A Comprehensive Transcriptomics Analysis Reveals Long Non-Coding RNA to Be Involved in the Key Metabolic Pathway in Response to Waterlogging Stress in Maize


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. Quantification and Standardization of the PCgenes and lncRNAs in the Transcriptome
2.4. Differential Expression and Pathway Analysis
2.5. Expression Network Construction
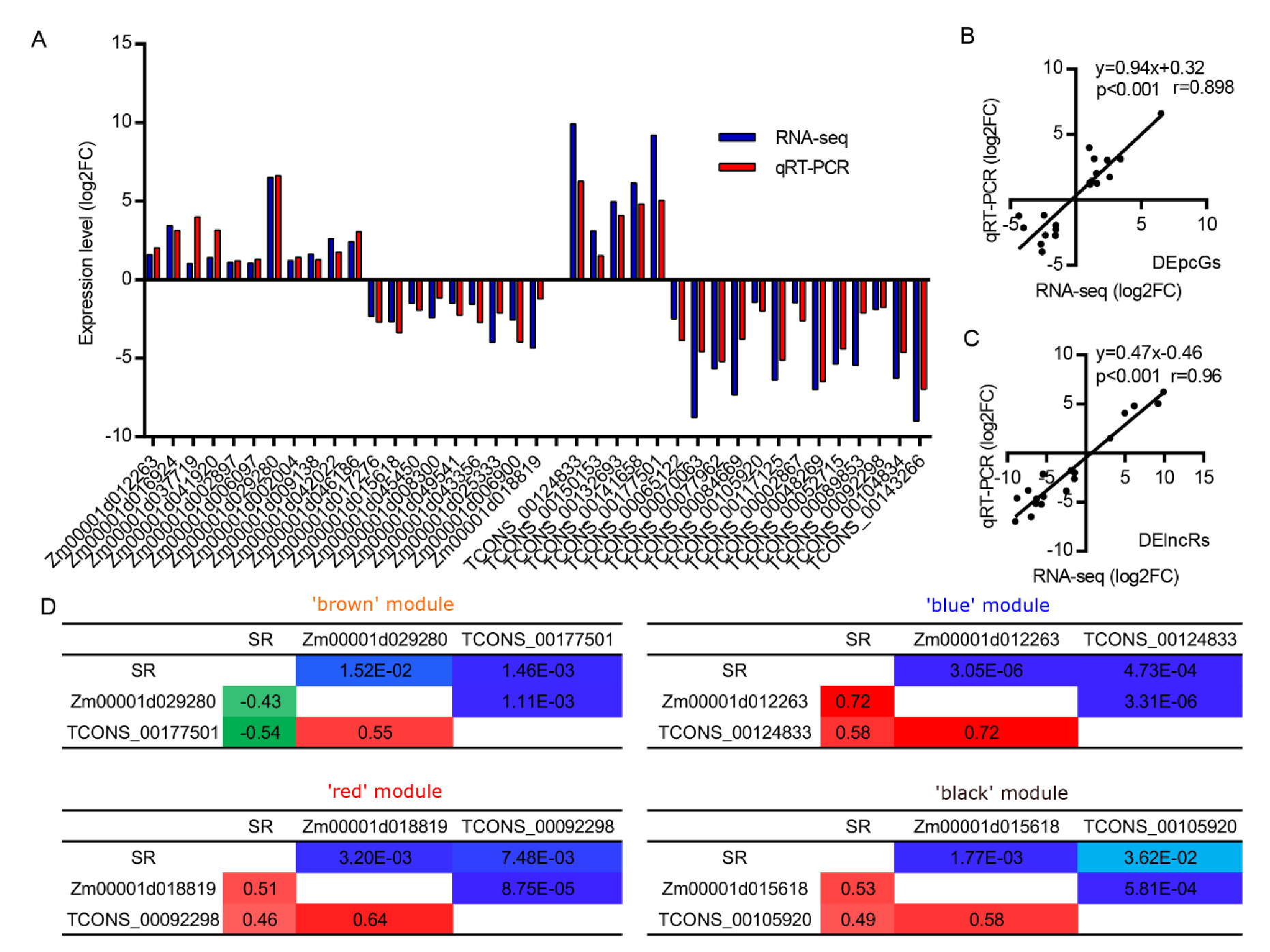
2.6. Quantitative Real-Time (RT) PCR
2.7. Verification of the Co-Expression Modules of DEpcGs and DElncRs in Different Inbred Lines
2.8. Conserved Motif Discovery
3. Results
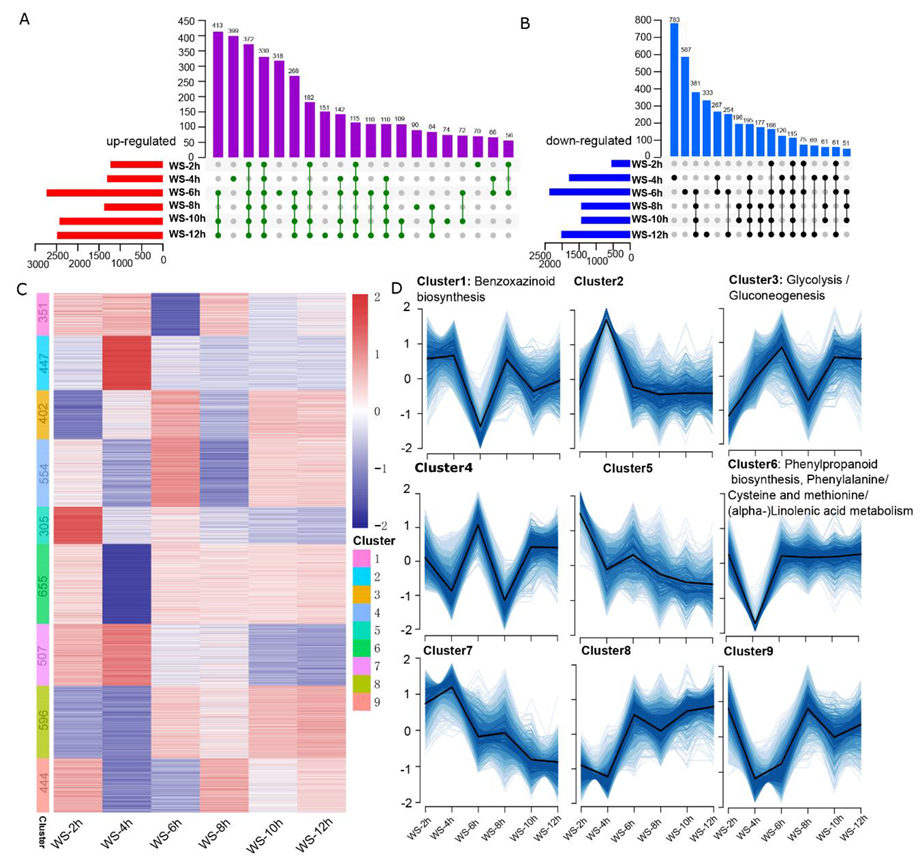
3.1. The Time-Course Transcriptomic Profiles of Seedling Root Tips Exposed to WS Conditions
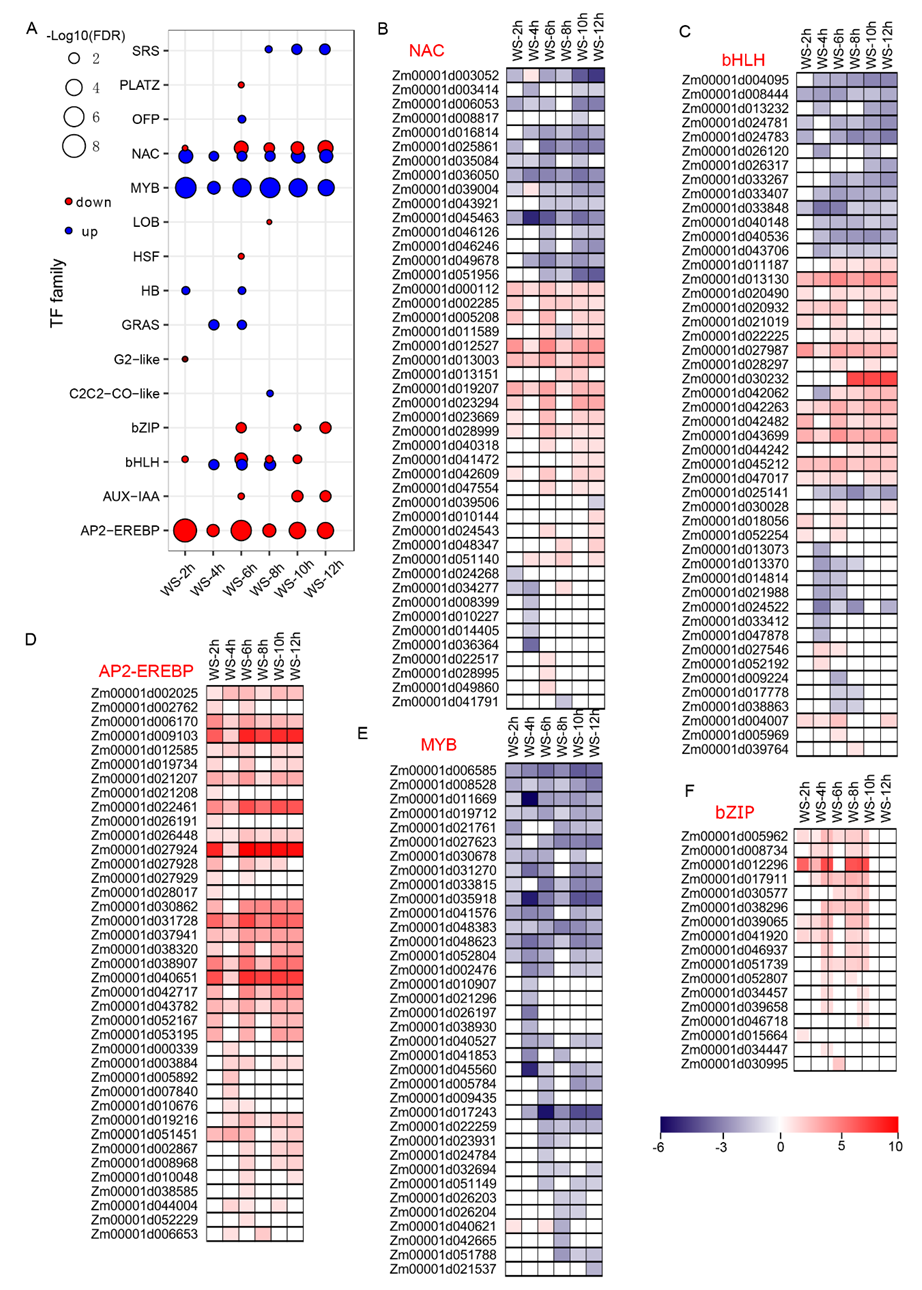
3.2. Differential Response of the Transcription Factor Families Involved in WS Conditions
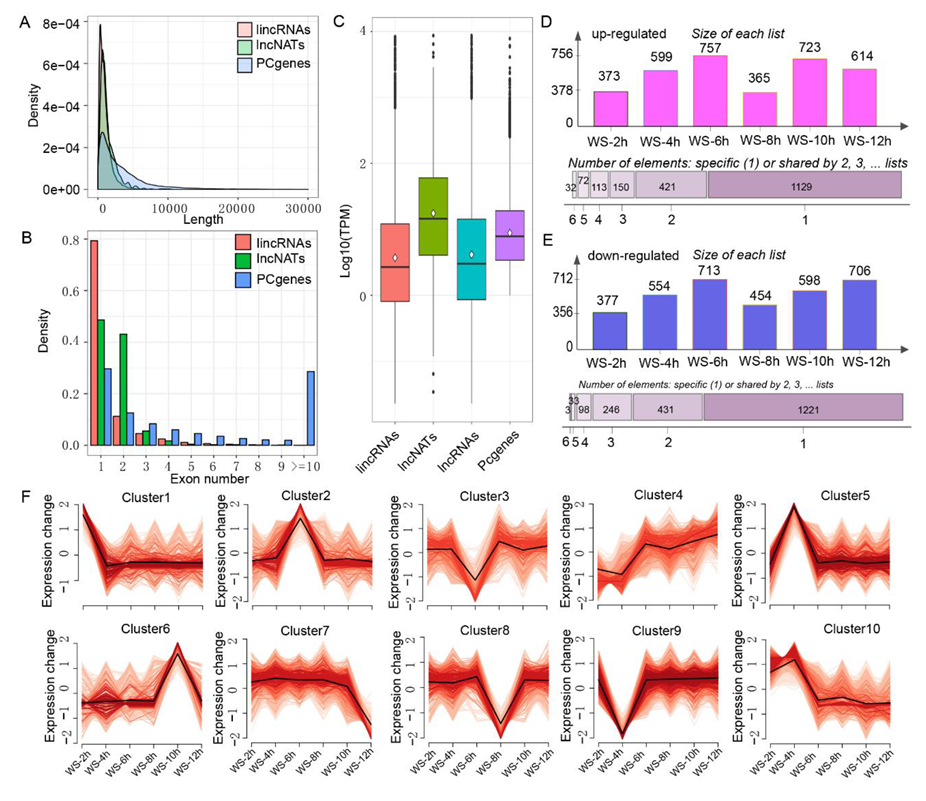
3.3. Identification and Characterization of lncRNAs Responding to WS in the Root Tips of Maize Seedling
3.4. Association of the Expression Between lncRNAs and DEpcGs
3.5. The Expression of DElncRs Is Positively Associated with Waterlogging Tolerance
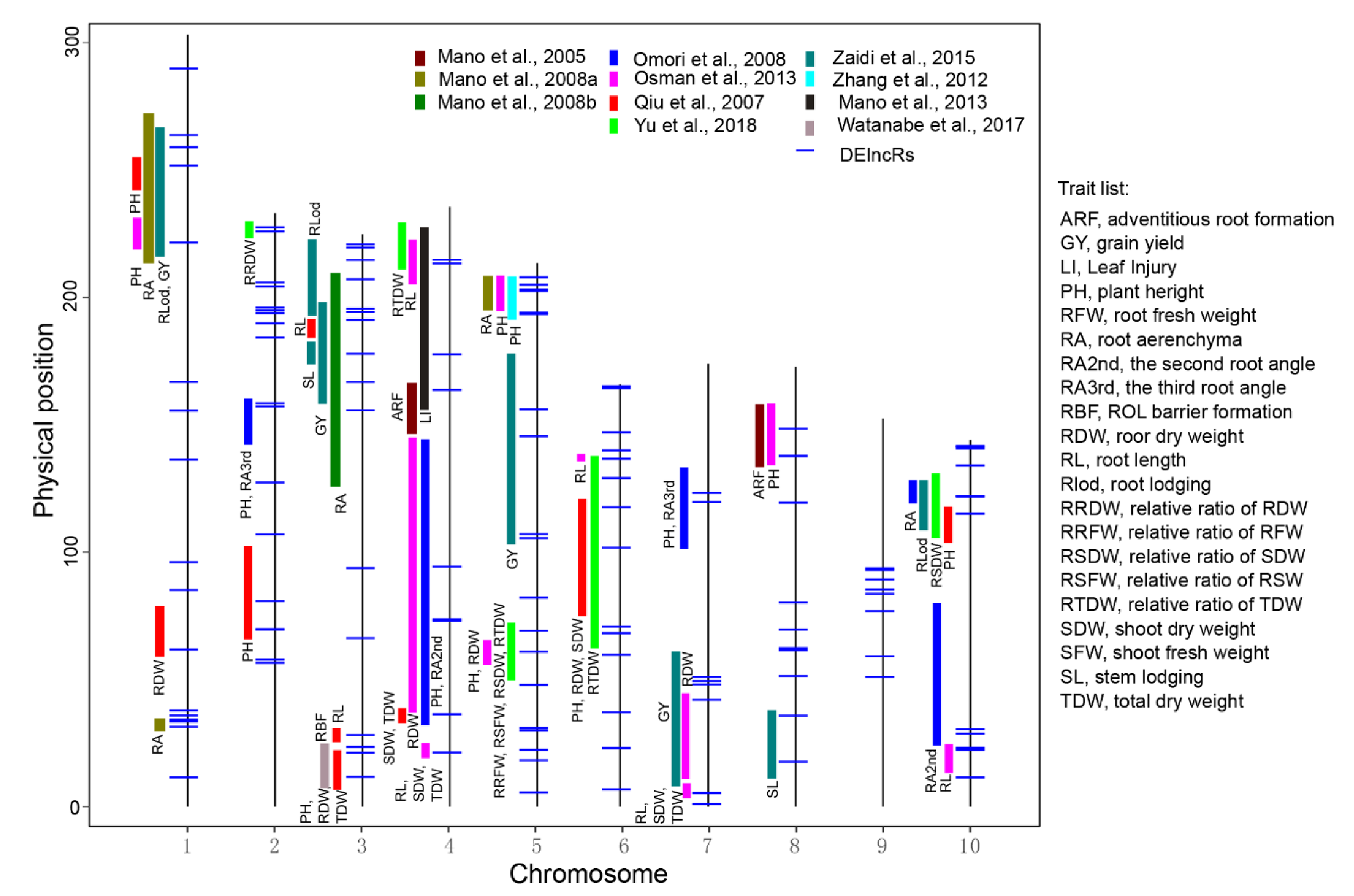
3.6. Most of the DElncRs Were Localized within the Previously Mapped Quantitative Trait Loci (QTL)
3.7. Conserved Anoxic Motif in the DElncR Promoter
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bailey-serres, J.; Fukao, T.; Gibbs, D.J.; Holdsworth, M.J.; Lee, S.C.; Licausi, F.; Perata, P.; Van Dongen, J.T. Making sense of low oxygen sensing. Trends Plant Sci. 2012, 17, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Mahendran, R.; Koirala, S.; Konoshima, L.; Yamazaki, D.; Watanabe, S.; Kim, H.; Kanae, S. Global flood risk under climate change. Nat. Clim. Chang. 2013, 3, 816–821. [Google Scholar] [CrossRef]
- Voesenek, L.A.; Bailey-Serres, J. Flood adaptive traits and processes: An overview. New Phytol. 2015, 206, 57–73. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Cheng, Y.; Linghu, J.; Yang, X.; Kang, L.; Zhang, Z.; Zhang, J.; He, C.; Du, X.; Peng, Z.; et al. RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 2013, 4, 2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Yuan, D.; Tu, L.; Gao, W.; He, Y.; Hu, H.; Wang, P.; Liu, N.; Lindsey, K.; Zhang, X. Long noncoding RNAs and their proposed functions in fibre development of cotton (Gossypium spp.). New Phytol. 2015, 207, 1181–1197. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Mustroph, A.; Sasidharan, R.; Vashisht, D.; Pedersen, O.; Oosumi, T.; Voesenek, L.A.; Bailey-Serres, J. Molecular characterization of the submergence response of the Arabidopsis thaliana ecotype Columbia. New Phytol. 2011, 190, 457–471. [Google Scholar] [CrossRef]
- Van Veen, H.; Vashisht, D.; Akman, M.; Girke, T.; Mustroph, A.; Reinen, E.; Hartman, S.; Kooiker, M.; Tienderen, P.; Schranz, M.; et al. Transcriptomes of Eight Arabidopsis thaliana Accessions Reveal Core Conserved, Genotype- and Organ-Specific Responses to Flooding Stress. Plant Physiol. 2016, 172, 668–689. [Google Scholar] [CrossRef]
- Vashisht, D.; van Veen, H.; Akman, M.; Sasidharan, R. Variation in Arabidopsis flooding responses identifies numerous putative “tolerance genes”. Plant Signal. Behav. 2016, 11, e1249083. [Google Scholar] [CrossRef] [Green Version]
- Minami, A.; Yano, K.; Gamuyao, R.; Nagai, K.; Kuroha, T.; Ayano, M.; Nakamori, M.; Koike, M.; Kondo, Y.; Niimi, Y.; et al. Time-course transcriptomics analysis reveals key responses of submerged deepwater rice to flooding. Plant Physiol. 2018, 176, 3081–3102. [Google Scholar] [CrossRef] [Green Version]
- Arora, K.; Panda, K.K.; Mittal, S.; Mallikarjuna, M.G.; Rao, A.R.; Dash, P.K.; Thirunavukkarasu, N. RNAseq revealed the important gene pathways controlling adaptive mechanisms under waterlogged stress in maize. Sci. Rep. 2017, 7, 10950. [Google Scholar] [CrossRef] [Green Version]
- Zou, X.; Jiang, Y.; Liu, L.; Zhang, Z.; Zheng, Y. Identification of transcriptome induced in roots of maize seedlings at the late stage of waterlogging. BMC Plant Biol. 2010, 10, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuzwieser, J.; Hauberg, J.; Howell, K.A.; Carroll, A.; Rennenberg, H.; Millar, A.H.; Whelan, J. Differential response of gray poplar leaves and roots underpins stress adaptation during hypoxia. Plant Physiol. 2009, 149, 461–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christianson, J.A.; Llewellyn, D.J.; Dennis, E.S.; Wilson, I.W. Global gene expression responses to waterlogging in roots and leaves of cotton (Gossypiumhirsutum L.). Plant Cell Physiol. 2010, 51, 21–37. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, A.; Lee, S.C.; Oosumi, T.; Zanetti, M.E.; Yang, H.; Ma, K.; Yaghoubi-Masihi, A.; Fukao, T.; Bailey-Serres, J. Cross-kingdom comparison of transcriptomic adjustments to low-oxygen stress highlights conserved and plant- specific responses. Plant Physiol. 2010, 152, 1484–1500. [Google Scholar] [CrossRef] [Green Version]
- Narsai, R.; Rocha, M.; Geigenberger, P.; Whelan, J.; van Dongen, J.T. Comparative analysis between plant species of transcriptional and metabolic responses to hypoxia. New Phytol. 2011, 190, 472–487. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Arenas-Huertero, C.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.C.; Kean, M.J.; Xu, J.; Chua, N.H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, R.; Mao, B.; Zhao, B.; Wang, J. Identification of lncRNAs involved in rice ovule development and female gametophyte abortion by genome-wide screening and functional analysis. BMC Genom. 2019, 20, 90. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Liao, J.Y.; Li, Z.Y.; Yu, Y.; Zhang, J.P.; Li, Q.F.; Qu, L.H.; Shu, W.S.; Chen, Y.Q. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014, 15, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Eichten, S.R.; Shimizu, R.; Petsch, K.; Yeh, C.T.; Wu, W.; Chettoor, A.M.; Givan, S.A.; Cole, R.A.; Fowler, J.E.; et al. Genome-wide discovery and characterization of maize long non-coding RNAs. Genome Biol. 2014, 15, R40. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, T.; Wang, B.; Han, B.; Zhou, H.; Wang, Y.; Li, D.Z.; Liu, A. Differential expression networks and inheritance patterns of long non-coding RNAs in castorbean seeds. Plant J. 2018, 95, 324–340. [Google Scholar] [CrossRef] [PubMed]
- Deng, P.C.; Liu, S.; Nie, X.J.; Weining, S.; Wu, L. Conservation analysis of long non-coding RNAs in plants. Sci. China Life Sci. 2018, 61, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I. Evolution to the rescue: Using comparative genomics to understand long non-coding RNAs. Nat. Rev. Genet. 2016, 17, 601–614. [Google Scholar] [CrossRef] [PubMed]
- Swiezewski, S.; Liu, F.; Magusin, A.; Dean, C. Cold-induced silencing by long antisense transcripts of an Arabidopsis Polycomb target. Nature 2009, 462, 799–802. [Google Scholar] [CrossRef]
- Heo, J.B.; Sung, S. Vernalization-mediated epigenetic silencing by a long intronic noncoding RNA. Science 2010, 331, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; García, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef]
- Rymarquis, L.A.; Kastenmayer, J.P.; Huttenhofer, A.G.; Green, P. Diamonds in the rough: mRNA-like non-coding RNAs. Trends Plant Sci. 2008, 13, 329–334. [Google Scholar] [CrossRef]
- Campalans, A.; Kondorosi, A.; Crespi, M. Enod40, a short open reading framecontaining mRNA, induces cytoplasmic localization of a nuclear RNA binding protein in Medicago truncatula. Plant Cell 2004, 16, 1047–1059. [Google Scholar] [CrossRef] [Green Version]
- Gultyaev, A.P.; Roussis, A. Identification of conserved secondary structures and expansion segments in enod40 RNAs reveals new enod40 homologues in plants. Nucleic Acids Res. 2007, 35, 3144–3152. [Google Scholar] [CrossRef]
- Ding, J.; Lu, Q.; Ouyang, Y.; Mao, H.; Zhang, P.; Yao, J.; Xu, C.; Li, X.; Xiao, J.; Zhang, Q. A long noncoding RNA regulates photoperiodsensitive male sterility, an essential component of hybrid rice. Proc. Natl. Acad. Sci. USA 2012, 109, 2654–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Luo, X.J.; Sun, F.; Hu, J.H.; Zha, X.J.; Su, W.; Yang, J.S. Overexpressing lncRNA LAIR increases grain yield and regulates neighbouring gene cluster expression in rice. Nat. Commun. 2018, 9, 3516. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Zhang, F.; Wang, H.; Wang, W.; Zhao, F.; Li, Z.; Sun, C.; Chen, F.; Xu, F.; Chang, S.; et al. Ef-cdlocus shortens rice maturity duration without yield penalty. Proc. Natl. Acad. Sci. USA 2019, 116, 18717–18722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Meng, X.; Dobrovolskaya, O.B.; Orlov, Y.L.; Chen, M. Non-coding RNA and their roles in stress response in plants. Genom. Proteom. Bioinf. 2017, 15, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhou, Y.F.; Feng, Y.Z.; He, H.; Lian, J.P.; Yang, Y.W.; Lei, M.Q.; Zhang, Y.C.; Chen, Y.Q. Transcriptional landscape of pathogen-responsive lncRNA in rice unveils the role of ALEX1 in jasmonate pathway and disease resistance. Plant Biotechnol. J. 2019. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Zhao, H.; Cui, P.; Albesher, N.; Xiong, L. A nucleus-localized long non-coding RNA enhances drought and salt stress tolerance. Plant Physiol. 2017, 175, 1321–1336. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Shi, S.; Jiang, N.; Khanzada, H.; Wassan, G.M.; Zhu, C.; Peng, X.; Xu, J.; Chen, Y.; Yu, Q.; et al. Genome-wide analysis of long non-coding RNAs affecting roots development at early stage in the rice response to cadmium stress. BMC Genom. 2018, 19, 460. [Google Scholar] [CrossRef]
- Chen, M.; Wang, C.; Bao, H.; Chen, H.; Wang, Y. Genome-wide identification and characterization of novel lncRNA in Populus under nitrogen deficiency. Mol. Genet. Genom. 2015, 291, 1663–1680. [Google Scholar] [CrossRef]
- Gong, L.C.; Xu, H.M.; Guo, G.L.; Zhang, T.; Shi, J.W.; Chang, C. Long Non-coding RNA H19 protects H9c2 cells against hypoxia-induced injury by targeting MicroRNA-139. Cell Physiol. Biochem. 2017, 44, 857–869. [Google Scholar] [CrossRef]
- Li, L.; Wang, Q.; Yuan, Z.; Chen, A.; Liu, Z.; Li, H.; Wang, Z. Long non-coding RNA H19 contributes to hypoxia-induced CPC injury by suppressing Sirt1 through miR-200a-3p. Acta Biochim. Biophys. Sin. 2018, 50, 950–959. [Google Scholar] [CrossRef] [Green Version]
- Bhan, A.; Deb, P.; Shihabeddin, N.; Ansari, K.I.; Brotto, M.; Mandal, S.S. Histone methylase MLL1 coordinates with HIF and regulate lncRNA HOTAIR expression under hypoxia. Gene 2017, 629, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhao, S.; Li, C.; Liu, C. LncRNA TUG1 serves an important role in hypoxia-induced myocardial cell injury by regulating the miR-145-5p-Binp3 axis. Mol. Med. Rep. 2018, 17, 2422–2430. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, S.J.; Zhu, S.; Jin, Y.; Cui, S.P.; Chen, J.Y.; Xiang, C.; Li, Q.Y.; He, C.; Zhao, S.F.; et al. Hypoxia-induced lncRNA-NUTF2P3-001 contributes to tumorigenesis of pancreatic cancer by derepressing the miR-3923/KRAS pathway. Oncotarget 2016, 7, 6000–6014. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Okada, T.; Fukushima, T.; Tsudzuki, T.; Sugiura, M.; Yukawa, Y. A novel hypoxic stress-responsive long non-coding RNA transcribed by RNA polymerase III in Arabidopsis. RNA Biol. 2012, 9, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirunavukkarasu, N.; Hossain, F.; Mohan, S.; Shiriga, K.; Mittal, S.; Sharma, R.; Singh, R.K.; Gupta, H.S. Genome-wide expression of transcriptomes and their co-expression pattern in subtropical maize (Zea mays L.) under waterlogging stress. PLoS ONE 2013, 8, e70433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Liang, K.; Fang, T.; Zhao, H.; Han, X.; Cai, M.; Qiu, F. A group VII ethylene response factor gene, ZmEREB180, coordinates waterlogging tolerance in maize seedlings. Plant Biotechnol. J. 2019, 17, 2286–2298. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Boil. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kumar, L.; Futschik, M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Zhou, X.; Ling, X.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38, W64–W70. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; De Peer, Y.V.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, D.J.; Conde, J.V.; Berckhan, S.; Prasad, G.; Mendiondo, G.M.; Holdsworth, M.J. Group VII ethylene response factors coordinate oxygen and nitric oxide signal transduction and stress responses in plants. Plant Physiol. 2015, 169, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Mano, Y.; Omori, F. Verification of QTL controlling root aerenchyma formation in a maize × teosinte” Zea nicaraguensis” advanced backcross population. Breed. Sci. 2008, 58, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Mano, Y.; Omori, F.; Kindiger, B.; Takahashi, H. A linkage map of maize × teosinte Zea luxurians and identification of QTLs controlling root aerenchyma formation. Mol. Breed. 2008, 21, 327–337. [Google Scholar] [CrossRef]
- Omori, F.; Mano, Y. QTL mapping of root angle in F2 populations from maize ‘B73′ × teosinte ‘Zea luxurians’. Plant Root 2008, 1, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Zheng, Y.; Zhang, Z.; Xu, S. Mapping of QTL associated with waterlogging tolerance during the seedling stage in maize. Ann. Bot. 2007, 99, 1067–1081. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, B.; Yu, F.; Li, L.; Wang, M.; Xue, Y.; Zhang, Z.; Yan, J.; Yue, B.; Zheng, Y.; et al. Identification of major QTL for waterlogging tolerance using genome-wide association and linkage mapping of maize seedlings. Plant Mol. Biol. Report. 2012, 31, 594–606. [Google Scholar] [CrossRef]
- Zaidi, P.H.; Rashid, Z.; Vinayan, M.T.; Almeida, G.D.; Phagna, R.K.; Babu, R. QTL mapping of agronomic waterlogging tolerance using recombinant inbred lines derived from tropical maize (Zea mays L.) germplasm. PLoS ONE 2015, 10, e0124350. [Google Scholar] [CrossRef]
- Zhang, Y.; Kong, X.; Dai, J.; Luo, Z.; Li, Z.; Lu, H.; Xu, S.; Tang, W.; Zhang, D.; Li, W.; et al. Global gene expression in cotton (Gossypium hirsutum L.) leaves to waterlogging stress. PLoS ONE 2017, 12, e0185075. [Google Scholar] [CrossRef]
- Lin, Y.; Li, W.; Zhang, Y.; Xia, C.; Liu, Y.; Wang, C.; Xu, R.; Zhang, L. Identification of genes/proteins related to submergence tolerance by transcriptome and proteome analyses in soybean. Sci. Rep. 2019, 9, 14688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapusta, A.; Feschotte, C. Volatile evolution of long noncoding RNA repertoires: Mechanisms and biological implications. Trends Genet. 2014, 30, 439–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Mcclung, C.R.; Zhang, C. Tick Tock: Circadian Regulation of Plant Innate Immunity. Annu. Rev. Phytopathol. 2017, 55, 287–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenham, K.; Mcclung, C.R. Integrating circadian dynamics with physiological processes in plants. Nat. Rev. Genet. 2015, 16, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tsai, H.; Joanito, I.; Wu, Y.; Chang, C.; Li, Y.; Wang, Y.; Hong, J.; Chu, J.; Hsu, C.; et al. LWD–TCP complex activates the morning gene CCA1 in Arabidopsis. Nat. Commun. 2016, 7, 13181. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Responsiveness | Element | Number of DElncRs | Ratio (%) |
|---|---|---|---|
| Anaerobic | ARE, GC-motif | 128 | 88.3 |
| Drought | MBS | 63 | 43.4 |
| Low-temperature | LTR | 45 | 31.0 |
| Abscisic acid | ABRE | 113 | 77.9 |
| Auxin | AuxRR-core, TGA-element | 63 | 43.4 |
| Gibberellin acid | TATC-box, GARE-motif, P-box | 75 | 51.7 |
| MeJA | TGACG-motif | 99 | 68.3 |
| Salicylic acid | TCA-element | 31 | 21.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, F.; Tan, Z.; Fang, T.; Tang, K.; Liang, K.; Qiu, F. A Comprehensive Transcriptomics Analysis Reveals Long Non-Coding RNA to Be Involved in the Key Metabolic Pathway in Response to Waterlogging Stress in Maize. Genes 2020, 11, 267. https://doi.org/10.3390/genes11030267
Yu F, Tan Z, Fang T, Tang K, Liang K, Qiu F. A Comprehensive Transcriptomics Analysis Reveals Long Non-Coding RNA to Be Involved in the Key Metabolic Pathway in Response to Waterlogging Stress in Maize. Genes. 2020; 11(3):267. https://doi.org/10.3390/genes11030267
Chicago/Turabian StyleYu, Feng, Zengdong Tan, Tian Fang, Kaiyuan Tang, Kun Liang, and Fazhan Qiu. 2020. "A Comprehensive Transcriptomics Analysis Reveals Long Non-Coding RNA to Be Involved in the Key Metabolic Pathway in Response to Waterlogging Stress in Maize" Genes 11, no. 3: 267. https://doi.org/10.3390/genes11030267
APA StyleYu, F., Tan, Z., Fang, T., Tang, K., Liang, K., & Qiu, F. (2020). A Comprehensive Transcriptomics Analysis Reveals Long Non-Coding RNA to Be Involved in the Key Metabolic Pathway in Response to Waterlogging Stress in Maize. Genes, 11(3), 267. https://doi.org/10.3390/genes11030267