Transcriptomic Analysis Reveals the Molecular Adaptation of Three Major Secondary Metabolic Pathways to Multiple Macronutrient Starvation in Tea (Camellia sinensis)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Stress Treatments
2.2. Library Construction and Sequencing
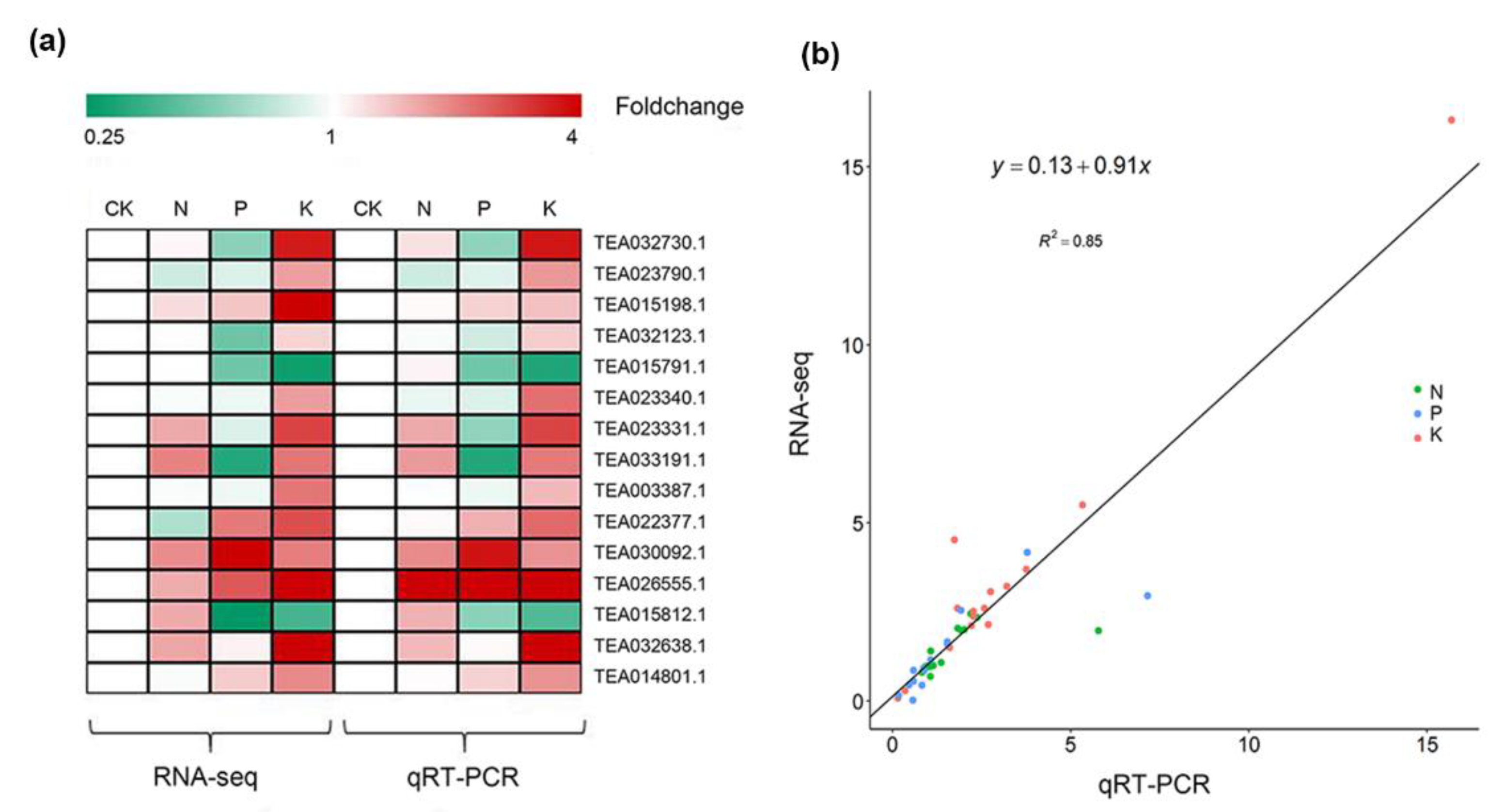
2.3. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Analysis
2.4. Extraction and Determination of Catechins, l-theanine, and Caffeine in Tea
3. Results
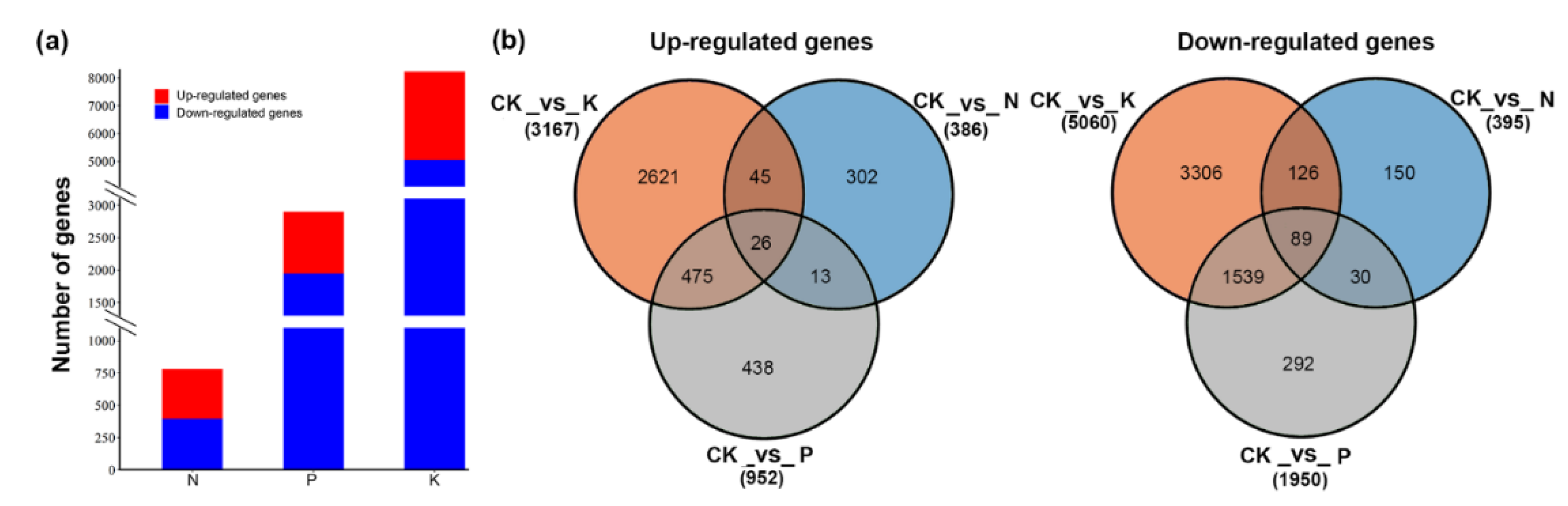
3.1. Transcriptome Analysis of Tea Plant Responses to Macronutrient Deficiency
3.2. Identification of Differentially Expressed Genes under Nitrogen (N), Phosphate (P), and Potassium (K) Starvation Conditions
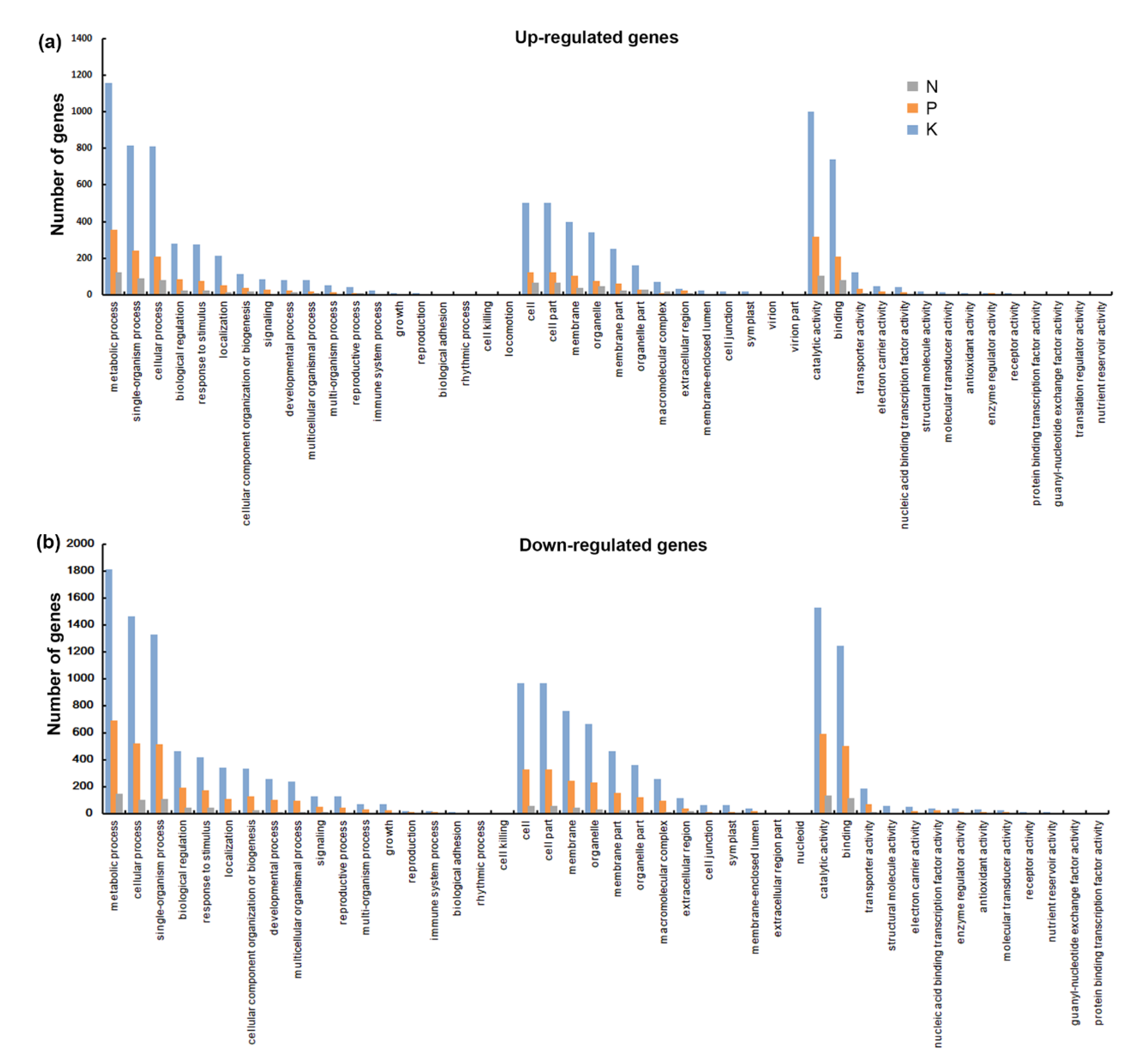
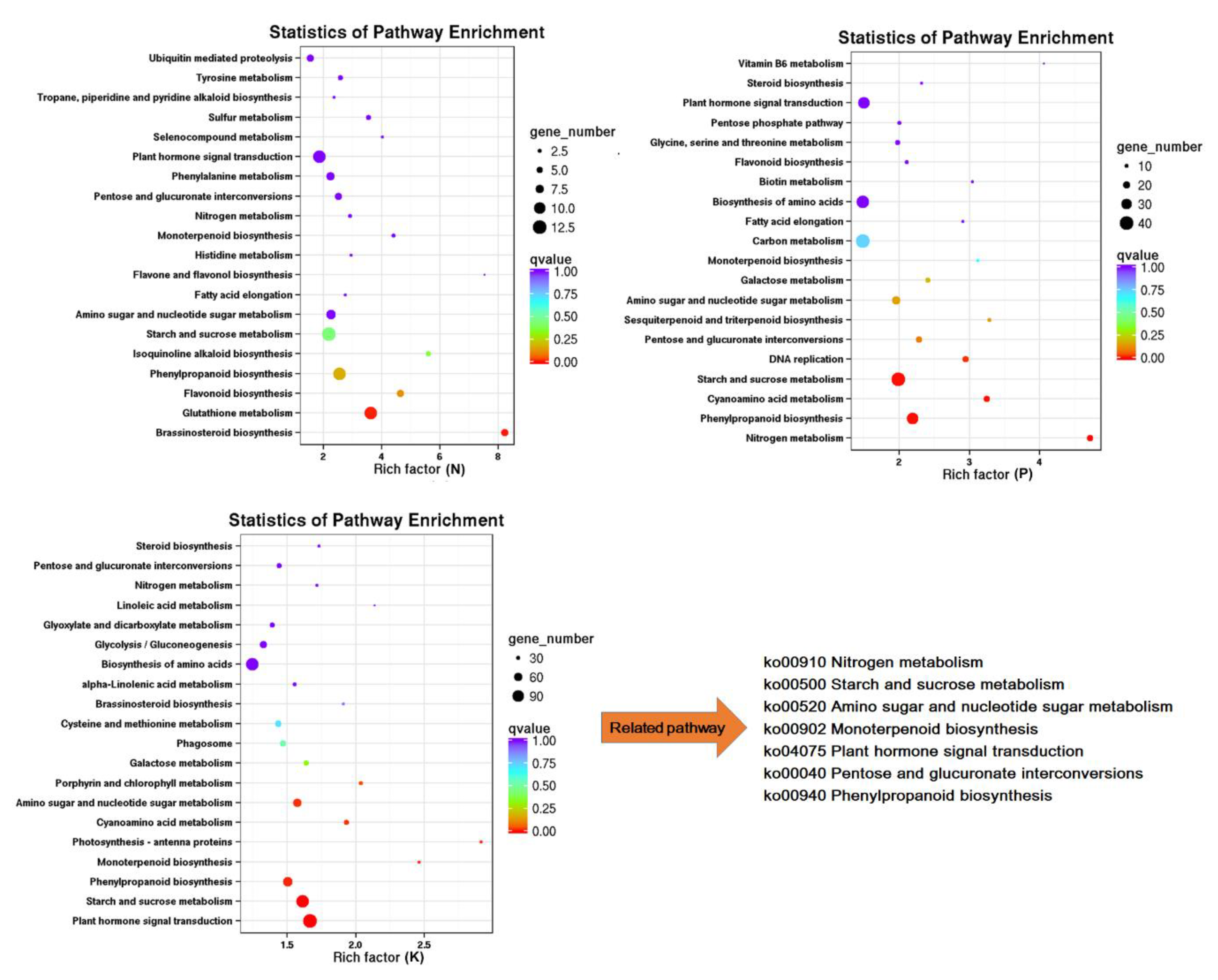
3.3. Function Enrichment Analysis of Common Differentially Expressed Genes (DEG)
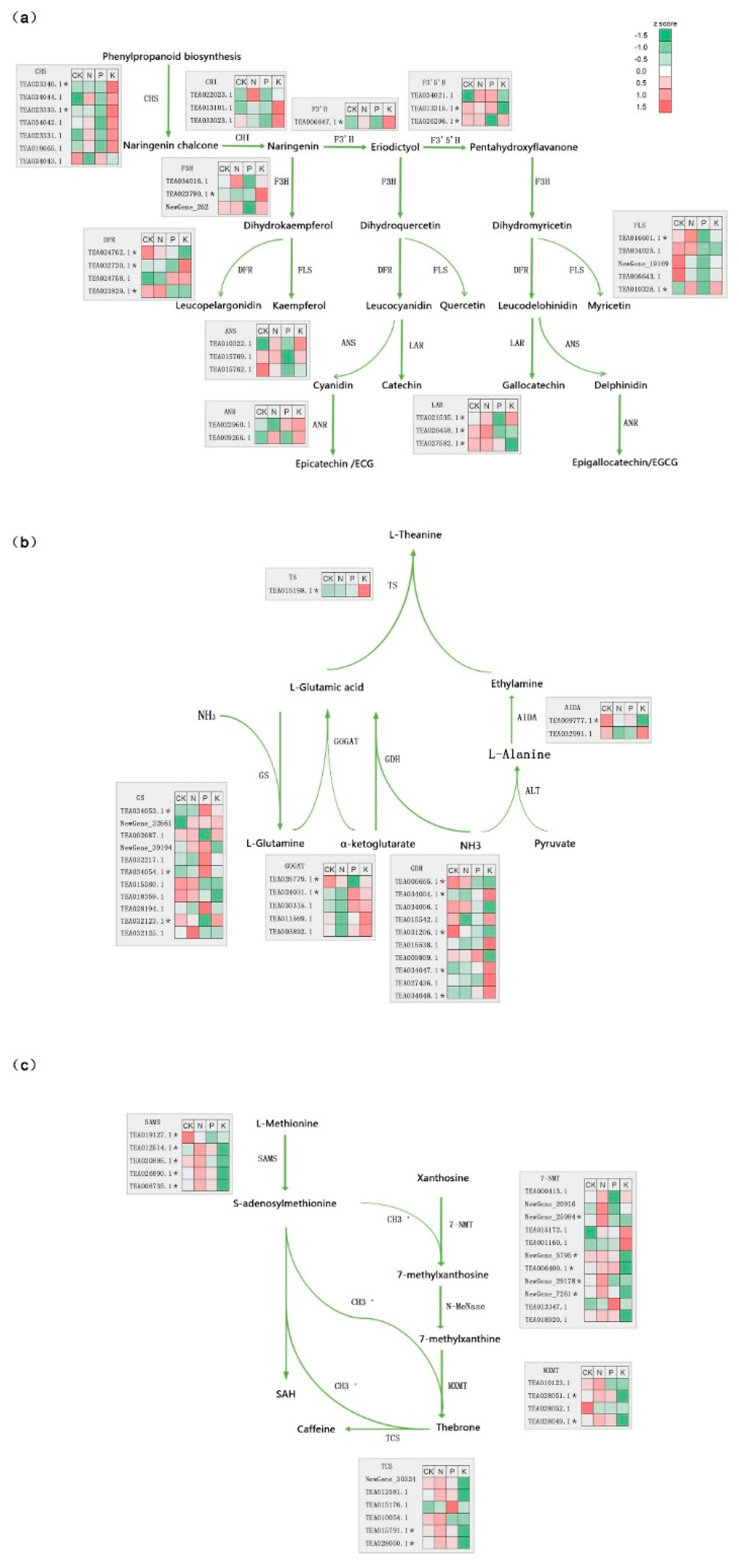
3.4. The Effect of N, P, and K Starvation on Three Major Secondary Metabolite Biosynthesis Pathways in Tea
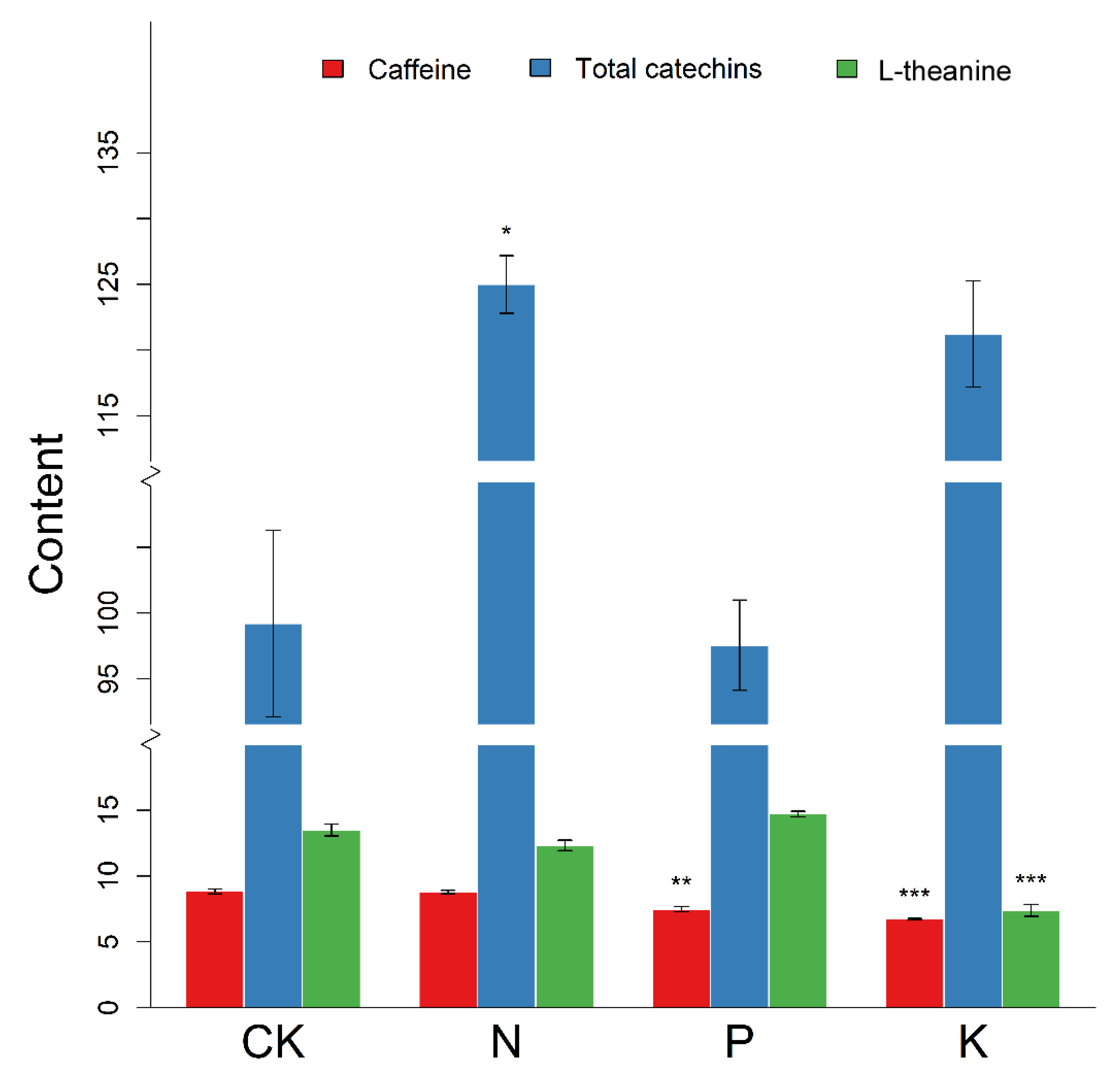
3.5. Changes in Major Secondary Metabolite Content Following N, P, and K Starvation
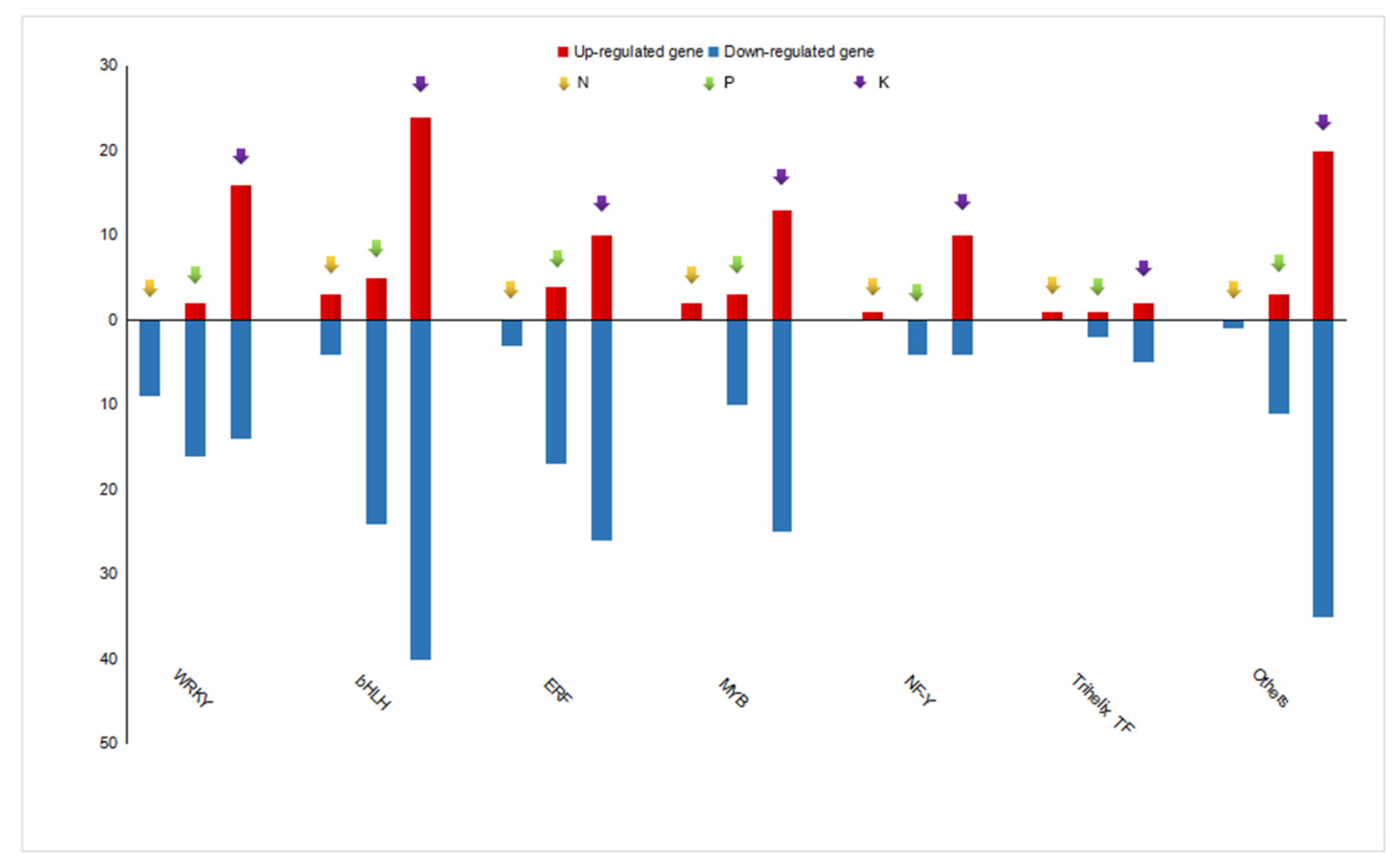
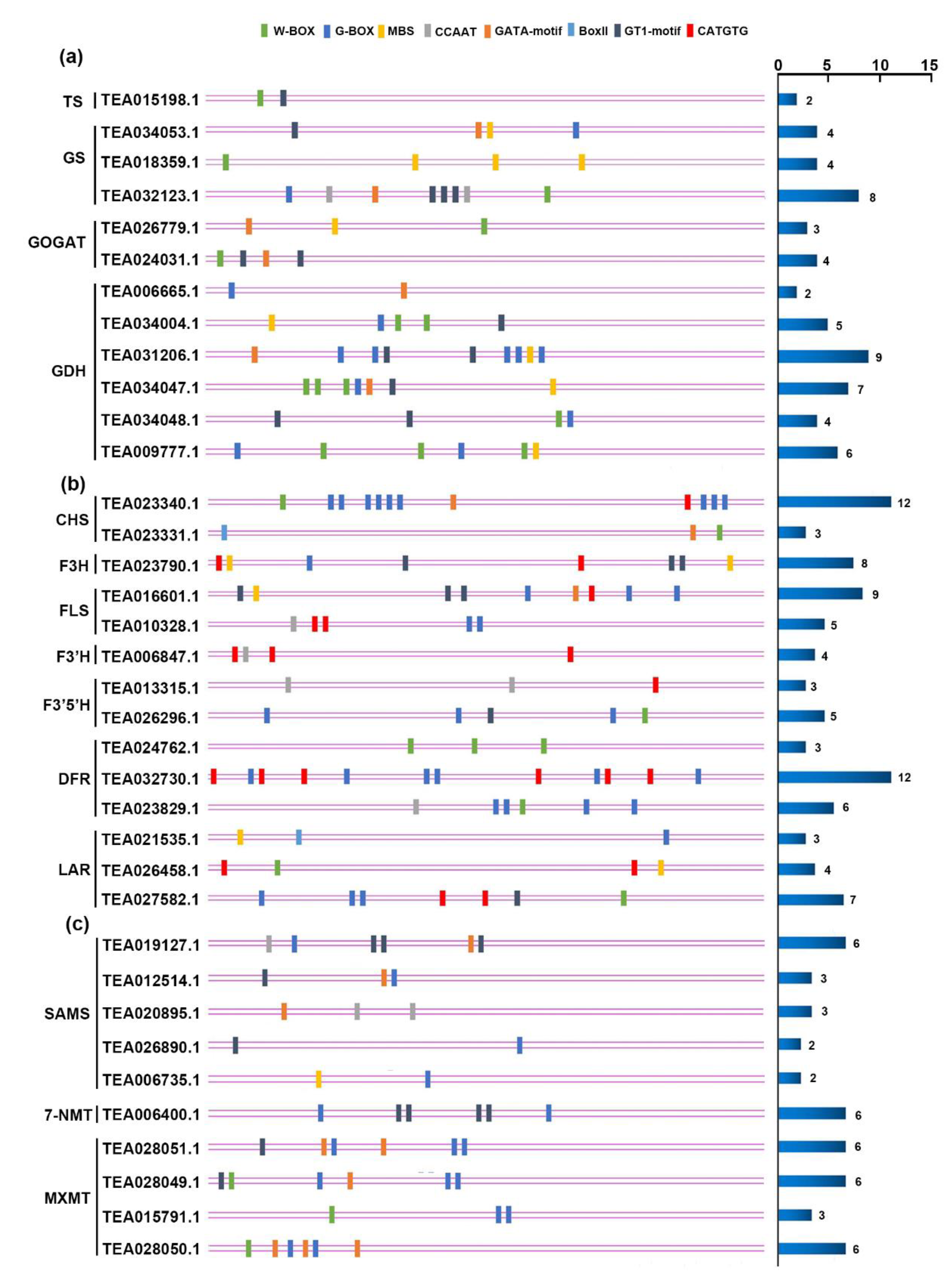
3.6. Analysis of Potential Regulators Involved in Metabolite Biosynthesis under N, P, and K Starvation Stress
4. Discussion
4.1. Global Gene Transcriptional Changes under N, P, and K Starvation
4.2. N, P, and K Starvation Differently Affect Three Major Secondary Metabolites in Tea
4.3. Differential Expressed Transcription Factors May Regulate Metabolite Biosynthesis under N, P, and K Starvation Stress
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Maathuis, F.J. Physiological functions of mineral macronutrients. Curr. Opin. Plant Biol. 2009, 12, 250–258. [Google Scholar] [CrossRef]
- Takehisa, H.; Sato, Y.; Antonio, B.A.; Nagamura, Y. Global transcriptome profile of rice root in response to essential macronutrient deficiency. Plant Signal. Behav. 2013, 8, e24409. [Google Scholar] [CrossRef]
- Zhong, Y.J.; Wang, Y.G.; Guo, J.F.; Zhul, X.L.; Shi, J.; He, Q.J.; Liu, Y.; Wu, Y.R.; Zhang, L.; Lv, Q.D.; et al. Rice SPX6 negatively regulates the phosphate starvation response through suppression of the transcription factor PHR2. New Phytol. 2018, 219, 135–148. [Google Scholar] [CrossRef]
- Schachtman, D.P.; Shin, R. Nutrient sensing and signaling: NPKS. Annu. Rev. Plant Biol. 2007, 58, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Medici, A.; Krouk, G. The primary nitrate response: A multifaceted signalling pathway. J. Exp. Bot. 2014, 65, 5567–5576. [Google Scholar] [CrossRef] [PubMed]
- Chien, P.S.; Chiang, C.P.; Leong, S.J.; Chiou, T.J. Sensing and Signaling of Phosphate Starvation—From Local to Long Distance. Plant Cell Physiol. 2018, 59, 1714–1722. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, W.H. Regulation of potassium transport and signaling in plants. Curr. Opin. Plant Biol. 2017, 39, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Bouguyon, E.; Gojon, A.; Nacry, P. Nitrate sensing and signaling in plants. Semin. Cell Dev. Biol. 2012, 23, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Ruffel, S.; Freixes, S.; Balzergue, S.; Tillard, P.; Jeudy, C.; Martin-Magniette, M.L.; van der Merwe, M.J.; Kakar, K.; Gouzy, J.; Fernie, A.R.; et al. Systemic signaling of the plant nitrogen status triggers specific transcriptome responses depending on the nitrogen source in Medicago truncatula. Plant Physiol. 2008, 146, 2020–2035. [Google Scholar] [CrossRef]
- Chiou, T.J.; Lin, S.I. Signaling network in sensing phosphate availability in plants. Annu. Rev. Plant Biol. 2011, 62, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, H.D.; Chen, L.Q.; Wang, Y.; Liu, L.L.; He, L.; Wu, W.H. A protein kinase, interacting with two calcineurin B-like proteins, regulates K+ transporter AKT1 in Arabidopsis. Cell 2006, 125, 1347–1360. [Google Scholar] [CrossRef] [PubMed]
- Medici, A.; Szponarski, w.; Dangeville, P.; Safi, A.; Dissanayake, I.M.; Saenchai, C.; Emanuel, A.; Rubio, V.; Lacombe, B.; Ruffel, S.; et al. Identification of molecular integrators shows that nitrogen actively controls the phosphate starvation response in plants. Plant Cell 2019, 31, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jiang, Z.; Wang, W.; Qiu, Y.; Zhang, Z.; Liu, Y.; Li, A.; Gao, X.; Liu, L.; Qian, Y.; et al. Nitrate–NRT1.1B–SPX4 cascade integrates nitrogen and phosphorus signalling networks in plants. Nat. Plants. 2019, 5, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Peng, J.S.; He, Y.N.; Zhang, G.B.; Yi, H.Y.; Fu, Y.L.; Gong, J.M. Arabidopsis NRT1.5 Mediates the Suppression of Nitrate Starvation-Induced Leaf Senescence by Modulating Foliar Potassium Level. Mol. Plant 2016, 9, 461–470. [Google Scholar] [CrossRef]
- Xia, E.-H.; Zhang, H.-B.; Sheng, J.; Li, K.; Zhang, Q.-J.; Kim, C.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W.; et al. The Tea Tree Genome Provides Insights into Tea Flavor and Independent Evolution of Caffeine Biosynthesis. Mol. Plant 2017, 10, 866–877. [Google Scholar] [CrossRef]
- Salehi, S.Y.; Hajiboland, S. A high internal phosphorus use efficiency in tea (Camellia sinensis L.) plant. Asian J. Plant Sci. 2008, 7, 30–36. [Google Scholar] [CrossRef]
- Dong, F.; Hu, J.; Shi, Y.; Liu, M.; Zhang, Q.; Ruan, J. Effects of nitrogen supply on flavonol glycoside biosynthesis and accumulation in tea leaves (Camellia sinensis). Plant Physiol. Biochem. 2019, 138, 48–57. [Google Scholar] [CrossRef]
- Chandran, A.K.; Priatama, R.A.; Kumar, V.; Xuan, Y.; Je, B.I.; Kim, C.M.; Jung, K.H.; Han, C.D. Genome-wide transcriptome analysis of expression in rice seedling roots in response to supplemental nitrogen. J. Plant Physiol. 2016, 200, 62–75. [Google Scholar] [CrossRef]
- Wei, H.R.; Yordanov, Y.S.; Georgieva, T.; Li, X.; Busov, V. Nitrogen deprivation promotes Populus root growth through global transcriptome reprogramming and activation of hierarchical genetic networks. New Phytol. 2013, 200, 483–497. [Google Scholar] [CrossRef]
- Secco, D.; Jabnoune, M.; Walker, H.; Shou, H.X.; Wu, P.; Poirier, Y.; Whelan, J. Spatio-temporal transcript profiling of rice roots and shoots in response to pStarvation and recovery. Plant Cell 2013, 25, 4285–4304. [Google Scholar] [CrossRef]
- Luo, B.; Ma, P.; Nie, Z.; Zhang, X.; He, X.; Ding, X.; Feng, X.; Lu, Q.; Ren, Z.; Lin, H.; et al. Metabolite profiling and genome-wide association studies reveal response mechanisms of phosphorus deficiency in maize seedling. Plant J. 2018, 97, 947–969. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.M.; Chen, Y.; Lu, L.; Lu, Y.F.; Li, L.Q. Transcriptome analysis reveals dynamic changes in the gene expression of tobacco seedlings under low potassium stress. J. Genet. 2015, 94, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Li, N.N.; Yang, Y.P.; Ye, J.H.; Lu, J.L.; Zheng, X.Q.; Liang, Y.R. Effects of sunlight on gene expression and chemical composition of light-sensitive albino tea plant. Plant Growth Regul. 2016, 78, 253–262. [Google Scholar] [CrossRef]
- Khan, N.; Mukhtar, H. Tea polyphenols for health promotion. Life Sci. 2007, 81, 519–533. [Google Scholar] [CrossRef]
- Lepiniec, L.; Debeaujon, I.; Routaboul, J.M.; Baudry, A.; Pourcel, L.; Nesi, N.; Caboche, M. Genetics and biochemistry of seed flavonoid. Annu. Rev. Plant Biol. 2006, 57, 405–430. [Google Scholar] [CrossRef]
- Zhou, T.S.; Zhou, R.; Yu, Y.B.; Xiao, Y.; Li, D.H.; Xiao, B.; Yu, O.; Yang, Y.J. Cloning and Characterization of a Flavonoid 3′-Hydroxylase Gene from Tea Plant (Camellia sinensis). Int. J. Mol. Sci. 2016, 17, 261. [Google Scholar] [CrossRef]
- Wang, Y.S.; Xu, Y.J.; Gao, L.P.; Yu, O.; Wang, X.Z.; He, X.J.; Jiang, X.L.; Liu, Y.J.; Xia, T. Functional analysis of Flavonoid 3′,5′-hydroxylase from Tea plant (Camellia sinensis): Critical role in the accumulation of catechins. BMC Plant Biol. 2014, 14, 347. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Wei, K.; Li, H.L.; Wang, L.Y.; Ruan, L.; Pang, D.D.; Cheng, H. Identification of key genes involved in catechin metabolism in tea seedlings based on transcriptomic and HPLC analysis. Plant Physiol. Bioch. 2018, 133, 107–115. [Google Scholar] [CrossRef]
- Kaufmann, K.; Pajoro, A.; Angenent, G.C. Regulation of transcription in plants: Mechanisms controlling developmental switches. Nat. Rev. Genet. 2010, 11, 830–842. [Google Scholar] [CrossRef]
- Nemie-Feyissa, D.; Olafsdottir, S.M.; Heidari, B.; Lillo, C. Nitrogen depletion and small R3-MYB transcription factors affecting anthocyanin accumulation in Arabidopsis leaves. Phytochemistry 2014, 98, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.C.; Song, L.; Zhang, Y.; Zheng, Z.; Liu, D. Arabidopsis PHL2 and PHR1 Act Redundantly as the Key Components of the Central Regulatory System Controlling Transcriptional Responses to Phosphate Starvation. Plant Physiol. 2016, 170, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Reddy, V.R. Potassium Starvation Limits Soybean Growth More than the Photosynthetic Processes across CO2 Levels. Front. Plant Sci. 2017, 8, 991. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Yang, H.; Wang, S.; Zhao, J.; Liu, C.; Gao, L.; Xia, E.; Lu, Y.; Tai, Y.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, F.; Wan, Q.; Ruan, J. Transcriptome analysis using RNA-Seq revealed the effects of nitrogen form on major secondary metabolite biosynthesis in tea (Camellia sinensis) plants. Acta Physiol. Plant 2018, 40, 127. [Google Scholar] [CrossRef]
- Ding, Z.; Jia, S.; Wang, Y.; Xiao, J.; Zhang, Y. Phosphate stresses affect ionome and metabolome in tea plants. Plant Physiol. Biochem. 2017, 120, 30–39. [Google Scholar] [CrossRef]
- Li, Y.; Wang, W.; Wei, K.; Ruan, L.; Wang, L.; Cheng, H.; Zhang, F.; Wu, L.; Bai, P. Differential transcriptomic changes in low-potassium sensitive and low-potassium tolerant tea plant (Camellia sinensis) genotypes under potassium deprivation. Sci. Hortic. 2019, 256, 108570. [Google Scholar] [CrossRef]
- Forieri, I.; Sticht, C.; Reichelt, M.; Grerz, N.; Hawkesford, M.J.; Malagoli, M.; Wirtz, M.; Hell, R. System analysis of metabolism and the transcriptome in Arabidopsis thaliana roots reveals differential co-regulation upon iron, sulfur and potassium deficiency. Plant Cell Environ. 2017, 40, 95–107. [Google Scholar] [CrossRef]
- Sinha, S.K.; Sevanthi, V.A.; Chaudhary, S.; Tyagi, P.; Venkadesan, S.; Rani, M.; Mandal, P.K. Transcriptome Analysis of Two Rice Varieties Contrasting for Nitrogen Use Efficiency under Chronic N Starvation Reveals Differences in Chloroplast and Starch Metabolism-Related Genes. Genes Basel 2018, 9, 206. [Google Scholar] [CrossRef]
- Xue, Y.; Zhuang, Q.; Zhu, S.; Xiao, B.; Liang, C.; Liao, H.; Tian, J. Genome Wide Transcriptome Analysis Reveals Complex Regulatory Mechanisms Underlying Phosphate Homeostasis in Soybean Nodules. Int. J. Mol. Sci. 2018, 19, 2924. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Jiang, H.; Wang, H.; Cui, J.; Wang, J.H.; Hu, J.; Guo, L.B.; Qian, Q.; Xue, D.W. Transcriptome Analysis of Rice Seedling Roots in Response to Potassium Deficiency. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Yang, X.; Yu, H.; Jiang, W.; Sun, N.; Liu, X.; Liu, X.; Zhang, X.; Wang, Y.; Gu, X. RNA-Seq-based transcriptome profiling of early nitrogen deficiency response in cucumber seedlings provides new insight into the putative nitrogen regulatory network. Plant Cell Physiol. 2015, 56, 455–467. [Google Scholar] [CrossRef]
- Park, M.R.; Baek, S.H.; de Los Reyes, B.G.; Yun, S.J.; Hasenstein, K.H. Transcriptome profiling characterizes phosphate deficiency effects on carbohydrate metabolism in rice leaves. J. Plant Physiol. 2012, 169, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Francisco, L.; Roberto, S.; Ana, C.; Martín, l.; Joaquín, I.; Antonio, L.; Javier, P.A. A conserved MYB transcription factor. Genes Dev. 2001, 15, 2122–2133. [Google Scholar]
- Wang, J.; Sun, J.; Miao, J.; Guo, J.; Shi, Z.; He, M.; Chen, Y.; Zhao, X.; Li, B.; Han, F.; et al. A phosphate starvation response regulator Ta-PHR1 is involved in phosphate signalling and increases grain yield in wheat. Ann. Bot. 2013, 111, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiao, F.; Wu, Z.; Li, Y.; Wang, X.; He, X.; Zhong, W.; Wu, P. OsPHR2 is involved in phosphate-starvation signaling and excessive phosphate accumulation in shoots of plants. Plant Physiol. 2008, 146, 1673–1686. [Google Scholar] [CrossRef]
- Lv, Q.; Zhong, Y.; Wang, Y.; Wang, Z.; Zhang, L.; Shi, J.; Wu, Z.; Liu, Y.; Mao, C.; Yi, K.; et al. SPX4 Negatively Regulates Phosphate Signaling and Homeostasis through Its Interaction with PHR2 in Rice. Plant Cell 2014, 26, 1586–1597. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Ruan, W.Y.; Shi, J.; Zhang, L.; Xiang, D.; Yang, C.; Li, C.Y.; Wu, Z.C.; Liu, Y.; Yu, Y.A.; et al. Rice SPX1 and SPX2 inhibit phosphate starvation responses through interacting with PHR2 in a phosphate-dependent manner. Proc. Natl. Acad. Sci. USA 2014, 111, 14953–14958. [Google Scholar] [CrossRef]
- Mudge, S.R.; Rae, A.L.; Diatloff, E.; Smith, F.W. Expression analysis suggests novel roles for members of the Pht1 family of phosphate transporters in Arabidopsis. Plant J. 2002, 31, 341–353. [Google Scholar] [CrossRef]
- Ma, T.L.; Wu, W.H.; Wang, Y. Transcriptome analysis of rice root responses to potassium deficiency. BMC Plant Biol. 2012, 12, 161. [Google Scholar] [CrossRef]
- Li, C.F.; Zhu, Y.; Yu, Y.; Zhao, Q.Y.; Wang, S.J.; Wang, X.C.; Yao, M.Z.; Luo, D.; Li, X.; Chen, L.; et al. Global transcriptome and gene regulation network for secondary metabolite biosynthesis of tea plant (Camellia sinensis). BMC Genom. 2015, 16, 560. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Li, M.-Y.; Wu, X.-J.; Huang, Y.; Ma, J.; Xiong, A.-S. Genome-wide analysis of basic helix−loop−helix family transcription factors and their role in responses to abiotic stress in carrot. Mol. Breed. 2015, 35, 125. [Google Scholar] [CrossRef]
- Quan, X.; Zeng, J.; Ye, L.; Chen, G.; Han, Z.; Shah, J.M.; Zhang, G. Transcriptome profiling analysis for two Tibetan wild barley genotypes in responses to low nitrogen. BMC Plant Biol. 2016, 16, 30. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.M.; Liu, Y.; Liu, X.; Jiang, J. Comparative Transcriptome Profiling of Two Tomato Genotypes in Response to Potassium-Deficiency Stress. Int. J. Mol. Sci. 2018, 19, 2402. [Google Scholar] [CrossRef]
- Bai, F.X.; Chen, C.L.; An, J.Y.; Xiao, S.Y.; Deng, X.X.; Pan, Z.Y. Transcriptome responses to phosphate deficiency in Poncirus trifoliata (L.) Raf. Acta Physiol. Plant 2014, 36, 3207–3215. [Google Scholar] [CrossRef]
- Licausi, F.; Ohme-Takagi, M.; Perata, P. APETALA2/Ethylene Responsive Factor (AP2/ERF) transcription factors: Mediators of stress responses and developmental programs. New Phytol. 2013, 199, 639–649. [Google Scholar] [CrossRef]
- Ramaiah, M.; Jain, A.; Raghothama, K.G. Ethylene Response Factor070 regulates root development and phosphate starvation-mediated responses. Plant Physiol. 2014, 164, 1484–1498. [Google Scholar] [CrossRef]
- Kim, M.J.; Ruzicka, D.; Shin, R.; Schachtman, D.P. The Arabidopsis AP2/ERF transcription factor RAP2.11 modulates plant response to low-potassium conditions. Mol. Plant 2012, 5, 1042–1057. [Google Scholar] [CrossRef]
- Liang, J.; He, J. Protective role of anthocyanins in plants under low nitrogen stress. Biochem. Biophys. Res. Commun. 2018, 498, 946–953. [Google Scholar] [CrossRef]
- Lea, U.S.; Slimestad, R.; Smedvig, P.; Lillo, C. Nitrogen deficiency enhances expression of specific MYB and bHLH transcription factors and accumulation of end products in the flavonoid pathway. Planta 2007, 225, 1245–1253. [Google Scholar] [CrossRef]
- Wang, R.; Jing, W.; Xiao, L.; Jin, Y.; Shen, L.; Zhang, W. The rice high-affinity potassium transporter1;1 is involved in salt tolerance and regulated by an MYB-Type transcription factor. Plant Physiol. 2015, 168, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.H.; Nimmo, G.A.; Jenkins, G.I.; Nimmo, H.G. BHLH32 modulates several biochemical and morphological processes that respond to Pi starvation in Arabidopsis. Biochem. J. 2007, 405, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Dugassa, N.F.; Trond, L.; Kristine, M.O.; Rune, S.; Cathrine, L. The endogenous GL3, but not EGL3, gene is necessary for anthocyanin accumulation as induced by nitrogen depletion in Arabidopsis rosette stage leaves. Planta 2009, 230, 747–754. [Google Scholar]
- Devaiah, B.N.; Karthikeyan, A.S.; Raghothama, K.G. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in Arabidopsis. Plant Physiol. 2007, 143, 1789–1801. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, R.; Zheng, M.; Liu, X.; Meng, F.; Wu, H.; Yao, Y.; Xin, M.; Peng, H.; Ni, Z.; et al. TaWRKY51 promotes lateral root formation through negative regulation of ethylene biosynthesis in wheat (Triticum aestivum L.). Plant J. 2018, 96, 372–388. [Google Scholar] [CrossRef]
- Dai, X.Y.; Wang, Y.Y.; Zhang, W.H. OsWRKY74, a WRKY transcription factor, modulates tolerance to phosphate starvation in rice. J. Exp. Bot. 2016, 67, 947–960. [Google Scholar] [CrossRef]
- Su, T.; Xu, Q.; Zhang, F.C.; Chen, Y.; Li, L.Q.; Wu, W.H.; Chen, Y.F. WRKY42 modulates phosphate homeostasis through regulating phosphate translocation and acquisition in Arabidopsis. Plant Physiol. 2015, 167, 1579–1591. [Google Scholar] [CrossRef]
- Wang, P.; Xu, X.; Tang, Z.; Zhang, W.; Huang, X.Y.; Zhao, F.J. OsWRKY28 Regulates Phosphate and Arsenate Accumulation, Root System Architecture and Fertility in Rice. Front. Plant Sci. 2018, 9, 1330. [Google Scholar] [CrossRef]
- Wang, P.; Yue, C.; Chen, D.; Zheng, Y.; Zhang, Q.; Yang, J.; Ye, N. Genome-wide identification of WRKY family genes and their response to abiotic stresses in tea plant (Camellia sinensis). Gene Genom. 2019, 41, 17–33. [Google Scholar] [CrossRef]
- Zhao, H.; Wu, D.; Kong, F.; Lin, K.; Zhang, H.; Li, G. The Arabidopsis thaliana Nuclear Factor Y Transcription Factors. Front. Plant Sci. 2016, 7, 2045. [Google Scholar] [CrossRef]
- Cui, X.; Wang, Y.X.; Liu, Z.W.; Wang, W.L.; Li, H.; Zhuang, J. Transcriptome-wide identification and expression profile analysis of the bHLH family genes in Camellia sinensis. Funct. Integr. Genom. 2018, 18, 489–503. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhao, X.; Gao, L.; Shi, X.; Dai, X.; Liu, Y.; Xia, T.; Wang, Y. Isolation and Characterization of Key Genes that Promote Flavonoid Accumulation in Purple-leaf Tea (Camellia sinensis L.). Sci. Rep. 2018, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhu, Z.; Cao, P.; Chen, H.; Chen, C.; Zhou, X.; Mao, Y.; Lei, J.; Jiang, Y.; Meng, W.; et al. Purple foliage coloration in tea (Camellia sinensis L.) arises from activation of the R2R3-MYB transcription factor CsAN1. Sci. Rep. 2016, 6, 32534. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hou, H.; Jiang, X.; Wang, P.; Dai, X.; Chen, W.; Gao, L.; Xia, T. A WD40 Repeat Protein from Camellia sinensis Regulates Anthocyanin and Proanthocyanidin Accumulation through the Formation of MYB-bHLH-WD40 Ternary Complexes. Int. J. Mol. Sci. 2018, 19, 1686. [Google Scholar] [CrossRef]
- Chen, W.; Hao, W.J.; Xu, Y.X.; Zheng, C.; Ni, D.J.; Yao, M.Z.; Chen, L. Isolation and Characterization of CsWRKY7, a Subgroup IId WRKY Transcription Factor from Camellia sinensis, Linked to Development in Arabidopsis. Int. J. Mol. Sci. 2019, 20, 2815. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, H.; Zhang, X.; He, Y.; Li, L.; Wang, Y.; Hong, G.; Xu, P. Transcriptomic Analysis Reveals the Molecular Adaptation of Three Major Secondary Metabolic Pathways to Multiple Macronutrient Starvation in Tea (Camellia sinensis). Genes 2020, 11, 241. https://doi.org/10.3390/genes11030241
Su H, Zhang X, He Y, Li L, Wang Y, Hong G, Xu P. Transcriptomic Analysis Reveals the Molecular Adaptation of Three Major Secondary Metabolic Pathways to Multiple Macronutrient Starvation in Tea (Camellia sinensis). Genes. 2020; 11(3):241. https://doi.org/10.3390/genes11030241
Chicago/Turabian StyleSu, Hui, Xueying Zhang, Yuqing He, Linying Li, Yuefei Wang, Gaojie Hong, and Ping Xu. 2020. "Transcriptomic Analysis Reveals the Molecular Adaptation of Three Major Secondary Metabolic Pathways to Multiple Macronutrient Starvation in Tea (Camellia sinensis)" Genes 11, no. 3: 241. https://doi.org/10.3390/genes11030241
APA StyleSu, H., Zhang, X., He, Y., Li, L., Wang, Y., Hong, G., & Xu, P. (2020). Transcriptomic Analysis Reveals the Molecular Adaptation of Three Major Secondary Metabolic Pathways to Multiple Macronutrient Starvation in Tea (Camellia sinensis). Genes, 11(3), 241. https://doi.org/10.3390/genes11030241