis
2.4. RNA-Seq
2.5. Enrichment Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. Statistics
2.8. Ethical Statement
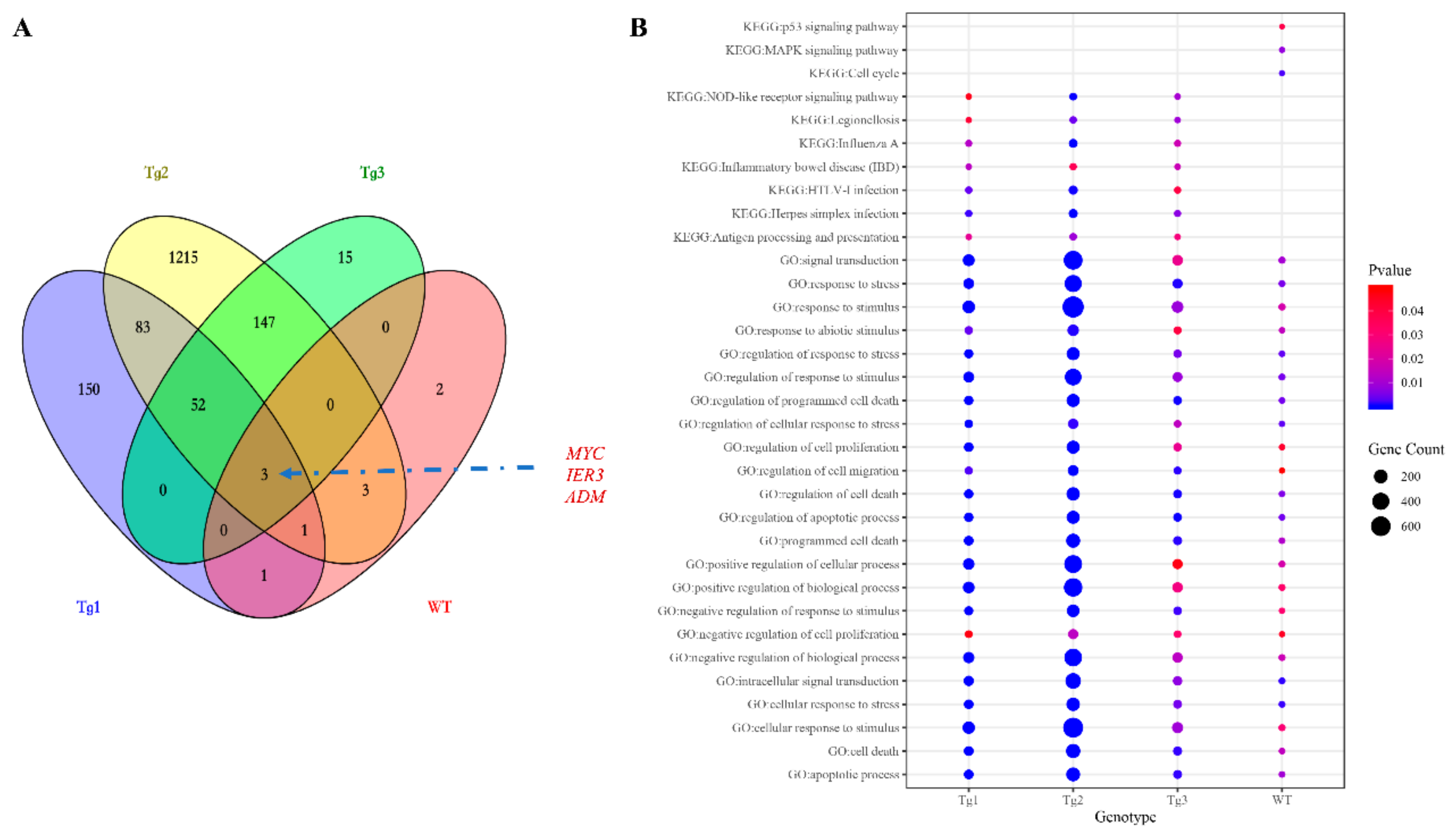
3. Results and Discussion
4. Conclusions
Abbreviations:
| ADPKD | Autosomal dominant polycystic kidney disease |
| PKD1/PKD2 | Polycystic kidney disease gene 1/2 |
| PC1/2 | Polycystin-1/2 |
| DEG | Differentially expressed gene |
| GO | Gene ontology; KEGG |
| Kyoto | Encyclopedia of Genes and Genomes |
| WT | Wild type |
| Tg | Transgenic |
| RNA-seq | mRNA sequencing |
| MAPK | Mitogen-activated protein kinase |
| cDNA | Complementary deoxyribonucleic acid |
| cAMP | Cyclic adenosin monophosphate |
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torres, V.E.; Harris, P.C. Autosomal dominant polycystic kidney disease: The last 3 years. Kidney Int. 2009, 76, 149–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabow, P.A. Autosomal Dominant Polycystic Kidney Disease—More Than a Renal Disease. Am. J. Kidney Dis. 1990, 16, 403–413. [Google Scholar] [CrossRef]
- Torres, V.E.; Higashihara, E.; Devuyst, O.; Chapman, A.B.; Gansevoort, R.T.; Grantham, J.J.; Perrone, R.D.; Ouyang, J.; Blais, J.D.; Czerwiec, F.S. Effect of Tolvaptan in Autosomal Dominant Polycystic Kidney Disease by CKD Stage: Results from the TEMPO 3:4 Trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, P.M.; Pound, S.E.; Carothers, A.D.; MacNicol, A.M.; Allan, P.L.; Watson, M.L.; Wright, A.F. Multipoint mapping of adult onset polycystic kidney disease (PKD1) on chromosome. J. Med. Genet. 1992, 29, 638–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a Gene for Polycystic Kidney Disease That Encodes an Integral Membrane Protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Chapin, H.C.; Caplan, M.J. The cell biology of polycystic kidney disease. J. Cell Biol. 2010, 191, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeders, S.T. Multilocus polycystic disease. Nat. Genet. 1992, 1, 235–237. [Google Scholar] [CrossRef]
- Pei, Y. A ‘two-hit’ model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol. Med. 2001, 7, 151–156. [Google Scholar] [CrossRef]
- Bastos, A.P.; Piontek, K.; Silva, A.M.; Martini, D.; Menezes, L.F.; Fonseca, J.M.; Fonseca, I.I.; Germino, G.G.; Onuchic, L.F. Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J. Am. Soc. Nephrol. 2009, 20, 2389–2402. [Google Scholar] [CrossRef] [Green Version]
- Takakura, A.; Contrino, L.; Zhou, X.; Bonventre, J.V.; Sun, Y.; Humphreys, B.D.; Zhou, J. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum. Mol. Genet. 2009, 18, 2523–2531. [Google Scholar] [CrossRef] [Green Version]
- Bell, P.D.; Fitzgibbon, W.; Sas, K.; Stenbit, A.E.; Amria, M.; Houston, A.; Reichert, R.; Gilley, S.; Siegal, G.P.; Bissler, J.; et al. Loss of primary cilia upregulates renal hypertrophic signaling and promotes cystogenesis. J. Am. Soc. Nephrol. 2011, 22, 839–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eccles, M.R.; Stayner, C.A. Polycystic kidney disease—Where gene dosage counts. F1000 Prime Rep. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.-F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ong, A.C.M.; Ward, C.J.; Butler, R.J.; Biddolph, S.; Bowker, C.; Torra, R.; Pei, Y.; Harris, P.C. Coordinate Expression of the Autosomal Dominant Polycystic Kidney Disease Proteins, Polycystin-2 And Polycystin-1, in Normal and Cystic Tissue. Am. J. Pathol. 1999, 154, 1721–1729. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.J.; Turley, H.; Ong, A.C.M.; Comley, M.; Biddolph, S.; Chetty, R.; Ratcliffe, P.J.; Gattner, K.; Harris, P.C. Polycystin, the polycystic kidney disease 1 protein, is expressed by epithelial cells in fetal, adult, and polycystic kidney. Proc. Natl. Acad. Sci. USA 1996, 93, 1524–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagao, S.; Kugita, M.; Yoshihara, D.; Yamaguchi, T. Animal models for human polycystic kidney disease. Exp. Anim. 2012, 61, 477–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.D. Mouse Models of Polycystic Kidney Disease. Curr. Top. Dev. Biol. 2008, 84, 311–350. [Google Scholar]
- He, J.; Ye, J.; Li, Q.; Feng, Y.; Bai, X.; Chen, X.; Wu, C.; Yu, Z.; Zhao, Y.; Hu, X.; et al. Construction of a transgenic pig model overexpressing polycystic kidney disease 2 (PKD2) gene. Transgenic Res. 2013, 22, 861–867. [Google Scholar] [CrossRef]
- Li, A.; Tian, X.; Zhang, X.; Huang, S.; Ma, Y.; Wu, D.; Moeckel, G.; Somlo, S.; Wu, G. Human polycystin-2 transgene dose-dependently rescues ADPKD phenotypes in Pkd2 mutant mice. Am. J. Pathol. 2015, 185, 2843–2860. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Fedeles, S.V.; Dong, K.; Anyatonwu, G.; Onoe, T.; Mitobe, M.; Gao, J.-D.; Okuhara, D.; Tian, X.; Gallagher, A.-R.; et al. Altered trafficking and stability of polycystins underlie polycystic kidney disease. J. Clin. Investig. 2014, 124, 5129–5144. [Google Scholar] [CrossRef] [Green Version]
- Ruan, J.; Li, H.; Xu, K.; Wu, T.; Wei, J.; Zhou, R.; Liu, Z.; Mu, Y.; Yang, S.; Ouyang, H.; et al. Highly efficient CRISPR/Cas9-mediated transgene knockin at the H11 locus in pigs. Sci. Rep. 2015, 5, 14253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; He, J.; Li, Q.; Feng, Y.; Bai, X.; Chen, X.; Zhao, Y.; Hu, X.; Yu, Z.; Li, N. Generation of c-Myc transgenic pigs for autosomal dominant polycystic kidney disease. Transgenic Res. 2013, 22, 1231–1239. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon: Fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [Green Version]
- Love, I.M.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunnen, S.J.; Malas, T.B.; Semeins, C.M.; Bakker, A.D.; Peters, D.J.M. Comprehensive transcriptome analysis of fluid shear stress altered gene expression in renal epithelial cells. J. Cell. Physiol. 2018, 233, 3615–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunnen, S.J.; Malas, T.B.; Formica, C.; Leonhard, W.N.; Hoen, P.A.T.; Peters, D.J. Comparative transcriptomics of shear stress treated Pkd1−/− cells and pre-cystic kidneys reveals pathways involved in early polycystic kidney disease. Biomed. Pharmacother. 2018, 108, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Devuyst, O.; Beauwens, R. Ion transport and cystogenesis: The paradigm of autosomal dominant polycystic kidney disease. Adv. Nephrol. Necker Hosp. 1998, 28, 439. [Google Scholar]
- Hull, R.N.; Cherry, W.R.; Weaver, G.W. The origin and characteristics of a pig kidney cell strain, LLC-PK. In Vitro 1976, 12, 670–677. [Google Scholar] [CrossRef]
- Autosomal Dominant Polycystic Kidney Disease Mutation Database: PKDB. Available online: http://pkdb.mayo.edu (accessed on 30 October 2019).
- Koulen, P.; Cai, Y.; Geng, L.; Maeda, Y.; Nishimura, S.; Witzgall, R.; Ehrlich, B.E.; Somlo, S. Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 2002, 4, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000, 408, 990–994. [Google Scholar] [CrossRef] [PubMed]
- Losekoot, M.; Ruivenkamp, C.A.L.; Tholens, A.P.; Grimbergen, J.E.M.A.; Vijfhuizen, L.; Vermeer, S.; Dijkman, H.B.; Cornelissen, E.A.M.; Bongers, E.M.H.F.; Peters, D.J.M. Neonatal onset autosomal dominant polycystic kidney disease (ADPKD) in a patient homozygous for a PKD2 missense mutation due to uniparental disomy. J. Med. Genet. 2012, 49, 37–40. [Google Scholar] [CrossRef]
- Grieben, M.; Pike, A.C.W.; Shintre, C.A.; Venturi, E.; El-Ajouz, S.; Tessitore, A.; Shrestha, L.; Mukhopadhyay, S.; Mahajan, P.; Chalk, R.; et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 2017, 24, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Trudel, M.; D’Agati, V.; Costantini, F. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int. 1991, 39, 665–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanoix, J.; D’Agati, V.; Szabolcs, M.; Trudel, M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 1996, 13, 1153–1160. [Google Scholar]
- Kurbegovic, A.; Côté, O.; Couillard, M.; Ward, C.J.; Harris, P.C.; Trudel, M. Pkd1 transgenic mice: Adult model of polycystic kidney disease with extrarenal and renal phenotypes. Hum. Mol. Genet. 2010, 19, 1174–1189. [Google Scholar] [CrossRef] [Green Version]
- Kurbegovic, A.; Trudel, M. Progressive development of polycystic kidney disease in the mouse model expressing Pkd1 extracellular domain. Hum. Mol. Genet. 2013, 22, 2361–2375. [Google Scholar] [CrossRef] [Green Version]
- Burtey, S.; Riera, M.; Ribe, E.; Pennekamp, P.; Passage, E.; Rance, R.; Dworniczak, B.; Fontés, M. Overexpression of PKD2 in the mouse is associated with renal tubulopathy. Nephrol. Dial. Transpl. 2008, 23, 1157–1165. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Sung, Y.H.; Yang, M.H.; Noh, J.Y.; Park, S.Y.; Lee, T.Y.; Yook, Y.J.; Yoo, K.H.; Roh, K.J.; Kim, I.; et al. Cyst formation in kidney via B-raf signaling in the PKD2 transgenic mice. J. Biol. Chem. 2009, 284, 7214–7222. [Google Scholar] [CrossRef] [Green Version]
- Parrot, C.; Kurbegovic, A.; Yao, G.; Couillard, M.; Côté, O.; Trudel, M. C-Myc is a regulator of the PKD1 gene and PC1-induced pathogenesis. Hum. Mol. Genet. 2019, 28, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, C.E.; Zhao, B.; Li, W.; Ouyang, Z.; Liu, Z.; Yang, H.; Fan, P.; O’Neill, A.; Gu, W.; et al. Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum. Mol. Genet. 2010, 19, 3983–3994. [Google Scholar] [CrossRef] [PubMed]
- Garrick, D.; Fiering, S.; Martin, D.I.K.; Whitelaw, E. Repeat-induced gene silencing in mammals. Nat. Genet. 1998, 18, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, P.; Rival-Gervier, S.; Houdebine, L.-M.; Montoliu, L. The potential benefits of insulators on heterologous constructs in transgenic animals. Transgenic Res. 2003, 12, 751–755. [Google Scholar] [CrossRef]
- McBurney, M.W.; Mai, T.; Yang, X.; Jardine, K. Evidence for Repeat-Induced Gene Silencing in Cultured Mammalian Cells: Inactivation of Tandem Repeats of Transfected Genes. Exp. Cell Res. 2002, 274, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-C.; Li, H.-P.; Hung, Y.-H.; Leu, Y.-W.; Wu, W.-H.; Wang, F.-S.; Lee, K.-D.; Chang, P.-J.; Wu, C.-S.; Lu, Y.-J.; et al. Targeted methylation of CMV and E1A viral promoters. Biochem. Biophys. Res. Commun. 2010, 402, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.R.; Harkins, R.N.; Wang, P.; Qian, H.S.; Liu, P.; Rubanyi, G.M. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J. Gene Med. 2004, 6, 395–404. [Google Scholar] [CrossRef]
- Koupepidou, P.; Felekkis, K.N.; Kränzlin, B.; Sticht, C.; Gretz, N.; Deltas, C. Cyst formation in the PKD2 (1-703) transgenic rat precedes deregulation of proliferation-related pathways. BMC Nephrol. 2010, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Verma, S.P.; Pandey, P. Profiling conserved biological pathways in Autosomal Dominant Polycystic Kidney Disorder (ADPKD) to elucidate key transcriptomic alterations regulating cystogenesis: A cross-species meta-analysis approach. Gene 2017, 627, 434–450. [Google Scholar] [CrossRef]
- Husson, H.; Manavalan, P.; Akmaev, V.R.; Russo, R.J.; Cook, B.; Richards, B.; Barberio, D.; Liu, D.; Cao, X.; Landes, G.M.; et al. New insights into ADPKD molecular pathways using combination of SAGE and microarray technologies. Genomics 2004, 84, 497–510. [Google Scholar] [CrossRef]
- Dweep, H.; Sticht, C.; Kharkar, A.; Pandey, P.; Gretz, N. Parallel Analysis of mRNA and microRNA Microarray Profiles to Explore Functional Regulatory Patterns in Polycystic Kidney Disease: Using PKD/Mhm Rat Model. PLoS ONE 2013, 8, e53780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, Y.M.; Kim, D.Y.; Koo, N.J.; Kim, Y.-M.; Lee, S.; Ko, J.Y.; Shin, Y.; Kim, B.H.; Mun, H.; Choi, S.; et al. Profiling of miRNAs and target genes related to cystogenesis in ADPKD mouse models. Sci. Rep. 2017, 7, 14151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dieplinger, B.; Mueller, T.; Kollerits, B.; Struck, J.; Ritz, E.; Von Eckardstein, A.; Haltmayer, M.; Kronenberg, F. Pro-A-type natriuretic peptide and pro-adrenomedullin predict progression of chronic kidney disease: The MMKD Study. Kidney Int. 2009, 75, 408–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishimitsu, T.; Nishikimi, T.; Saito, Y.; Kitamura, K.; Eto, T.; Kangawa, K.; Matsuo, H.; Omae, T.; Matsuoka, H. Plasma levels of adrenomedullin, a newly identified hypotensive peptide, in patients with hypertension and renal failure. J. Clin. Investig. 1994, 94, 2158–2161. [Google Scholar] [CrossRef] [Green Version]
- Dai, B.; David, V.; Martin, A.; Huang, J.; Li, H.; Jiao, Y.; Gu, W.; Quarles, L.D. A Comparative Transcriptome Analysis Identifying FGF23 Regulated Genes in the Kidney of a Mouse CKD Model. PLoS ONE 2012, 7, e44161. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Huo, Y.; Chen, S.; Xu, D.; Yang, B.; Xue, C.; Fu, L.; Bu, L.; Song, S.; Mei, C. Identification of Key Genes and Candidated Pathways in Human Autosomal Dominant Polycystic Kidney Disease by Bioinformatics Analysis. Kidney Blood Press. Res. 2019, 44, 533–552. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Du, P.; Fan, B.; Han, H.; Zhen, J.; Shang, J.; Wang, X.; Li, X.; Shi, W.; Tang, W.; Bao, C.; et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int. 2013, 84, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Magenheimer, B.S.; Xia, S.; Johnson, T.; Wallace, D.P.; Calvet, J.P.; Li, R. A tumor necrosis factor-α-mediated pathway promoting autosomal dominant polycystic kidney disease. Nat. Med. 2008, 14, 863–868. [Google Scholar] [CrossRef] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Dang, Y.; Wang, Z.; Wang, H.; Pan, Y.; He, J. Identification of ADPKD-Related Genes and Pathways in Cells Overexpressing PKD2. Genes 2020, 11, 122. https://doi.org/10.3390/genes11020122
Zhang Z, Dang Y, Wang Z, Wang H, Pan Y, He J. Identification of ADPKD-Related Genes and Pathways in Cells Overexpressing PKD2. Genes. 2020; 11(2):122. https://doi.org/10.3390/genes11020122
Chicago/Turabian StyleZhang, Zhe, Yanna Dang, Zizengceng Wang, Huanan Wang, Yuchun Pan, and Jin He. 2020. "Identification of ADPKD-Related Genes and Pathways in Cells Overexpressing PKD2" Genes 11, no. 2: 122. https://doi.org/10.3390/genes11020122